

Summary
The mitotic checkpoint ensures proper segregation of chromosomes by delaying anaphase until all kinetochores are bound to microtubules. This inhibitory signal is composed of a complex containing Mad2, which inhibits anaphase progression. The complex can be disassembled by p31comet and TRIP13; however, TRIP13 knockdown has been shown to cause only a mild mitotic delay. Overexpression of checkpoint genes, as well as TRIP13, is correlated with chromosomal instability (CIN) in cancer, but the initial effects of Mad2 overexpression are prolonged mitosis and decreased proliferation. Here we show that TRIP13 overexpression significantly reduced, and TRIP13 reduction significantly exacerbated, the mitotic delay associated with Mad2 overexpression but not that induced by microtubule depolymerization. The combination of Mad2 overexpression and TRIP13 loss reduced the ability of checkpoint complexes to disassemble and significantly inhibited the proliferation of cells in culture and tumor xenografts. These results identify an unexpected dependency on TRIP13 in cells overexpressing Mad2.
Keywords: Mitotic checkpoint, spindle assembly checkpoint, Mad2, TRIP13, cancer, cell cycle, mitosis
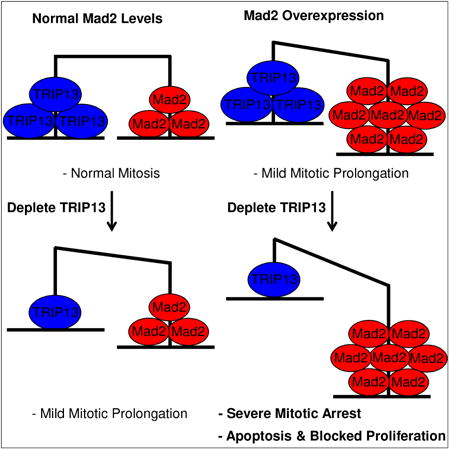
Graphical abstract
TRIP13 is a putative mitotic checkpoint silencing protein. However, depletion of TRIP13 causes only mild mitotic exit phenotypes. Marks et al. find that TRIP13 becomes critical for mitotic exit in Mad2-overexpressing cells. Both proteins are co-overexpressed in cancer, and TRIP13 may be a therapeutic target in Mad2-overexpressing tumors.
Introduction
The mitotic checkpoint ensures proper segregation of chromosomes during mitosis by generating a diffusible, inhibitory signal to the E3 ubiquitin ligase, Anaphase Promoting Complex (APC/C). Inhibition of the APC/C blocks anaphase progression until sister chromatids are properly attached to opposite spindle poles by microtubules bound to their kinetochores (Foley and Kapoor, 2012; Musacchio, 2015). Once the checkpoint signal is quenched, the APC/C can ubiquitinate and degrade key mitotic substrates, such as cyclin B1 and securin, which allows anaphase progression and mitotic exit. Kinetochores not bound by microtubules can generate this signal by templating the conversion of Mad2 from its inactive, open, conformer (O-Mad2), to its closed, active, conformer (C-Mad2) (De Antoni et al., 2005). Together with BubR1, Bub3, and Cdc20, C-Mad2 can form mitotic checkpoint complexes (MCC) that can bind to and inhibit APC/C activity (Sudakin et al., 2001). In addition to the formation of MCC at unoccupied kinetochores, recent work has shown that Mad1-Mad2 complexes present at nuclear pores during interphase also can induce the formation of MCC prior to mitosis, and that this process is critical for basal mitotic timing (Rodriguez-Bravo et al., 2014).
Once the checkpoint is satisfied, the signal is quenched, in part, by p31comet (Habu et al., 2002; Xia et al., 2004). p31comet mimics the structure of C-Mad2, which is thought to allow it to cap the templating reaction and prevent further production of C-Mad2 (Mapelli et al., 2006; Yang et al., 2007). Additionally, p31comet stimulates the disassembly of MCC in mitotically arrested cell extracts, and this process is dependent on ATP (Teichner et al., 2011; Westhorpe et al., 2011). Recently, an AAA+ ATPase, TRIP13, has been shown to aid in this disassembly reaction (Eytan et al., 2014; Wang et al., 2014). Mechanistically, TRIP13 is able to catalyze the conversion of C-Mad2 to inactive O-Mad2, with p31comet serving as an adaptor to recruit TRIP13 to C-Mad2 containing complexes (Ye et al., 2015). However, cells with TRIP13 transiently depleted by siRNA have only a mildly prolonged mitosis (Wang et al., 2014). Further, full knockout of TRIP13 in HeLa and HCT116 cells is tolerable, with knockout cells having a relatively normal unperturbed mitosis (Ma and Poon, 2016), inconsistent with a requirement for TRIP13 to quench the checkpoint. Surprisingly, these TRIP13 knockout cells could not activate a robust checkpoint in response to nocodazole, suggesting that TRIP13 may play a role in both activation and inactivation of checkpoint signaling. A defect in checkpoint signaling has also been seen in C. elegans when the TRIP13 homolog, PCH-2, is mutated (Nelson et al., 2015). In these mutants, there is a defect in Mad2 recruitment to kinetochores, which was partially reversible by p31comet disruption, but this was not observed in the human cell line knockout. Thus, TRIP13's role in the mitotic checkpoint remains incompletely understood.
Dysregulation of the mitotic checkpoint, and in turn the process of sister chromatid separation, has the potential to lead to chromosome mis-segregation and gains/losses of chromosomes in daughter cells. Aneuploidy, the state of having gained or lost chromosomes, is a hallmark of cancer. Surprisingly, the checkpoint is only rarely disrupted in cancer cells (Tighe et al., 2001). Conversely, mitotic checkpoint genes are frequently overexpressed in cancer and overexpression of multiple checkpoint genes is correlated with chromosomal instability in human tumors (Carter et al., 2006). Previous work, from our lab and others, has shown that the loss of major tumor suppressor pathways, such as the Rb or p53 pathways, can lead to transcriptional upregulation of checkpoint genes through E2F sites in their promoters (Hernando et al., 2004; Schvartzman et al., 2011). Overexpression of Mad2 is required for the high levels of the chromosomal instability in cells that have lost these tumor suppressors (Schvartzman et al., 2011). Further, overexpression of Mad2 by itself can lead to aneuploidy and tumorigenesis in mice (Sotillo et al., 2007). It has also been shown that overexpression can induce hyperstabilization of kinetochore-microtubule attachments and thus lead to chromosome mis-segregation (Kabeche and Compton, 2012). Finally, transient overexpression of Mad2 in a Kras-induced lung cancer model induced genomic instability, allowing these tumors to become independent of the initiating oncogene (Sotillo et al., 2010). For these reasons, we sought to identify potential vulnerabilities in Mad2-overexpressing cells.
