

Abstract
In recent years, Enteromorpha prolifera blooms had serious impacts on costal environments and fisheries in China. Nevertheless, the effects of E. prolifera on microbial ecology remain unknown. In this study, for the first time, an Illumina sequencing analysis was used to investigate bacterial communities in source water, aquaculture ponds with E. prolifera, and an aquaculture pond in which E. prolifera -free. Principal coordinate and phylogenic analyses revealed obvious differences among the bacterial communities in the pond water with and without E. prolifera. Abundant bacterial taxa in the E. prolifera-containing pond were generally absent from the pond without E. prolifera. Interestingly, pond water with E. prolifera was dominated by Actinomycetales (> 50%), as well as by anaerobic bacteria in the underlying sediment (Desulfobacterales and Desulfuromonadales (> 20%). Pond water in which E. prolifera-free was dominated by Rhodobacterales (58.19%), as well as aerobic and facultative anaerobic bacteria in the sediment. In addition, the ecological functions of other dominant bacteria, such as Candidatus Aquiluna, Microcella spp., and Marivita spp., should be studied in depth. Overall, massive growth of E. prolifera will have serious effects on bacterial communities, and, thus, it will have an important impact on the environment. The novel findings in this study will be valuable for understanding green tides.
Introduction
Green tides are colossal accumulations of green macroalgae. These tides are associated with eutrophication, and they have major ecological and economic impacts on coastal environments. In recent years, massive blooms of Enteromorpha prolifera have become increasingly frequent in the coastal waters of China. These blooms are characterized by their large scale, severe environmental effects, and long durations, which have led to serious impacts on coastal landscapes, fisheries, and tourism in China. In June 2008, the world’s largest green tide, covering approximately 600 km2, occurred along the coast of the Yellow Sea in China [1, 2]. The direct economic loss caused by this tide was 1.32 billion yuan.
The excessive growth of some species of green algae, such as E. prolifera and Ulva spp., has been reported in green tide events in many parts of the world [2–5]. Green macroalgal blooms have substantially changed marine community structures and functions, and they have had negative impacts on commercial fish farms by creating anoxic conditions in enclosed environments. These adverse effects are usually associated with shading, biomass decomposition, and anoxia [6]. The multiple adverse effects of macroalgal blooms may thoroughly alter the function and structure of affected ecosystems [7]. Microalgae have been shown to negatively affect other benthic communities [8], microbenthic communities [9], and macrofauna [10]. Nevertheless, the effect of E. prolifera on aquatic microbial ecology has not been reported.
Aquaculture systems are relatively complex ecosystems that often harbor diverse bacterial communities. Microbial communities play major roles in aquaculture ponds, as they are closely linked with productivity, nutrient cycling, and the nutrition status and disease resistance of cultured animals [11–15]. Microbial processes, both aerobic and anaerobic, impact other water quality factors and the content of inorganic nutrients [14]. Through the activity of heterotrophic decomposers, nitrogen and phosphorus are recycled to stimulate primary production [11].
The general aim of the present study was to analyze bacterial communities in environments with E. prolifera, as well as in environments in which E. prolifera-free, using an Illumina MiSeq sequencing analysis. Our results show that the presence of E. prolifera alters bacterial communities, and they also reveal the nature of the microbial community in the E. prolifera-free environment. These results are of great significance for studying the interactions between bacteria and E. prolifera, and they provide information that will help predict and prevent E. prolifera blooms in aquaculture environments.
Material and methods
Study areas and sampling
Samples were collected from shrimp farms in Putian, Fujian Province, China. The area of each aquaculture pond is approximately 66,600 m2. The investigated culture ponds have the same seawater source, with an inlet. During sampling, outbreaks of E. prolifera were observed in most of the culture ponds (Fig 1). One pond did not have an occurrence of E. prolifera. Samples included seawater sources (seawater, SWW; sediment, SWS), three culture ponds with E. prolifera (water, CPEW1, CPEW2, and CPEW3; sediment, CPES1, CPES2, and CPES3), and one culture pond in which E. prolifera-free (water, CPW; sediment, CPS). Water samples were collected at 0.5m depth using an organic glass hydrophore. After each sample collection, 500 mL of water was firstly filtered by silk yarn to remove large particles, then was filtered through 0.22-μm filters (EMD Millipore, Billerica, MA, USA) under low (2 m bar) vacuum pressure. After filtration, membranes were put inside 1.5 ml centrifuge tube immediately and frozen in liquid nitrogen. Sediment samples from the ponds were taken at a 0–4-cm depth with a corer sampler. When the sediments were retrieved, air-exposed outer surface of the sediments was discarded and the central cores were taken with a sterile stainless. All of the samples were kept at −80°C before DNA extraction.
Fig 1. Samples collected from pond water with (a) and without (b) E. prolifera.

Ethics statement
No specific permits were required for the described field studies. Our study area is not privately owned or protected in any way. Our field studies did not involve endangered or protected species.
DNA extraction and Illumina MiSeq sequencing
The filters were cut into small pieces and transferred into the extraction tubes using ethanol-flamed forceps and surgical scissors. For each sample, triplicate filters were mixed, and DNA was extracted using the DNA Extraction Kit (Omega, USA) for bacterial communities. One gram (wet weight) of the sediment samples from each frozen section was extracted using a soil DNA extraction kit (Omega, USA) according to the manufacturer’s instructions and sterile techniques to avoid cross contamination.
Then, next-generation sequencing library construction and Illumina MiSeq sequencing were undertaken. Fifty nanograms of DNA was used to polymerase chain reaction (PCR) amplify the V3-V4 hypervariable regions of the bacteria 16S rRNA gene using the primers 319F (5′–ACTCCTACGGGAGGCAGCAG–3′) and 806R (5′–GGACTACHVGGGTWTCTAAT–3′). Indexed adapters were added to the ends of the 16S rRNA amplicons by a limited-cycle PCR. DNA libraries were validated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and quantified by Qubit and real-time PCR (Applied Biosystems, Carlsbad, CA, USA). DNA libraries were multiplexed and loaded onto an Illumina MiSeq analyzer (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Sequencing was performed using a 2 × 250 paired-end configuration, and image analysis and base calling were undertaken by the MiSeq control software of the MiSeq analyzer. All of the sequences can be downloaded from the National Center for Biotechnology Sequence Read Archive Database under the accession number SRR5665810.
Data analysis
Primer sequences were trimmed from the reads, and reads that contained Ns, were shorter than 400 bp, or had primer mismatches were excluded. Singleton amplicons were removed. Unique sequences were aligned and clustered into operational taxonomic units (OTUs) at a 97% cutoff using the QIIME platform with the UCLUST method. Then, the sequences were assigned to a taxonomy by the RDP classifier with a confidence cutoff of 0.8. Shannon diversity and observed species metrics were assessed using the QIIME platform. A principal coordinate analysis (PCoA) was conducted by computing the unweighted UniFrac distances among the samples using QIIME. Following the output taxonomy file classification, a heatmap was generated to depict the relative percentage of each classification of bacteria (y-axis) within each sample (x-axis clustering) using the GENE-E module. A taxonomy assignment of tags and OTUs was performed using the Global Alignment for Sequence Taxonomy method.
Results
Diversity analysis
A total of 1,321,884 raw reads that correspond to the exact sequences of the samples were obtained. After tag merges and quality control, we obtained a total of 1,280,418 tags (S1 Table). The results revealed that the compositions of the microbial communities were significant different among the different samples (analysis of variance (ANOVA), p < 0.01). Diversity entails both taxon richness and evenness, and our results demonstrated that both parameters were highest in the SWW and SWS, and lowest in the CPW and CPS (S1 Table). OTUs and Chao1 estimator supported the aforementioned diversity results, and there were significant differences (ANOVA, p < 0.01) between water with and without E. prolifera. Our results suggest that the ponds with E. prolifera had higher bacterial richness and diversity than ponds without E. prolifera.
PCoA
To further analyze the effect of E. prolifera on bacterial community composition (beta diversity), we summarized the unweighted UniFrac distances among the samples via a PCoA (Fig 2). This analysis clustered the microbial communities according to different samples (analysis of similarity (ANOSIM), p < 0.001). The clustering results also indicated that groups of pond sediment communities were more similar to each other than to those of the pond water samples. In addition, pond water samples with E. prolifera clustered together, and they were separated from inlet water and pond water without E. prolifera, indicating that bacterial communities in the CPEW were affected by E. prolifera.
Fig 2. Principal coordinate analysis (PCoA) of unweighted UniFrac distances between pond samples.

Blue symbols represent pond water samples. Orange symbols represent pond sediment samples. The given percentages represent the amount of variance explained by the corresponding axis.
Taxonomic composition
All sequences were classified from phylum to genus according to the program QIIME using the default setting, and 53 different phyla or groups were identified in the samples. Phylogeny more intuitively illustrates community differences among the different samples. Node size is expressed as abundance; the green coverage area represents low abundance; and the red coverage area represents high abundance. Phylogeny displays the development and evolution of bacterial species from the outside seawater to the pond water.
Phylum-order taxonomic distribution
Water samples and sediment samples displayed dissimilar 16S rRNA profiles, even for phylum-order level distributions (Fig 3). The SWW was primarily composed of Gammaproteobacteria (45.71%), Alphaproteobacteria (16.55%), Bacteroidetes (6.89%), and Actinobacteria (7.34%), and it was mainly dominated by the orders Alteromonadales (13.10%), Rhodobacterales (12.97%), Oceanospirillales (5.80%), Actinomycetales (5.55%), and Flavobacteriales (5.51%). The CPEW1, CPEW2, and CPEW3 were primarily composed of Actinobacteria (32.35%–52.05%), Alphaproteobacteria (10.16–27.14%), Bacteroidetes (15.08%–26.10%), and Gammaproteobacteria (6.86%–8.50%), and they were mainly dominated by the orders Actinomycetales (28.17%–50.29%), Rhodobacterales (4.79%–23.02%), and Flavobacteriales (9.98%–25.11%). The CPW was mainly composed of Alphaproteobacteria (67.42%), Bacteroidetes (11.63%), and Actinobacteria (13.63%), and it was mainly dominated by the orders Rhodobacterales (58.19%), Actinomycetales (13.61%), Sphingobacteriales (5.78%), and Flavobacteriales (5.01%).
Fig 3. Differences in bacterial communities between the source water (SWW and SWS), pond water with E. prolifera (CPEW and CPES), and pond water in which E. prolifera-free (CPW and CPS).

a, Phylum taxonomic level; b, Order taxonomic level.