Mad2 overexpression can cause a prolonged mitosis and aneuploidy in mouse embryonic fibroblasts (Sotillo et al., 2007). In normal cells, prolonged mitosis can lead to p53 dependent G1 arrest or mitotic cell death (Gascoigne and Taylor, 2008; Vogel et al., 2004), so it is surprising that Mad2 overexpression is so well tolerated in cancer. Interestingly, TRIP13 is also overexpressed in cancer (Banerjee et al., 2014) and is part of the same gene signature correlated with chromosomal instability (Carter et al., 2006). Given that overexpression of TRIP13 could oppose the effects of Mad2 overexpression, we hypothesized that TRIP13 may be of increased importance for mitotic exit in Mad2-overexpressing cells. We found that TRIP13 overexpression had minimal effects on basal mitotic timing or nocodazole arrest, but was able to antagonize the cell cycle effects of Mad2 overexpression. Additionally, we found that while reducing levels of TRIP13 in normal cells had only modest effects on mitotic timing, in the presence of Mad2 overexpression TRIP13 reduction caused extremely prolonged mitoses and significantly reduced the ability of Mad2-overexpressing cells to proliferate. The combination of TRIP13 reduction with Mad2 overexpression also significantly reduced the ability of these cells to form tumors in a xenograft model. These results demonstrate the increased importance of TRIP13 in the context of Mad2 overexpression, and further define the roles of TRIP13 in the mitotic checkpoint. The results also suggest TRIP13 may be a therapeutic target for Mad2-overexpressing tumors.
Results
TRIP13 is overexpressed in Mad2 overexpressing tumors
We investigated the correlation between the expression of TRIP13 and Mad2 in human tumors using the cBioPortal database (cBioPortal.org (Cerami et al., 2012; Gao et al., 2013)). Across multiple datasets from different tumor types, we found that levels of TRIP13 correlated closely with those of Mad2 (Figure 1A). Many mitotic checkpoint genes, including Mad2, have a core E2F/CDE transcription factor binding site in their promoters, and this motif is also found near the transcription start site for TRIP13 (Figure 1B). Further, ChIP-seq data from the Encode database, on the UCSC genome browser (https://genome.ucsc.edu/ENCODE/), showed that for three different E2F proteins (E2F1, E2F4, E2F6), each could bind near the start site of TRIP13 in HeLa cells (Figure 1C). Mouse embryonic fibroblasts deficient for the 3 Rb family members (Rb, p107, p130) have elevated levels of Mad2, and we found that these cells also have elevated levels of TRIP13 (Figure 1D). Together, this suggests that expression of TRIP13 is potentially regulated similarly to Mad2 and other checkpoint genes. This is consistent with a recent finding using single-cell transcriptome analysis, identifying correlated Mad2 and TRIP13 expression in mouse 3T3 cells (Macosko et al., 2015). Thus, while TRIP13 is broadly overexpressed in cancer, it is most commonly overexpressed in the context of Mad2 overexpression.
Figure 1. TRIP13 and Mad2 expression are correlated in cancer.
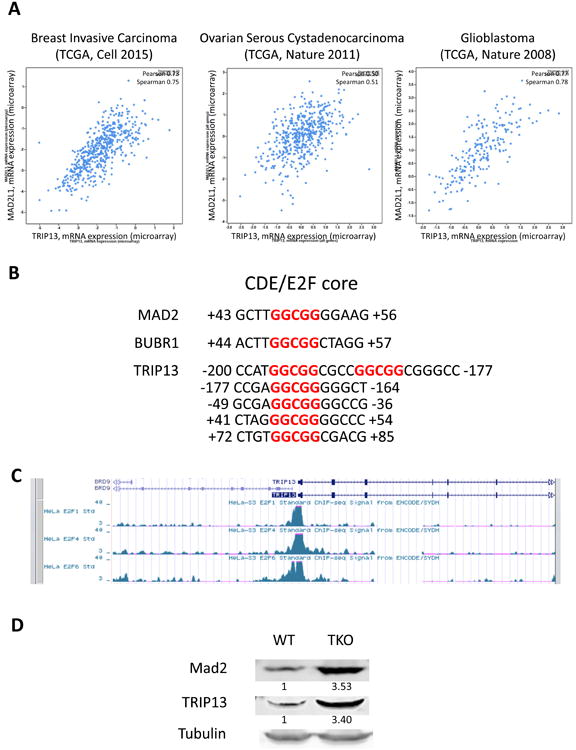
(A) Mad2 versus TRIP13 microarray expression data in three representative datasets from cBioPortal.org. (B) GGCGG CDE/E2F core sites present between -200 and +100 from the transcription start sites of MAD2, BUBR1, and TRIP13. (C) ChIP-Seq data from Encode database (https://genome.ucsc.edu/ENCODE/) for E2F1, E2F4, E2F6 binding near TRIP13 transcription start site in HeLa cells. (D) Mad2 and TRIP13 expression are both elevated by Western blot in Wild Type (WT) and Rb-family (Rb, p107, p130) deficient TKO MEFs. Quantification is shown below corresponding bands.
TRIP13 overexpression blocks the effects of Mad2 overexpression
While TRIP13 has been shown to aid in mitotic checkpoint complex disassembly, little is known about the effect of TRIP13 overexpression on a normal versus perturbed mitosis. To study the effect of TRIP13 overexpression in the context of Mad2 overexpression, we used human retinal pigment epithelial cells (RPE2) containing a doxycycline inducible HA-Mad2 construct (Sotillo et al., 2007), which we called RPE-M. Induction of Mad2 is observed in these cells by 24 hours (Figure 2A). As expected, the overexpression of Mad2 caused an approximately 4-fold increase in mitotic duration from an average of 44 minutes to 217 minutes (Figure 2B). We transduced this RPE-M cell line with a constitutively expressing TRIP13-GFP vector (Figure 2A). Alone, cells with TRIP13 overexpression had a similar basal mitotic duration compared to parental RPE cells. However, TRIP13 overexpression significantly reduced the prolongation of mitosis induced by Mad2 overexpression from an increase of 173 minutes (217-44 minutes) to an increase of only 10 minutes (48-38 minutes) (Figure 2B).
Figure 2. TRIP13 overexpression blunts Mad2 overexpression phenotypes.
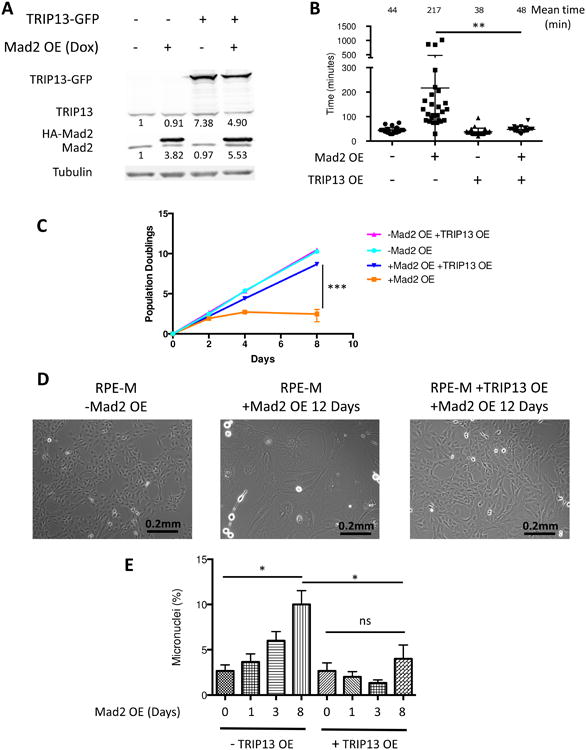
(A) Western blot of RPE2 cells transduced with dox-inducible HA-Mad2 construct and/or constitutive TRIP13-GFP construct with or without doxycycline addition for 24 hours. Total quantification (endogenous + exogenous) is shown below corresponding bands. (B) Mitotic timing of RPE-M or T13G cells, +/- 24 hours doxycycline, measured by cell rounding in time-lapse images. n ≥20 cells for each condition. (C) Equal numbers of RPE-M and T13G cells were plated with or without doxycycline on day 0 and were counted and replated on day 2 and day 4, and counted on day 8. Population doublings of RPE-M and T13G cells is plotted over time. n=3 independent experiments (D) Bright field imaging of RPE-M cells without doxycycline treatment and RPE-M and T13G cells with doxycycline treatment for 12 days. (E) Micronuclei were counted in 100 cells in triplicate with DAPI staining in RPE-M or T13G cells treated with doxycycline for 0, 1, 3, or 8 days. * indicates p < 0.05, ** p < 0.01, *** p < 0.001, ns indicates not significant (p > 0.05); Unpaired t test. Data are represented as mean± SD.
Prolonged mitosis leads to a p53-dependent G1 arrest (Vogel et al., 2004). Mad2 overexpression in p53 wild type RPE cells led to an accumulation of large, flat, cells of senescent appearance, and caused a dramatic decrease in proliferation over the course of 8 days (Figures 2C and 2D). TRIP13 overexpression prevented the changes in cell morphology and significantly rescued the ability of Mad2-overexpressing cells to proliferate (Figures 2C and 2D). Mad2 overexpression also induced an accumulation of cells with abnormal nuclear morphologies such as micronuclei, indicative of abnormal chromosome segregation at mitotic exit, and these phenotypes were also blocked by TRIP13 overexpression (Figure 2E). Thus while TRIP13 overexpression has little effect on mitotic duration or micronuclei formation in a normal mitosis, it is able to strongly antagonize these effects in Mad2-overexpressing cells. We speculate that tumor-sustaining proliferation rates and chromosome mis-segregation events in Mad2-overexpressing cells requires a finely tuned balance of Mad2 and TRIP13 levels (see below and Discussion).
TRIP13 is critical for mitotic exit in Mad2-overexpressing cells
Given the ability of TRIP13 overexpression to block a Mad2-overexpression induced arrest, we wanted to determine if these cells had an increased dependence on TRIP13 for mitotic exit. To study the ability of Mad2-overexpressing cells to proliferate over time, we knocked down p53 to allow them to continue to divide in the presence of prolonged mitosis (Figure S1). We tested the effect of TRIP13 knockdown in these cells using two siRNAs previously used to study the effects of TRIP13 loss (Wang et al., 2014). Cells were transfected with either TRIP13 siRNA or control siRNA and were subsequently treated with or without doxycycline to induce Mad2 overexpression. We followed the effect of TRIP13 knockdown on mitotic duration by time-lapse imaging and counted cell numbers to assess the effects on proliferation (Figure 3A).
Figure 3. TRIP13 knockdown causes a severely prolonged mitosis in Mad2-overexpressing cells.

(A) Schematic showing transfection of RPE-M p53 knockdown cells with TRIP13 or control siRNAs followed by doxycycline addition after 3 days followed by analysis by cell counting and time-lapse imaging. (B) Western blot of TRIP13 knockdown with two independent siRNAs with or without 24 hours of doxycycline treatment. Total quantification (endogenous + exogenous) is shown below corresponding bands. (C) Mitotic timing and fate of RPE-M cells transfected with control or TRIP13 siRNA +/- 24 hours doxycycline measured by cell rounding in time-lapse images. n ≥20 cells for each condition. *** indicates p < 0.001; Unpaired t test. Data are represented as mean± SD. See also Figures S1, S2, S3 and S4
Both siRNAs were capable of substantially reducing the levels of TRIP13 in inducible Mad2-overexpressing cells (Figure 3B). Consistent with previous studies (Wang et al., 2014), knockdown of TRIP13 with either siRNA caused a mild prolongation of mitosis (1.5-2 fold) in the absence of Mad2 overexpression (Figure 3C). However, while Mad2 overexpression alone caused an average ∼3-4 fold increase in mitotic duration, these cells were extremely dependent on TRIP13 for mitotic exit. Knockdown of TRIP13 with either siRNA in Mad2-overexpressing cells caused a severe mitotic arrest, with cells arresting in mitosis on average for more than 20-fold longer than an unperturbed mitosis (Figures 3C and S2).
To test the effect of TRIP13 knockdown in a system where Mad2 is overexpressed due to loss of the Rb pathway, we used a vector expressing the HPV E6 and E7 proteins, which inhibit p53 and Rb function (Halbert et al., 1992; Yim and Park, 2005). Transduction of parental RPE cells with the vector led to increased Mad2 and TRIP13 expression, as expected for E2F target genes (Figure S3A). Further, expression of the E6/E7 proteins led to an increase in mitotic timing, from an average of 39 minutes to 53 minutes (Figure S3B). TRIP13 knockdown further prolonged mitosis in E6/E7 transduced cells to an average of 118 minutes. These results demonstrate the ability of TRIP13 loss to exacerbate mitotic arrest in a model of oncogene-induced Mad2 overexpression.
To generalize our findings to other cell lines, we tested the effects of TRIP13 knockdown and transient Mad2 overexpression in two breast cancer cell lines, MCF7 and MDA-MB-436 (Figure S4A). To transiently overexpress Mad2, we transfected cells with a Myc-FLAG-tagged human Mad2 expression vector. Neither cell line had significantly elevated Mad2 or TRIP13 levels compared to RPE cells (data not shown). High Mad2 levels may be selected against in cell lines grown in culture for prolonged periods. Both knockdown of TRIP13 and overexpression of Mad2 led to statistically significant increases in mitotic duration in both cell lines (Figure S4B). The combination of Mad2 overexpression and TRIP13 knockdown led to extremely prolonged mitoses, similar to our results in RPE cells (Figure S4B). Thus, the dependence of Mad2-overexpressing cells on TRIP13 is not specific to RPE cells.
TRIP13 knockdown is synthetic lethal with Mad2 overexpression
We tested the growth of cells with TRIP13 knockdown with or without Mad2 overexpression to confirm that this increased mitotic arrest was associated with a decrease in proliferation. While either TRIP13 knockdown or Mad2 overexpression alone caused only a mild decrease in proliferation, both TRIP13 siRNAs induced an almost complete block of cell proliferation upon Mad2 induction (Figure 4A). Time-lapse imaging showed evidence of cells undergoing a mitotic cell death (Figure S2), and we observed an increase in γH2AX and cleaved caspase 3 by Western blot, only in cells with the combination of TRIP13 siRNA and Mad2 overexpression (Figure 4B).
Figure 4. TRIP13 knockdown and Mad2 overexpression combine to reduce cell growth in vitro and in xenografts.

(A) Equal numbers of RPE-M cells transfected with TRIP13 or control siRNAs were plated with or without doxycycline on day 0 and were counted and replated on day 1 and counted on day 3. Population doublings of cells is plotted over time. n=3 independent experiments (B) Western blot of γH2AX and cleaved caspase 3 on RPE-M cells transfected with siRNAs for 6 days +/- Mad2 overexpression for 3 days. (C) Western blot of Mad2 and TRIP13 on parental RPE and Mad2-inducible RPE-M cells transduced with a doxycycline-inducible TRIP13 or control shRNA construct. Total quantification (endogenous + exogenous) is shown below corresponding bands. (D) Tumor volumes over time of RPE and RPE-M cells transduced with TRIP13 or control shRNA injected subcutaneously into nude mice (n=10). Mice were continuously fed with doxycycline-containing feed. Tumor volumes and incidence at week 5 are shown on the right. ** indicates p < 0.01; Unpaired t test. Data are represented as mean± SD.