Sediment samples had significantly different abundances of bacterial taxa (ANOVA, p < 0.01). The SWS had higher levels of Thiotrichales (10.03%) and Chromatiales (5.42%) than the CPES and CPS. The CPES had higher Desulfobacterales (13.73%–21.07%) abundances than the other sediment samples. The CPS had the highest abundances of Sphingobacteriales (11.50%) and Rhodobacterales (7.47%), while it had a relatively low abundance (2.37%) of Desulfobacterales.
Genus-level distribution
Heatmaps were used to intuitively display the differences in the relative abundances of different genera (Fig 4). The genera with the highest contribution to the ordination were selected. More than 60% of the total OTUs corresponded to Proteobacteria in each library. The most dominant groups of the total Proteobacteria tags were Alphaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria. The next most dominant groups of the total bacterial tags were Bacteroidetes and Actinobacteria. The dominant genera and families in the SWW were Vibrio (8.24%), Rhodobacteraceae (7.97%), Lactococcus (3.90%), and Piscirickettsiaceae (3.54%). The CPEW was characterized by Microbacteriaceae (11.87%–22.12%), Candidatus Aquiluna (8.64–23.02%), and Rhodobacteraceae (4.01%–14.41%). The CPW was dominated by Rhodobacteraceae (mainly Marivita 33.31%), Rhodobacteraceae (12.34%), Roseobacter (4.95%), Microbacteriaceae (8.19%), and Candidatus Aquiluna (5.29%). The CPES was dominated by many genera that were affiliated with Desulfobacterales (16.15%–22.10%) and Gammaproteobacteria incertae sedis (4.81%–7.83%). The CPS was dominated by Gammaproteobacteria incertae sedis (10.37%), Aliifodinibius (6.00%), Marinobacter (5.64%), Rhodobacteraceae (4.59%), and Chitinophagaceae (3.83%).
Fig 4. Heatmap showing the phylogenetic distribution between the source water (SWW and SWS), pond water with E. prolifera (CPEW and CPES), and pond water in which E. prolifera -free (CPW and CPS).
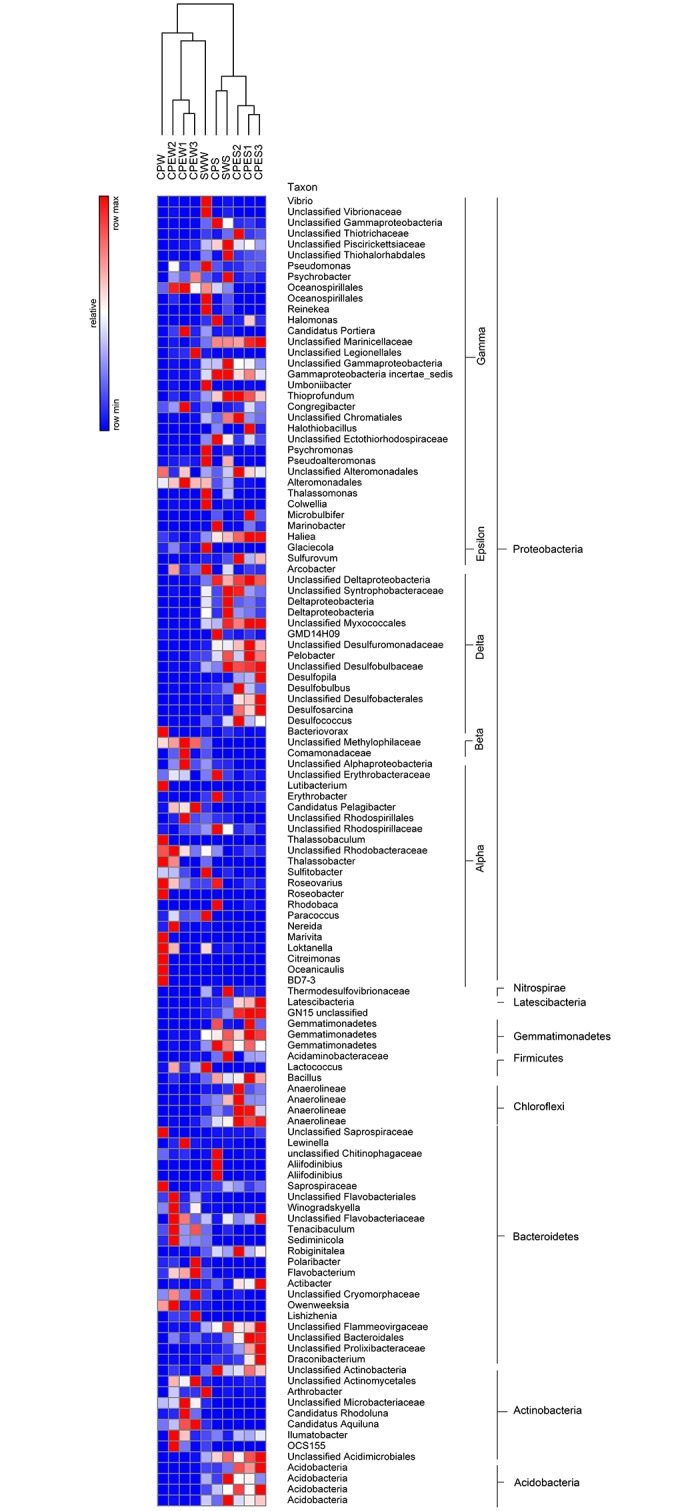
The relative percentage of each bacterial genus (y-axis) in each sample (x-axis clustering) is shown. Colored bars represent relative percentages.