To investigate the long-term effects of TRIP13 knockdown in the presence or absence of Mad2 overexpression, we inserted a dox-inducible TRIP13 shRNA-expressing construct into the parental RPE si-p53 cells or the dox-inducible, Mad2-overexpressing, RPE-M si-p53 cells. Cells expressing this hairpin had substantial knockdown of TRIP13 after doxycycline treatment (Figure 4C). Nude mice were injected with either RPE with control or TRIP13 shRNA, or RPE-M cells with control or TRIP13 shRNA. All mice were maintained on doxycycline for 5 weeks. At 5 weeks, the majority of mice injected with control cells, or cells with only Mad2 overexpression or TRIP13 knockdown alone, grew large tumors (Figure 4D). However, tumor formation was almost completely blocked for cells that had combined overexpression of Mad2 and TRIP13 knockdown, with only 2 of 10 animals having very small (<10 mm3) tumors present after 5 weeks (Figure 4D).
Divergent roles of TRIP13 in Mad2-overexpression arrest versus nocodazole arrest
Prior studies have shown that complete knockout of TRIP13 is not lethal to cells, but, surprisingly, leads to a defect in the ability to arrest in nocodazole (Ma and Poon, 2016). TRIP13 knockout cells still had the ability to recruit Mad2 to kinetochores in the presence of nocodazole but were significantly reduced in their ability to form Mad2-Cdc20 complexes. Notably, the vast majority of the Mad2 found in these TRIP13 knockout cells was in the closed conformer, suggesting that open Mad2 may be necessary for a robust nocodazole arrest.
We generated TRIP13 knockout RPE-M si-p53 cells with CRISPR-Cas9 by inducing frameshift mutations in the first exon (Figure 5A). Similar to results seen in HeLa or HCT116 cells (Ma and Poon, 2016), the unperturbed mitotic timing of these cells was similar to those of parental cells, and also had a decreased ability to arrest in nocodazole (Figure 5B). However, overexpression of Mad2 in these cells was able to induce mitotic arrests of over 20 hours (Figures 5C and S5). The doxycycline inducible HA-Mad2 construct we used in these experiments, and in previous work (Sotillo et al., 2007), was of mouse origin. Even though there is extensive homology between mouse and human Mad2, we sought to confirm Mad2 of human origin would behave similarly. We transiently transfected RPE and TRIP13 knockout cells with a human Myc-FLAG-Mad2 construct and achieved similar mitotic timing results as with the mouse construct (Figures S6A and S6B).
Figure 5. Divergent roles of TRIP13 in Mad2-overexpression arrest versus unperturbed or nocodazole arrest.
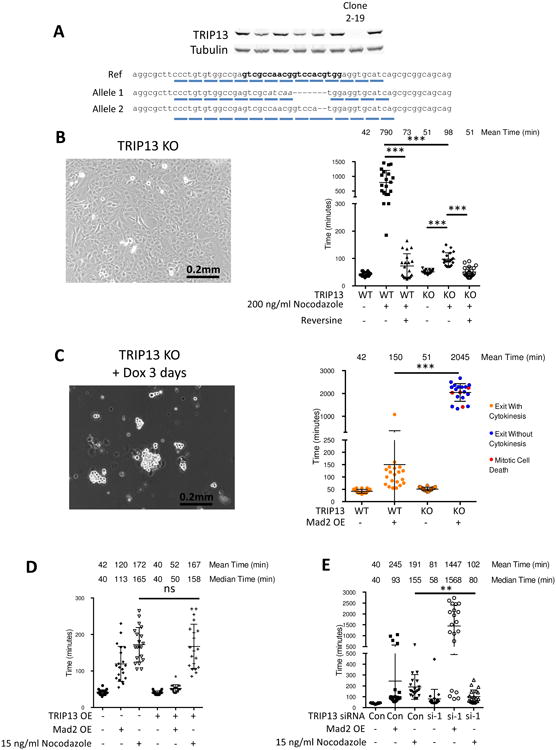
(A) Validation of CRISPR-Cas9 knockout of TRIP13. Cell clones were screened for TRIP13 loss by Western blot. DNA from clone 2-19 was cloned and sequenced, with one allele having net deletion of seven base pairs and the second allele having a deletion of two base pairs, both leading to loss of reading frame. The gRNA sequence is indicated in bold. (B) Morphology of TRIP13 knockout cells by bright field microscopy (Left). Quantification of mitotic timing in RPE-M and TRIP13 knockout cells treated with +/- 200 ng/ml nocodazole 4 hours prior to imaging +/- 0.5 uM reversine 1 hour prior to imaging (Right). n ≥20 cells for each condition. (C) Morphology of TRIP13 knockout cells after 3 days Mad2 overexpression by bright field microscopy (Left). Quantification of mitotic timing and fate in RPE-M and TRIP13 knockout cells treated with +/- doxycycline 16 hours prior to imaging (Right). n ≥20 cells for each condition. (D) Mitotic timing of RPE-M or T13G cells treated with doxycycline for 24 hours or a low dose of nocodazole (15 ng/ml) 4 hours prior to imaging. n ≥20 cells for each condition. (E) Mitotic timing of RPE-M cells transfected with control siRNA or TRIP13 siRNA #1 4 days prior to imaging and treated with either doxycycline (24 hours) or low-dose (15 ng/ml) nocodazole (4 hours) prior to imaging. n ≥20 cells for each condition. ** indicates p < 0.01, *** p < 0.001, ns indicates not significant (p > 0.05); Unpaired t test. Data are represented as mean± SD. See also Figures S5 and S6
We sought to determine how TRIP13 knockdown and overexpression affected the timing of low dose nocodazole arrests of a duration similar to Mad2 overexpression. RPE cells treated with 15 ng/ml nocodazole had a 191 minute average mitotic duration and 155 minute median mitotic duration, compared to 245 minute and 93 minute respective mean and median mitotic durations for Mad2-overexpressing cells (Figure 5E). While TRIP13 knockdown induced severely prolonged mitosis when Mad2 was overexpressed (1447 and 1568 mean and median mitotic durations) it did not cause similarly severely prolonged mitoses in low-dose nocodazole cells (102 and 80 minute mean and median mitotic durations) (Figure 5E). Additionally, we found TRIP13 overexpression, while able to blunt a Mad2-overexpression induced arrest, had no effect on a low dose nocodazole arrest (Figure 5D). This demonstrates there is a difference in the ability of TRIP13 to quench a kinetochore-produced nocodazole arrest versus a Mad2-overexpression arrest.
Additionally, we tested whether TRIP13 knockdown cells arrested with Mad2 overexpression would be able to escape this arrest upon treatment with the MPS1 inhibitor reversine. Continued MPS1 activity is necessary for the formation of MCC and maintenance of arrest in nocodazole (Hewitt et al., 2010; Maciejowski et al., 2010; Santaguida et al., 2010). We found that treatment with reversine was unable to rescue the mitotic arrest caused by Mad2 overexpression and TRIP13 knockdown (Figure 6A). Similarly, the arrest caused by Mad2 overexpression in TRIP13 knockout cells was largely resistant to reversine, with a statistically significant, but minor reduction in mitotic duration, from an average of 2045 minutes to 1625 minutes. (Figure 6A). This demonstrates that TRIP13 is necessary for reversine-induced disassembly of Mad2-overexpression produced complexes.
Figure 6. TRIP13 knockout and knockdown cells have reduced ability to disassemble MCC in the presence of reversine.