The most abundant bacterial species, and a detrended correspondence analysis (DCA)
To explore the effect of E. prolifera on the bacterial communities, highly abundant (> 1%) species were illustrated with a heatmap (Fig 5) and a DCA (Fig 6). The DCA was performed to discern possible linkages between the samples and the dominant bacterial species. The eigenvalues for the first two multivariate axes were 0.7523 and 0.2094, respectively. The DCA plots showed a clear separation among the samples from different environments. The samples obtained from pond water with E. prolifera were mainly composed of Microcella, Candidatus Aquiluna rubra, Rhodobacteraceae, and Actinomycetales. In the pond water of CPW, the bacterial community was dominated by Marivita hallyeonensis (33.31%). Except for common bacterial classes, families, and genera (Gammaproteobacteria, Piscirickettsiaceae, and Marinicellaceae, respectively), some families affiliated with the order Desulfobacterales, such as Desulfuromonadaceae, Desulfobulbaceae, and Desulfobacteraceae, dominated the sediment samples from the E. prolifera-containing environments. These findings indicate that the samples obtained from pond water with E. prolifera had distinctly different species compositions, compared with the samples from the pond without E. prolifera (ANOVA, p < 0.01).
Fig 5. Heatmap showing the differences in the distributions of the most abundant bacterial species (> 1%) among the source water (SWW and SWS), pond water with E. prolifera (CPEW and CPES), and pond water in which E. prolifera -free (CPW and CPS).
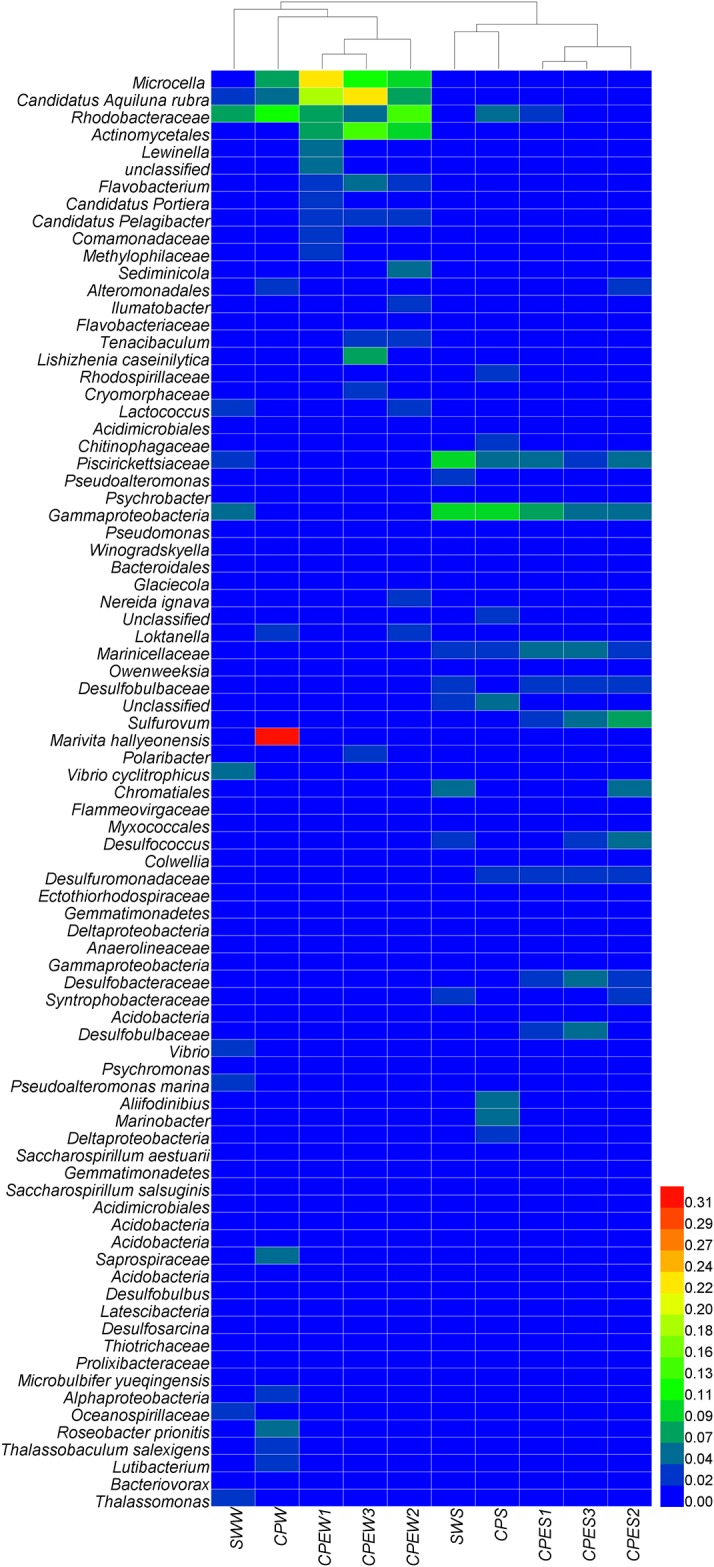
The relative percentage of each bacterial species (y-axis) in each sample (x-axis clustering) is shown. Colored bars represent relative percentages.
Fig 6. Detrended correspondence analysis (DCA) of bacterial community between the source of water (SWW and SWS), pond water with E. prolifera (CPEW and CPES), and pond water in which E. prolifera -free (CPW and CPS).