(A) Mitotic timing of TRIP13 wild-type, knockout (Left) or knockdown (Right) cells with or without Mad2-overexpression or reversine (0.5 uM) treatment. Mad2 induced 24 hours prior to imaging with doxycycline, reversine added 1 hour prior to imaging. n ≥20 cells for each condition. (B) Western blot of Cdc20 IPs from mitotic RPE-M and TRIP13 knockout cells +/- Mad2 overexpression +/- reversine. RPE-M and TRIP13 knockout cells were synchronized with 20 hours treatment of Cdk1 inhibitor RO-3306 +/- doxycycline 8 hours after 10 uM RO-3306 addition. Cells were released into fresh media for 30 minutes followed by treatment with 50 ng/ml nocodazole and 10 uM MG132 for 3 hours +/- 1 uM reversine for the last 1.5 hours. Mitotic cells were harvested by mitotic shakeoff. (C) Quantification of Mad2 in Cdc20 IPs in (B) normalized to Cdc20 for each condition (Left). Quantification of the reduction in normalized Mad2 levels in Cdc20 IP after reversine addition for each condition (Right). * indicates p < 0.05, ns indicates not significant (p > 0.05); Unpaired t test. Data are represented as mean± SD. See also Figure S7
To further investigate the effect of reversine on the disassembly of Mad2-Cdc20 complexes in mitotically arrested cells, we immunoprecipitated Cdc20 in cells arrested with nocodazole and the proteasome inhibitor MG132. As previously seen in TRIP13 knockout cells (Ma and Poon, 2016) there was a significant reduction in the amount of Mad2-Cdc20 complex formed in nocodazole in the absence of TRIP13 (lane 5 vs. lane 1 of Cdc20 IP, Figures 6B and 6C). However, when Mad2 was overexpressed, this rescued the ability of Mad2 and BubR1 to bind Cdc20 in TRIP13 knockout cells (lane 7 vs. lane 5 of Cdc20 IP, Figure 6B). Further, the ability of reversine to induce disassembly of complexes was reproducibly reduced in Mad2-overexpressing TRIP13 knockout cells compared to wild-type and Mad2-overexpressing cells (lane 8 vs. lane 7 and lane 6 vs. lane 5 of Cdc20 IP, Figures 6B and 6C). These results suggest that when Mad2 is overexpressed in TRIP13 knockout cells, Mad2-Cdc20 complexes can form, but have reduced ability to be disassembled, causing a buildup of inhibitory complexes, quantified in Figure 6C, which leads to an extremely severe mitotic arrest.
One reason previously proposed to be a cause of poor MCC formation in TRIP13 knockout cells was a deficiency in O-Mad2 (Ma and Poon, 2016). Using a previously described O-Mad2 specific antibody (Sedgwick et al., 2016), we find our TRIP13 knockout cells have lower levels of O-Mad2 than parental cells (Figure S7). This potentially explains their reduced ability to form checkpoint complexes and arrest in the presence of nocodazole (Figures 5B, 6B). Transient overexpression of Myc-FLAG-Mad2 was able to significantly increase the levels of O-Mad2 in TRIP13 knockout cells (Figure S7). Thus, overexpression of Mad2 is able to provide an additional source of O-Mad2 in these cells, which potentially explains their ability to now form sufficient mitotic checkpoint complexes (Figure 6B).
Discussion
TRIP13 has been identified as a putative mitotic checkpoint quenching protein, working in concert with p31comet to inactivate C-Mad2-containing mitotic checkpoint complexes. However, the effects others have observed upon TRIP13 depletion have been mild on an unperturbed mitosis, and the effects of overexpression on mitosis have not been adequately investigated. While TRIP13 is overexpressed in cancer and correlated with chromosomal instability, it is frequently co-overexpressed with Mad2. Thus, we do not believe that TRIP13 overexpression causes chromosomal instability by inducing a weakened checkpoint. Instead, as TRIP13 expression is co-regulated with other checkpoint genes, its overexpression potentially buffers the effects of checkpoint gene overexpression. p31comet has also been shown to be regulated similarly to Mad2 and buffer Mad2 overexpression effects on proliferation (Date et al., 2013).
To date, there has been limited investigation into the effects of TRIP13 overexpression. Wang et al. demonstrated that TRIP13 overexpression potentially causes a decrease in Mad1-Mad2 interaction in interphase cells (Wang et al., 2014), but the consequences on mitotic timing in different conditions had not been studied. Our results show that TRIP13 overexpression can block many of the effects of Mad2 overexpression, such as prolongation of mitosis, and p53-dependent growth arrest (Figures 2B and 2C). We did not, however, see evidence of a dramatically weakened checkpoint with TRIP13 overexpression, either on basal mitotic timing (Figure 2B) or in response to a low dose of nocodazole (Figure 5D). This again suggests that TRIP13 overexpression in cancer cells may not be causing a weakened checkpoint, but may buffer the effects of elevated checkpoint gene expression. We speculate that such buffering facilitates a state whereby Mad2 overexpression is not dramatically impairing proliferation but at the same time can still induce chromosome instability. While we have not identified that precise balance in RPE cells, we believe that cancer cells undergo strong selective pressure to achieve such a state of sustained growth and extensive tumor heterogeneity.
Banerjee et al. also investigated the effects of TRIP13 overexpression in cancer, but focused on a potential DNA repair role for TRIP13 in promoting non-homologous end joining (NHEJ) (Banerjee et al., 2014). They found TRIP13 overexpression could accelerate head and neck squamous cell carcinoma (HNSCC) cell line growth but with increased dependency on NHEJ. Thus, TRIP13 overexpression may have multiple roles in the context of cancer.
Given Mad2 is frequently overexpressed in tumors and is correlated with chromosomal instability (Carter et al., 2006; Schvartzman et al., 2010), we were intrigued by the possibility that Mad2-overexpressing cells would be at increased dependency on TRIP13. While in an unperturbed mitosis TRIP13 knockdown caused only a mild mitotic delay, as has been observed previously (Wang et al., 2014), in the presence of Mad2 overexpression TRIP13 knockdown caused a synergistically prolonged mitotic arrest frequently of >20 hours (Figures 3C and S4B). We also knocked down TRIP13 in RPE cells transduced to express the HPV E6/E7 proteins, which led to Mad2 overexpression through inhibition of Rb and p53 pathways. Our results are consistent with elevated Mad2 creating an increased dependency on TRIP13 for mitotic exit. However, the mitotic prolongation in these cells, both with and without TRIP13 knockdown, was less than what we observed in Mad2 inducible overexpressing cells (Figures S3B and 3C). This is probably explained by the lower amount of Mad2 overexpression in these cells compared to the inducible overexpressing cells (Figures S3A and 2A). We note that additional pathways dysregulated in cancer, such as the Myc pathway, may play a role in causing the very high levels of Mad2 seen in cancer (Menssen et al., 2007).
There are potentially multiple pathways for quenching mitotic checkpoint complexes in addition to TRIP13 and/or p31comet dependent pathways. For instance, APC15 has been shown to be critical for removing MCC that is already bound to APC/C (Mansfeld et al., 2011). Recently, CCT chaperonin (chaperonin - containing TCP1) has been shown to be able to stimulate the disassembly of certain types of MCC (Kaisari et al., 2017). TRIP13 does not appear required for quenching of the checkpoint in an unperturbed mitosis, as knockdown causes only mild mitotic delays. Thus, it is possible that the alternative pathways are largely sufficient for dealing with checkpoint complexes formed during a normal mitosis. However, when Mad2 is overexpressed, it appears that these pathways are no longer adequate to cope with this excessive inhibitory signal, and TRIP13 becomes necessary in this situation.