Discussion
The purpose of this study was to explore the bacterial diversity from source seawater to the aquaculture environment, and especially to compare bacterial communities in environments with or without E. prolifera. By applying large-scale 16S rRNA gene sequencing, the microbial populations of these environments were systematically investigated. Alphaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria, Actinobacteria, and Bacteroidetes were the dominant phyla, and they exhibited distinct distributions among the aquaculture environments (ANOVA, p < 0.01). Overall, our results indicate that E. prolifera changes the species composition and abundance of key bacterial taxa. As a result, E. prolifera would has a significant impact on the microbial community of aquaculture environments.
The diversity of the microbial community were lower in the culture ponds than those in source water. Similar with our previous study through clone library method [16], aquaculture environment will reduce the bacterial diversity and species number, leading to a simplified structure of the microbial community. That is mainly because culture environment had relatively steady of phy-chemical characteristic than external water body. For pond water with E. prolifera, bacterial community had higher diversity and richness than those in E. prolifera-free ponds. Macroalgal biomass not only represents an extremely large source of organic matter, and it is likely to compete with other organisms for inorganic nutrients. In this study, nutrients concentration (nitrogen and phosphorus nutrients) in E. prolifera ponds were much lower than those in E. prolifera-free ponds (S1 Fig). Other studies showed that in relatively low-nutrient conditions, bacterial communities exhibit relatively high diversities than those on concentrated media or environment [17, 18]. That should be mainly explained the reason that high bacterial diversity of bacterial community were found in E. prolifera ponds.
When we consider the dominant individual taxa, a distinct difference was observed between the CPE and CP environments (ANOVA, p < 0.01). The most prominent example of this difference is the presence in the CPW and CPS of many species that are rare in the CPEW and CPES samples (Fig 6). This result is in agreement with many studies that showed that phytoplankton affect the microbial community structure [19–21]. Another study suggested that macroalgae species may control their epibiotic bacterial communities [22]. Therefore, the distinct differences of the bacterial communities between the CPEW and CPW were caused by the outbreak of E. prolifera.
Heterotrophic bacteria were the major microbes in the CPEW, and they were dominated by Actinomycetales (49.80%), which mainly included the family Microbacteriaceae (22.12%) and Candidatus Aquiluna (19.66%). Blast results indicated that these Actinomycetales sequences shared 99% similarity with Microcella and Candidatus Aquiluna rubra (Fig 6). Candidatus Aquiluna rubra is represented by an aerobic, red-pigmented strain, which was isolated previously from a eutrophic pond [23]. Genome information suggests that Candidatus Aquiluna rubra is a photoheterotroph carrying actinorhodopsin, and that it has the smallest genome ever reported for a free-living member of the Actinobacteria [24]. The genome sequence of Actinorhodopsin-encoding marine bacterium suggested that this bacteria could be capable of both carbon fixation and rhodopsin-based phototrophy. This highlight the vast potential adaptation for novel metabolic strategies involving rhodopsins [25]. This high potential for photoheterotrophy could lead to an enhanced carbon turnover and/or nutrient acquisition and might be relevant also in terms of carbon sequestration through the microbial carbon pump [26]. Microcella is a Gram-positive, chemoorganotrophic bacterium [27]. Thus, the high abundances of Candidatus Aquiluna rubra and Microcella that were observed in the environment with E. prolifera make the ecology and evolution of this lineage interesting. However, information regarding these two bacteria only includes their taxonomic classifications, while their ecological functions have not been reported.
Heterotrophic microbes from E. prolifera habitats readily use the available dissolved organic matter [28]. However, a recent study showed that the active microbial community was strongly correlated with specific dissolved organic matter molecules, specifically compounds that are easily degradable [29]. The results of Gomez-Consarnau et al. (2012) support the view that specific carbon compounds trigger the growth of certain bacterial strains[30]. When E. prolifera decays, it produces large amounts of organic matter, which is preferred by certain heterotrophic bacteria. This increases the abundance of these bacteria, which ultimately alters the overall bacterial community. This phenomenon was illustrated by a study that showed that the microbenthic community metabolism clearly shifted to heterotrophic, while the photoautotrophic activity was located in the macroalgal canopy [31]. On the other hand, E. prolifera water will increase the abundance of Flavobacteriales genera, such as Sediminicola, Flavobacterium, Tenacibaculum, Owenweeksia and Winogradskyella (Fig 4). Some species of these genera are potential pathogens [32–34], and increase the infection chance for aquaculture animal. The annual economic losses caused by some Flavobacterium pathogens is extremely serious.
The CPW was dominated by Rhodobacterales (58.19%), which mainly included Marivita (33.31%), Rhodobacteraceae (12.34%), and Roseobacter (4.95%). The OTUs (31.8% of total sequence) of the genus Marivita shared 99% similarity with M. hallyeonensis. Recently, M. hallyeonensis has been described as a member of the genus Marivita, and it is often found in coastal environments [35]. Although some bacteria, such as Marivita, had very high abundances in pond water, their ecological functions remain unknown. There were also many dominant bacterial taxa, such as Roseobacter, Saprospiraceae, Thalassobaculum, Loktanella, and Lutibacterium, in the pond water of CPW, which were rare in the CPEW pond. In addition, the Rhodobacteraceae family is known to exhibit a diverse range of metabolic activity. These Rhodobacterales genera could undergo aerobic anoxygenic photosynthesis, sulfur oxidation, carbon monoxide oxidation, and DMSP demethylation [36]. They also can improve water quality through reducing concentration of ammonia [37], nitrate [36], and hydrogen sulfide [38]. Roseobacter strains can possibly establish an antagonistic beneficial bacterial community in the rearing environment of turbot larva and thereby limit the survival of pathogenic bacteria [39], and can act as probiotic microorganisms in aquaculture environment. In addition, a few roseobacters strains are capable of producing algaecidal substances, so-called roseobacticides [40], which are deadly to some algal strains. Rhodobacteraceae may therefore have played an important role in maintaining the health of the culture system [41], and maybe play important role in inhibiting the occurrence of E. prolifera. These are very meaningful questions that require in-depth study.