Considering the differential requirement for TRIP13 in an unperturbed mitosis versus a Mad2-prolonged mitosis, we tested if TRIP13 would be similarly important in the context of other mitotic arrests. We found that modulation of TRIP13 did not affect nocodazole arrests of similar duration in the same way as it did Mad2-overexpression induced arrests (Figures 5D and 5E). This suggests that there is a qualitative difference in the role of TRIP13 in a nocodazole-dependent kinetochore produced arrest versus a Mad2-overexpression induced arrest. Others have shown that there can be different requirements for disassembly of different pools of MCC (Ma and Poon, 2011).
A potential source for non-kinetochore generated MCC is from cytosolic conversion of O-Mad2 to C-Mad2. Mad2 has previously been shown to spontaneously convert from O-Mad2 to C-Mad2 (Luo et al., 2004), although at slow rates that would not be sufficient for responsiveness to kinetochore attachment status. However, over time, this spontaneous conversion could produce a sufficient amount of kinetochore-independent inhibitory complexes and the cell may need a mechanism to disrupt them. Mad2 overexpression may, by mass action, cause an increase in the amount of spontaneous complex formation. Importantly, there is evidence in yeast that Mad2-overexpression can cause a kinetochore-independent arrest (Mariani et al., 2012). If TRIP13 were more critical for reversing the formation of this type of complex, it would explain our results showing a clear differential requirement for TRIP13 in a nocodazole versus Mad2-induced arrest (Figures 5D and 5E).
A surprising and interesting phenotype that has been reported in TRIP13 knockout cells is their deficiency in producing a robust checkpoint response to nocodazole (Ma and Poon, 2016). We corroborated those results in our TRIP13 knockout cells (Figure 5B), but found that when Mad2 was overexpressed, TRIP13 knockout cells strongly arrested and regained the ability to form checkpoint complexes (Figures 5C and 6B). The previously studied TRIP13 knockout cells showed the vast majority of Mad2 was present in the closed conformer (Ma and Poon, 2016), and the authors speculated that recycling of C-Mad2 to O-Mad2 by TRIP13 may be necessary for checkpoint complex formation in the presence of nocodazole. Our TRIP13 knockout cells also had significantly lower levels of O-Mad2, however Mad2 overexpression was able to provide a source of newly produced O-Mad2 (Figure S7), potentially accounting for the ability of these cells to now execute very prolonged mitotic arrests.
While further work needs to be done to fully understand the mechanism of the relatively mild effects of TRIP13 loss on an unperturbed mitosis versus its necessity in Mad2-overexpressing cells, we can take advantage of this relationship as a potential way to target Mad2-overexpressing cells. In normal cells, reduction of TRIP13 caused only a mild effect on proliferation in vitro (Figure 4A). Additionally, genetically engineered TRIP13 hypomorph mice with severe reductions in TRIP13 protein levels are viable and grossly normal, although born at non-mendelian ratios and sometimes presenting with smaller body size (Li et al., 2007; Roig et al., 2010). However, in Mad2-overexpressing cells, TRIP13 knockdown caused severe reductions in proliferative capacity in vitro (Figure 4A) and in tumor formation (Figure 4D). Others have shown that TRIP13 knockdown in cancer cell lines can reduce cancer cell growth (Banerjee et al., 2014; Tao et al., 2017). This suggests TRIP13 could be a therapeutic target in tumors with Mad2 overexpression, with potentially a better toxicity profile than typical antimitotic chemotherapeutics.
Overexpression of Mad2, while potentially important for generating chromosomal instability to initiate tumorigenesis or allow escape from oncogene addiction, may not be required for tumor maintenance (Sotillo et al., 2007; Sotillo et al., 2010). Thus, while its expression may be tied to the loss of tumor suppressors required for maintenance, it is possible that overexpression of Mad2 would be selected against, especially after targeting with inhibition of TRIP13. We have seen evidence in occasional surviving TRIP13 knockout Mad2-overexpressing cells that they have lost the inducible Mad2 overexpression (data not shown). We still believe that, although resistance may be possible, targeting TRIP13 may be beneficial in shrinking tumors either alone or in combination with other therapeutics.
In a previous effort to target Mad2-overexpressing cells, Bian et al. performed a synthetic genetic array screen in yeast for genes whose deletion specifically reduces the viability of Mad2-overexpressing cells, and identified PPP2R1A, a subunit of the phosphatase PP2A, as a potential target (Bian et al., 2014). As an AAA-ATPase, TRIP13 is potentially a more readily druggable target. Other AAA-ATPases, particularly p97, have been targeted with small molecules that show promise as cancer therapeutics (Anderson et al., 2015; Zhou et al., 2015). The strength of the synthetic lethality of TRIP13 loss with Mad2 overexpression makes targeting TRIP13 a new therapeutic opportunity in the management of aneuploid tumors.
Experimental Procedures
Cell culture and chemicals
RPE2 cells were cultured in a 1:1 mixture of DME and Ham's F12 and supplemented with 15 mM HEPES, 10% fetal bovine serum, L-glutamine, and 100 U/ml penicillin/streptomycin. MCF7 and MDA-MB-436 cells were cultured in DME supplemented with 10% fetal bovine serum, L-glutamine, and 100 U/ml penicillin/streptomycin. MEFs were cultured in DME supplemented with 10% fetal bovine serum, L-glutamine, 0.001% B-Mercaptoethanol, and 100 U/ml penicillin/streptomycin. Multiple nocodazole (15-200 ng/ml) (Sigma) and reversine (0.5-1 uM) (Sigma) concentrations were used, as indicated in figure legends. Other chemicals were used at the following concentrations: 10 uM MG132 (Enzo Life Sciences), 10 uM RO-3306 (Calbiochem), 5 ug/ml doxycycline (BD Biosciences), 0.2 ug/ml doxorubicin (Sigma).
For cell synchronization for Cdc20 immunoprecipitation, cells were synchronized with 20 hours treatment of Cdk1 inhibitor RO-3306 +/- doxycycline 8 hours after 10 uM RO-3306 addition. Cells were released into fresh media for 30 minutes followed by treatment with 50 ng/ml nocodazole and 10 uM MG132 for 3 hours +/- 1 uM reversine for the last 1.5 hours. Mitotic cells were harvested by mitotic shakeoff.
Description of constructs
Retroviral constructs were produced by transfecting 293GP2 cells and Lentiviral constructs were produced by transfecting 293T cells using standard Lipofectamine 2000 (Thermo Fisher) protocol. Target cells were transduced with 1:1 viral media to fresh media in 8 ug/ml polybrene.
All PCR amplifications was performed with Phusion Hot Start II polymerase (New England BioLabs)
Mad2-overexpressing RPE-M cells were made by co-transfecting RPE2 cells with pTRE-HA-Mad2 (mouse) and pCAGGS-rtTA (puromycin) and subsequent single cell clones were tested for Mad2 induction by Western blot. Transient human Mad2 overexpressing cells were made by transfecting target cells with Myc-DDK (FLAG) tagged Mad2 (Origene, #RC203273) using Lipofectamine 3000 (Thermo Fisher). The TRIP13 overexpression construct was made by cloning TRIP13 cDNA (Open Biosystems) into pBabe-GFP. TRIP13 cDNA was amplified and cloned into a pBabe-GFP (blasticidin) construct with XhoI and MfeI/EcoRI downstream of the GFP tag. The p53 siRNA construct was cloned from pLVTH-sip53 (addgene #12239). GFP was removed, and neomycin resistance cloned in using MluI and XmaI.