Major obvious effects of macroalgae are due to light attenuation of photosynthetic communities and the production of hypoxic or anoxic conditions. This leads to the production and accumulation of hydrogen sulfide in sediments below macroalgal mats [42, 43]. The CPES had high abundances of sulfate-reducing bacteria (Desulfobacterales and Desulfuromonadales, > 18% of the total sequences), while few such bacteria were detected in the CPS. Desulfobacterales are sulfate-reducing bacteria that reduce sulfates to sulfides to obtain energy. They are strictly anaerobic. Numerous members of the Desulfuromonadales are capable of anaerobic respiration by utilizing a variety of compounds as electron acceptors, including sulfur [44]. Desulfobacterales (Desulfobulbaceae and Desulfobacteraceae) have also been identified as the major sulfate-reducing bacteria in sediments beneath intensive aquaculture ponds [45, 46]. Asami et al. (2005) demonstrated that the sulfur cycle is accelerated in sediments beneath shellfish aquaculture ponds, and that certain taxa of sulfur-reducing bacteria were more abundant in these environments [45]. The dominance of anaerobic bacteria might be due to the generation of hypoxic or anoxic conditions by E. prolifera. This also explains the differences in the relative bacterial abundances, as well as the DCA, between the CPES and CPS samples. These sulfate-reducing bacteria could transform sulfur to hydrogen sulfide under anoxic conditions, which causes serious harm to aquaculture animals and environment.
Regarding bacterial taxa, the CPS was mainly characterized by aerobic and facultatively anaerobic species. These bacterial taxa included Aliifodinibius, Marinobacter, Rhodobacteraceae, and Chitinophagaceae, of which a few were detected in the CPES samples. Aliifodinibius, a genus of novel Gram-negative rods that are facultatively anaerobic, oxidase-negative, and catalase-positive, is a member of the phylum Bacteroidetes [47]. Nevertheless, its ecological function is unknown. Marinobacter has nitrous oxide reductase (nosZ) genes, and the capability of utilizing nitrite and nitrate to produce N2 as the aerobic denitrification product [48]. The CPS was mainly characterized by aerobic bacteria, which can reduce the damage caused by nitrite. These bacteria have the potential to improve the quality of the environment, which is important in aquaculture.
In conclusion, the results from this study demonstrated that there were obvious differences in the bacterial community structures between pond water with and without E. prolifera. The pond water with E. prolifera was dominated by Actinomycetales, and anaerobic sulfur-reducing bacteria were highly abundant in the underlying sediment. The pond water without E. prolifera was dominated by Rhodobacterales, and aerobic and anaerobic bacteria were highly abundant in the underlying sediment. These findings indicate that the massive growth of E. prolifera had a significant effect on the microbial community, and that E. prolifera has the potential to harm the environment.
Supporting information
(TIF)
(DOCX)
Data Availability
All of the sequences can be downloaded from the National Center for Biotechnology Sequence Read Archive Database under the accession number SRR3714433.
Funding Statement
This research was supported by the Project of Guangdong Science and Technology Department (2016A020221024 and 2017A020216008), the Project of Fujian Science and Technology Department (2015N0013 and 2016I1002), the National Natural Science Foundation of China (41406130), the national Spark Program project (2015GA720002), the Key Laboratory for Ecological Environment in Coastal Areas, State Oceanic Administration (201507), the Knowledge Innovation Program of the Chinese Academy of Sciences (SQ201220), and Science and Technology plan project of Nantong city (MS12016058).
References
- 1.China. Ocean News (2008).
- 2.Liu D, Keesing JK, Dong Z, Zhen Y, Di B, Shi Y, et al. Recurrence of the world’s largest green-tide in 2009 in Yellow Sea, China: Porphyra yezoensis aquaculture rafts confirmed as nursery for macroalgal blooms. Marine Pollution Bulletin. 2010;60(9):1423–32. 10.1016/j.marpolbul.2010.05.015 [DOI] [PubMed] [Google Scholar]
- 3.Morand P, Merceron M. Macroalgal population and sustainability. J Coastal Res. 2005:1009–20. [Google Scholar]
- 4.Valiela I, McClelland J, Hauxwell J, Behr PJ, Hersh D, Foreman K. Macroalgal blooms in shallow estuaries: controls and ecophysiological and ecosystem consequences. Limnol Oceanogr. 1997;42(5):1105–18. [Google Scholar]
- 5.Hu C, Li D, Chen C, Ge J, Muller-Karger FE, Liu J, et al. On the recurrent Ulva prolifera blooms in the Yellow Sea and East China Sea. J Geophys Res: Oceans. 2010;115(C5). [Google Scholar]
- 6.Nelson TA, Haberlin K, Nelson AV, Ribarich H, Hotchkiss R, Alstyne KLV, et al. Ecological and physiological controls of species composition in green macroalgal blooms. Ecology. 2008;89(5):1287–98. [DOI] [PubMed] [Google Scholar]
- 7.Valiela I, McClelland J, Hauxwell J, Behr PJ, Hersh D, Foreman K. Macroalgal blooms in shallow estuaries: controls and ecophysiological and ecosystem consequences. Limnology and Oceanography. 1997;42(5):1105–18. [Google Scholar]
- 8.Den Hartog C. Suffocation of a littoral Zostera bed by Enteromorpha radiata. Aquat Bot. 1994;47(1):21–8. [Google Scholar]
- 9.Sundback K, Jonsson B, Nilsson P, Lindstrom I. Impact of accumulating drifting macroalgae on a shallow-water sediment system: an experimental study. Mar Ecol- Prog Ser. 1990;58(3). [Google Scholar]
- 10.Norkko A, Bonsdorff E. Rapid zoobenthic community responses to accumulations of drifting algae. Mar Ecol- Prog ser. 1996;131(1):143–57. [Google Scholar]
- 11.Moriarty DJ. The role of microorganisms in aquaculture ponds. Aquaculture. 1997;151(1):333–49. [Google Scholar]
- 12.Verschuere L, Rombaut G, Sorgeloos P, Verstraete W. Probiotic Bacteria as Biological Control Agents in Aquaculture. Microbiol Molecul Biol R. 2000;64(4):655–71. 10.1128/mmbr.64.4.655-671.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatesoupe FJ. The use of probiotics in aquaculture. Aquaculture. 1999;180(1–2):147–65. 10.1016/S0044-8486(99)00187-8. [DOI] [Google Scholar]
- 14.Crab R, Avnimelech Y, Defoirdt T, Bossier P, Verstraete W. Nitrogen removal techniques in aquaculture for a sustainable production. Aquaculture. 2007;270(1–4):1–14. 10.1016/j.aquaculture.2007.05.006. [DOI] [Google Scholar]
- 15.Zhang XZ, Dai LP, Wu ZH, Jian JC, Lu YS. Expression pattern of heat shock protein 90 gene of humphead snapper Lutjanus sanguineus during pathogenic Vibrio harveyi stress. J Fish Biol. 2011;79(1):178–93. 10.1111/j.1095-8649.2011.03012.x [DOI] [PubMed] [Google Scholar]
- 16.Wang C-z, Lin G-r, Yan T, Zheng Z-p, Chen B, Sun F-l. The cellular community in the intestine of the shrimp Penaeus penicillatus and its culture environments. Fish Sci. 2014;80:1001–7. 10.1007/s12562-014-0765-3 [DOI] [Google Scholar]
- 17.Balkwill DL. Numbers, diversity, and morphological characteristics of aerobic, chemoheterotrophic bacteria in deep subsurface sediments from a site in South Carolina. Geomicrobiology Journal. 1989;7(1–2):33–52. [Google Scholar]
- 18.Connon SA, Giovannoni SJ. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Applied and environmental microbiology. 2002;68(8):3878–85. 10.1128/AEM.68.8.3878-3885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuhrman JA. Microbial community structure and its functional implications. Nature. 2009;459(7244):193–9. 10.1038/nature08058 [DOI] [PubMed] [Google Scholar]
- 20.Weisse T, Müller H, Pinto-Coelho RM, Schweizer A, Springmann D, Baldringer G. Response of the microbial loop to the phytoplankton spring bloom in a large prealpine lake. Limnology and Oceanography. 1990;35(4):781–94. [Google Scholar]
- 21.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336(6081):608–11. 10.1126/science.1218344 [DOI] [PubMed] [Google Scholar]
- 22.Lachnit T, Blümel M, Imhoff JF, Wahl M. Specific epibacterial communities on macroalgae: phylogeny matters more than habitat. Aquatic Biology. 2009;5(2):181–6. [Google Scholar]
- 23.Hahn MW. Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. International Journal of Systematic and Evolutionary Microbiology. 2009;59(1):112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang I, Lee K, Yang S-J, Choi A, Kang D, Lee YK, et al. Genome Sequence of “Candidatus Aquiluna” sp. Strain IMCC13023, a Marine Member of the Actinobacteria Isolated from an Arctic Fjord. Journal of Bacteriology. 2012;194(13):3550–1. 10.1128/JB.00586-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma AK, Sommerfeld K, Bullerjahn GS, Matteson AR, Wilhelm SW, Jezbera J, et al. Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. The ISME journal. 2009;3(6):726–37. 10.1038/ismej.2009.13 [DOI] [PubMed] [Google Scholar]
- 26.Jiao N, Herndl GJ, Hansell DA, Benner R, Kattner G, Wilhelm SW, et al. Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nature Reviews Microbiology. 2010;8(8):593–9. 10.1038/nrmicro2386 [DOI] [PubMed] [Google Scholar]
- 27.Veríssimo A. Microcella Bergey's Manual of Systematics of Archaea and Bacteria: John Wiley & Sons, Ltd; 2015. [Google Scholar]