The siRNAs were obtained from Dhamacon siGenome; siT13-1: TRIP13 ORF (D-016262-01); siT13-2: TRIP13 3′ UTR (D-016262-21); siCon: siGENOME non-targeting siRNA #2 (D-001210-02). Cells were transfected with 25 nM siRNAs with Lipofectamine 2000 (Thermo Fisher). Inducible shRNA constructs were cloned into the RT3REVIR shRNA backbone (derived from pQCXIX) (Fellmann et al., 2013). The antisense guide sequence for TRIP13 shRNA was ATCACCAAACACAATATTATGT, and the antisense guide sequence for the Control shRNA was TAGATAAGCATTATAATTCCTA. Cells were selected by growing single cell clones from Venus positive cells and testing for knockdown.
TRIP13 knockout cells were made by cloning TRIP13 gRNA (target sequence gtcgccaacggtccacgtgg) into pX330 expressing Cas9 (Addgene), by digesting the vector with BbsI and ligated to annealed and phosphorylated sgRNA oligonucleotides. RPE-M sip53-neo cells were nucleofected with the 5 ug px330-gRNA and 1 ug GFP in Amaxa buffer V using A-023 program on an Amaxa nucleofector as per manufacturer's protocol. Single cells were FACS sorted and clones were grown and tested for loss of TRIP13 expression by Western blot. DNA was extracted from clone 2-19 with DNeasy Blood & Tissue kit (Qiagen). DNA was amplified and sequenced for mutations at the predicted cut site by Topo cloning with the Zero Blunt TOPO PCR cloning kit (Thermo Fisher).
pLXSN-E6/E7 (Halbert et al., 1992) was a gift from the Prasad Jallepalli lab. Parental RPE cells were transduced with the vector and selected with G418 (500 ug/ml) for 10 days.
Time-lapse imaging and Immunofluorescence
Micronuclei were scored by counting the number of cells with DAPI signals outside of the main nucleus. 100 cells were counted per condition, in triplicate. Cells were fixed with 4% PFA for 20 minutes at room temperature, followed by blocking with 10% normal goat serum, incubation with primary antibody (CREST 1:100) at 4°C overnight, incubation with secondary 1:1000 goat anti-human Alexa Fluor 568 for 1 hour room temperature, DAPI 1 ug/ml 5 minutes room temperature, and mounted on coverslips with Aqua-Mount (Fisher).
For time-lapse imaging, cells were plated onto 6 or 12 well glass bottom plates (MatTek) and imaged with DIC imaging using a Zeiss Axis Observer Microscope with a Plan-apochromat 20×/0.8 M27 objective and Zen Acquisition 2.0 software. Images were taken every 5 minutes, with 7 z-stacks 3 microns apart taken for each point. Mitotic duration was measured by timing from when the cells rounded to when they flattened back on the plate.
Proliferation assays and Xenografts
For cell culture proliferation assays, cells were counted and plated in equal numbers, and subsequently counted and replated in equal numbers on each day of the analysis as indicated. Experiments were performed in three biological replicates. Cumulative fold was measured by multiplying the fold changes in cell number at each time point.
Xenografts were performed by injecting 1 million cells per condition subcutaneously into nude mice aged 5-6 weeks. Mice were maintained on doxycycline containing feed for the entirety of the experiment. Tumor sizes were measured with calipers every week. Volume was calculated by tumor volume = 0.5*length*width2. Tumor incidence was defined as the number of animals that had solid palpable tumors at the end of 5 weeks
Antibody Techniques
Mad2 (Clone 48): Mouse, BD Biosciences. BubR1 (A300-995A): Rabbit, Bethyl Laboratories. Actin (A2066): Rabbit, Sigma. Beta-Tubulin (T4026): Mouse, Sigma. TRIP13: Rabbit, Song-Tao Liu lab. p53 (DO-1): Mouse, Santa Cruz Biotechnology. Cleaved Caspase-3 (Asp175) (9661): Rabbit, Cell Signaling Technology. Total H2AX (07-627): Rabbit, EMD Millipore. pSer139 H2AX (JBW301): Mouse, EMD Millipore. Cdc20 (E-7): Mouse, Santa-Cruz. Normal Mouse IgG (sc-2025): Mouse, Santa Cruz Biotechnology. CREST anti-centromere HCT-0100: Human, Immunovision. Alexa Fluor 568 anti-human secondary: Goat, Life Technologies. O-Mad2: Mouse, Jakob Nilsson lab.
Cdc20 immunoprecipitation was performed as described previously (Rodriguez-Bravo et al., 2014). In brief, whole cell extracts were lysed by nitrogen cavitation (2000 psi, 5 min; Parr Instruments) in buffer B (140 mM NaCl, 30 mM HEPES, pH 7.8, 10 mM sodium pyrophosphate, 10 mM NaF, 10 mM PMSF, 0.3 mM sodium orthovanadate, 20 mM b-glycerophosphate, 1 mM DTT, 1× protease inhibitor cocktail). 300 ug total protein was loaded onto 30 uL Protein G Dynabeads (Fisher) conjugated to 2.5 ug Cdc20 or normal mouse IgG and incubated 2 hours at 4°C. Beads were washed 3× with buffer B and eluted in 2× sample buffer at 70C for 10 minutes.
O-Mad2 immunoprecipitations were performed as above, except cells were lysed with 50mM Tris-HCl, pH 8.0, 150mM NaCl, 0.5% NP40, 10% Glycerol, 0.5mM EDTA, 1× Protease Inhibitor Cocktail for 30 minutes at 4°C. 800 ug protein was loaded per sample on 4 ug antibody or normal mouse IgG antibody conjugated to beads and crosslinked with BS3 (Thermo Fisher). Washes were performed with PBS-T (0.02% Tween 20). Subsequent Western blots were probed using BD Biosciences Mad2 antibody.
Lysates for Western blot were prepared by resuspending whole cell extracts in lysis buffer (0.5% NP40, 50 mM TRIS HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1x complete mini protease inhibitor cocktail (Roche)) for 30 minutes on ice. Histones were extracted from remaining pellet by resuspending in 0.1 M HCl for 30 minutes at room temperature. Proteins were resolved by SDS-PAGE, and then transferred to a PVDF membrane, blocked in Odyssey blocking buffer, and incubated with primary antibodies in Odyssey blocking buffer + 0.05% Tween 20 overnight at 4°C. After washing and incubation with IRDye secondary antibodies (in Odyssey blocking buffer), membranes were imaged and quantified on a Li-Cor Odyssey scanner.
Statistical Methods
All statistical analyses were performed using an unpaired two-tailed t test. * denotes p < 0.05, ** denotes p < 0.01, *** denotes p< 0.001.
Supplementary Material
Highlights.
- TRIP13 and Mad2 are overexpressed and have correlated expression in cancer.
- TRIP13 is critical for mitotic exit in Mad2-overexpressing cells.
- TRIP13 is not required for exit from an unperturbed mitosis or nocodazole arrest.
- TRIP13 is a therapeutic target and its loss is synthetic lethal with Mad2 overexpression.
Acknowledgments
We wish to thank Yvette Chin, Peter Cook, Paulina Wojnarowicz, Yuji Shi, Shefali Krishna, Jens Hamann, and Jose Rodriguez-Rodriguez for technical assistance and Prasad Jallepalli for experimental advice and for help in editing this manuscript. We would also like to thank the Memorial Sloan Kettering Flow Cytometry, Molecular Cytologenetics, Molecular Cytology, and RNAi Core Facilities. Funding: NIH/NCI grant P30 CA008748. R.B. was supported in part by a Jesús Serra-CNIO Visiting Scientist Fellowship.