- 28.Pregnall A. Release of dissolved organic carbon from the estuarine intertidal macroalga Enteromorpha prolifera. Marine Biology. 1983;73(1):37–42. [Google Scholar]
- 29.Osterholz H, Singer G, Wemheuer B, Daniel R, Simon M, Niggemann J, et al. Deciphering associations between dissolved organic molecules and bacterial communities in a pelagic marine system. ISME J. 2016;10(7):1717–30. 10.1038/ismej.2015.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gómez-Consarnau L, Lindh MV, Gasol JM, Pinhassi J. Structuring of bacterioplankton communities by specific dissolved organic carbon compounds. Environmental Microbiology. 2012;14(9):2361–78. 10.1111/j.1462-2920.2012.02804.x [DOI] [PubMed] [Google Scholar]
- 31.Corzo A, Van Bergeijk S, Garcia-Robledo E. Effects of green macroalgal blooms on intertidal sediments: net metabolism and carbon and nitrogen contents. Marine Ecology Progress Series. 2009;380:81–93. [Google Scholar]
- 32.Ludwig W, Euzéby J, Whitman WB. Taxonomic outlines of the phyla Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes Bergey’s Manual® of Systematic Bacteriology: Springer; 2010. p. 21–4. [Google Scholar]
- 33.Duchaud E, Boussaha M, Loux V, Bernardet J-F, Michel C, Kerouault B, et al. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nature biotechnology. 2007;25(7):763–9. 10.1038/nbt1313 [DOI] [PubMed] [Google Scholar]
- 34.Romero M, Avendaño-Herrera R, Magariños B, Cámara M, Otero A. Acylhomoserine lactone production and degradation by the fish pathogen Tenacibaculum maritimum, a member of the Cytophaga–Flavobacterium–Bacteroides (CFB) group. FEMS microbiology letters. 2010;304(2):131–9. 10.1111/j.1574-6968.2009.01889.x [DOI] [PubMed] [Google Scholar]
- 35.Yoon J-H, Kang S-J, Lee J-S. Marivita geojedonensis sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2013;63(2):423–7. [DOI] [PubMed] [Google Scholar]
- 36.Brinkhoff T, Giebel H-A, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 2008;189(6):531–9. 10.1007/s00203-008-0353-y [DOI] [PubMed] [Google Scholar]
- 37.Voget S, Wemheuer B, Brinkhoff T, Vollmers J, Dietrich S, Giebel H-A, et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2015;9(2):371–84. 10.1038/ismej.2014.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenk S, Moraru C, Hahnke S, Arnds J, Richter M, Kube M, et al. Roseobacter clade bacteria are abundant in coastal sediments and encode a novel combination of sulfur oxidation genes. The ISME journal. 2012;6(12):2178–87. 10.1038/ismej.2012.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hjelm M, Bergh O, Riaza A, Nielsen J. Selection and identification of autochthonous potential probiotic bacteria from turbot larvae (Scophthalmus maximus) rearing units. Systematic and Applied Microbiology. 2004;27(3):360 10.1078/0723-2020-00256 [DOI] [PubMed] [Google Scholar]
- 40.Seyedsayamdost MR, Case RJ, Kolter R, Clardy J. The Jekyll-and-Hyde chemistry of Phaeobacter gallaeciensis. Nature chemistry. 2011;3(4):331–5. 10.1038/nchem.1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cardona E, Gueguen Y, Magré K, Lorgeoux B, Piquemal D, Pierrat F, et al. Bacterial community characterization of water and intestine of the shrimp Litopenaeus stylirostris in a biofloc system. Bmc Microbiology. 2016;16(1):157 10.1186/s12866-016-0770-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nedergaard RI, Risgaard-Petersen N, Finster K. The importance of sulfate reduction associated with Ulva lactuca thalli during decomposition: a mesocosm experiment. J Exp Mar Biol Ecol. 2002;275(1):15–29. [Google Scholar]
- 43.Lomstein BA, Guldberg LB, Neubauer A-TA, Hansen J, Donnelly A, Herbert RA, et al. Benthic decomposition of Ulva lactuca: a controlled laboratory experiment. Aquat Bot. 2006;85(4):271–81. [Google Scholar]
- 44.Garrity G, Staley JT, Boone DR, De Vos P, Goodfellow M, Rainey FA, et al. Bergey's Manual® of Systematic Bacteriology: Volume Two: The Proteobacteria: Springer Science & Business Media; 2006. [Google Scholar]
- 45.Asami H, Aida M, Watanabe K. Accelerated sulfur cycle in coastal marine sediment beneath areas of intensive shellfish aquaculture. Applied and Environmental Microbiology. 2005;71(6):2925–33. 10.1128/AEM.71.6.2925-2933.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kondo R, Mori Y, Sakami T. Comparison of sulphate-reducing bacterial communities in Japanese fish farm sediments with different levels of organic enrichment. Microbes and Environments. 2011;27(2):193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y-X, Liu J-H, Xiao W, Ma X-L, Lai Y-H, Li Z-Y, et al. Aliifodinibius roseus gen. nov., sp. nov., and Aliifodinibius sediminis sp. nov., two moderately halophilic bacteria isolated from salt mine samples. Int J Syst Evol Microbiol. 2013;63(8):2907–13. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Ai G-M, Miao L-L, Liu Z-P. Marinobacter strain NNA5, a newly isolated and highly efficient aerobic denitrifier with zero N2O emission. Bioresource Technol. 2016;206:9–15. 10.1016/j.biortech.2016.01.066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(DOCX)
Data Availability Statement
All of the sequences can be downloaded from the National Center for Biotechnology Sequence Read Archive Database under the accession number SRR3714433.