Footnotes
Author Contributions: D.H.M., R.T., and R.B. conceived of and designed the experiments. D.H.M., R.T., Y.C., R.S., and C.K. performed the experiments. D.H.M., R.T. and R.B. analyzed the data, and D.H.M, R.T., and R.B. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Russo N, Liu M, Basrur V, Bellile E, Palanisamy N, Scanlon CS, van Tubergen E, Inglehart RC, Metwally T, et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat Commun. 2014;5:4527. doi: 10.1038/ncomms5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Y, Kitagawa R, Bansal PK, Fujii Y, Stepanov A, Kitagawa K. Synthetic genetic array screen identifies PP2A as a therapeutic target in Mad2-overexpressing tumors. Proc Natl Acad Sci USA. 2014;111:1628–1633. doi: 10.1073/pnas.1315588111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date DA, Burrows AC, Venere M, Jackson MW, Summers MK. Coordinated regulation of p31(Comet) and Mad2 expression is required for cellular proliferation. Cell Cycle. 2013;12:3824–3832. doi: 10.4161/cc.26811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, et al. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15:214–225. doi: 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Eytan E, Wang K, Miniowitz-Shemtov S, Sitry-Shevah D, Kaisari S, Yen TJ, Liu ST, Hershko A. Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet) Proc Natl Acad Sci U S A. 2014;111:12019–12024. doi: 10.1073/pnas.1412901111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Biol. 2012;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T. Identification of a MAD2-binding protein, CMT2, and its role in mitosis. EMBO J. 2002;21:6419–6428. doi: 10.1093/emboj/cdf659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando E, Nahlé Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, Taylor SS. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol. 2010;190:25–34. doi: 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeche L, Compton DA. Checkpoint-independent stabilization of kinetochore-microtubule attachments by Mad2 in human cells. Curr Biol. 2012;22:638–644. doi: 10.1016/j.cub.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisari S, Sitry-Shevah D, Miniowitz-Shemtov S, Teichner A, Hershko A. Role of CCT chaperonin in the disassembly of mitotic checkpoint complexes. Proc Natl Acad Sci U S A. 2017;114:956–961. doi: 10.1073/pnas.1620451114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XC, Li X, Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet. 2007;3:e130. doi: 10.1371/journal.pgen.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol. 2004;11:338–345. doi: 10.1038/nsmb748. [DOI] [PubMed] [Google Scholar]
- Ma HT, Poon RYC. Orderly Inactivation of the Key Checkpoint Protein Mitotic Arrest Deficient 2 (MAD2) during Mitotic Progression. J Biol Chem. 2011;286:13052–13059. doi: 10.1074/jbc.M110.201897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Poon RYC. TRIP13 Regulates Both the Activation and Inactivation of the Spindle-Assembly Checkpoint. Cell Rep. 2016;14:1086–1099. doi: 10.1016/j.celrep.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, George KA, Terret ME, Zhang C, Shokat KM, Jallepalli PV. Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol. 2010;190:89–100. doi: 10.1083/jcb.201001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfeld J, Collin P, Collins MO, Choudhary JS, Pines J. APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat Cell Biol. 2011;13:1234–1243. doi: 10.1038/ncb2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Filipp FV, Rancati G, Massimiliano L, Nezi L, Stier G, Hagan RS, Confalonieri S, Piatti S, Sattler M, et al. Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J. 2006;25:1273–1284. doi: 10.1038/sj.emboj.7601033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani L, Chiroli E, Nezi L, Muller H, Piatti S, Musacchio A, Ciliberto A. Role of the Mad2 dimerization interface in the spindle assembly checkpoint independent of kinetochores. Curr Biol. 2012;22:1900–1908. doi: 10.1016/j.cub.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, Diebold J, Hermeking H. c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle. 2007;6:339–352. doi: 10.4161/cc.6.3.3808. [DOI] [PubMed] [Google Scholar]
- Musacchio A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol. 2015;25:R1002–1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- Nelson CR, Hwang T, Chen PH, Bhalla N. TRIP13PCH-2 promotes Mad2 localization to unattached kinetochores in the spindle checkpoint response. J Cell Biol. 2015;211:503–516. doi: 10.1083/jcb.201505114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Bravo V, Maciejowski J, Corona J, Buch HK, Collin P, Kanemaki MT, Shah JV, Jallepalli PV. Nuclear pores protect genome integrity by assembling a premitotic and mad1-dependent anaphase inhibitor. Cell. 2014;156:1017–1031. doi: 10.1016/j.cell.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig I, Dowdle JA, Toth A, de Rooij DG, Jasin M, Keeney S. Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Duijf PHG, Sotillo R, Coker C, Benezra R. Mad2 Is a Critical Mediator of the Chromosome Instability Observed upon Rb and p53 Pathway Inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick GG, Larsen MS, Lischetti T, Streicher W, Jersie-Christensen RR, Olsen JV, Nilsson J. Conformation-specific anti-Mad2 monoclonal antibodies for the dissection of checkpoint signaling. MAbs. 2016;8:689–697. doi: 10.1080/19420862.2016.1160988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–440. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–936. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Yang G, Yang H, Song D, Hu L, Xie B, Wang H, Gao L, Gao M, Xu H, et al. TRIP13 impairs mitotic checkpoint surveillance and is associated with poor prognosis in multiple myeloma. Oncotarget. 2017 doi: 10.18632/oncotarget.14957. ePub Feb 1, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichner A, Eytan E, Sitry-Shevah D, Miniowitz-Shemtov S, Dumin E, Gromis J, Hershko A. p31comet Promotes disassembly of the mitotic checkpoint complex in an ATP-dependent process. Proc Natl Acad Sci U S A. 2011;108:3187–3192. doi: 10.1073/pnas.1100023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Johnson VL, Albertella M, Taylor SS. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2001;2:609–614. doi: 10.1093/embo-reports/kve127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Kienitz A, Hofmann I, Müller R, Bastians H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene. 2004;23:6845–6853. doi: 10.1038/sj.onc.1207860. [DOI] [PubMed] [Google Scholar]
- Wang K, Sturt-Gillespie B, Hittle JC, Macdonald D, Chan GK, Yen TJ, Liu ST. Thyroid hormone receptor interacting protein 13 (TRIP13) AAA-ATPase is a novel mitotic checkpoint-silencing protein. J Biol Chem. 2014;289:23928–23937. doi: 10.1074/jbc.M114.585315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci. 2011;124:3905–3916. doi: 10.1242/jcs.093286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. EMBO J. 2004;23:3133–3143. doi: 10.1038/sj.emboj.7600322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Li B, Tomchick DR, Machius M, Rizo J, Yu H, Luo X. p31comet blocks Mad2 activation through structural mimicry. Cell. 2007;131:744–755. doi: 10.1016/j.cell.2007.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Rosenberg SC, Moeller A, Speir JA, Su TY, Corbett KD. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife. 2015;4 doi: 10.7554/eLife.07367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim EK, Park JS. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res Treat. 2005;37:319–324. doi: 10.4143/crt.2005.37.6.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, Rice J, Valle E, Soriano F, Menon MK, et al. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083) J Med Chem. 2015;58:9480–9497. doi: 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.