

Abstract
We attempt to correlate the clinical pharmacology of dopamine replacement therapy (DRT) in Parkinson Disease with known features of striatal dopamine actions. Despite its obvious impact, DRT does not normalize motor function, likely due to disrupted phasic dopaminergic signaling. The DRT Short Duration Response is likely a permissive-paracrine effect, possibly resulting from dopaminergic support of corticostriate synaptic plasticity. The DRT Long Duration Response may result from mimicry of tonic dopamine signaling regulation of movement vigor. Our understanding of dopamine actions does not explain important aspects of DRT clinical pharmacology. Reducing these knowledge gaps provides opportunities to improve understanding of dopamine actions and symptomatic treatment of Parkinson disease.
Introduction
Parkinson disease (PD) is a common neurodegenerative disorder and the most common serious movement disorder. The defining clinical features of bradykinesia, rigidity, and resting tremor, accompanied by characteristic alterations of posture, gait, and voice quality, are among the most striking phenomena in Neurology. Equally impressive is the marked improvement seen in many patients with dopamine replacement therapy (DRT). The discovery that dopamine is the primary neurotransmitter of the nigrostriatal projection, whose disruption causes the cardinal motor features of parkinsonism, focused attention on understanding striatal dopamine actions. The large literature in this field, while far from conclusive, indicates that striatal dopaminergic neurotransmission mediates important aspects of learning, motivation, and goal-directed behaviors.
The complex basic science literature on striatal dopamine suggests several important components of its actions.1 Disruption of striatal dopaminergic signaling should manifest in complex ways and it should be possible to correlate important features of the clinical response to DRT with basic aspects of striatal dopamine signaling. The goal of this Grand Rounds is to explore these potential correlations to assist identification of mechanisms relevant to the clinical actions of DRT. Similarly, failures of our present understanding of dopaminergic nigrostriatal signaling to explain important features of DRT clinical pharmacology point to important areas for future investigation. We suggest that disruption of three key functions of striatal dopaminergic signaling – phasic dopaminergic signaling, permissive-paracrine dopaminergic modulation of corticostriate synaptic plasticity, and tonic dopaminergic signaling that estimates the background rate of reward - explain important features of DRT clinical pharmacology. These potential correlations expose significant gaps between DRT clinical pharmacology and our present knowledge of dopaminergic nigrostriatal signaling.
Dopamine Replacement Therapy Has Ceiling Effects
It is widely recognized that DRT does not restore normal function. This is likely due to 2 features of PD. While most of the defining motor features of PD – tremor, bradykinesia, rigidity – are secondary to nigrostriatal degeneration, PD is a multifocal neurodegeneration affecting many brain regions, even in early disease. Dopamine replacement resistant clinical features, including some gait and postural control deficits, likely reflect pathologies outside the basal ganglia. But even with generally dopamine replacement responsive features, such as bradykinesia, it is unusual for DRT to normalize function. The Earlier versus Later Levodopa Therapy in Parkinson disease (ELLDOPA) trial provides a pertinent example.2 This trial enrolled mildly symptomatic subjects with mean total Unified Parkinson’s Disease Rating Scale (UPDRS) scores of approximately 27 (mean motor component scores approximately 19). At 9 weeks after study initiation, treatment with 300 or 600 mg of L-dopa per day resulted in an approximately 4 point change in total UPDRS scores, with most of the change attributable to motor score changes. Experienced clinicians recognize that even in optimally treated patients, movement speed rarely normalizes and finely coordinated movements continue to be significantly impaired. This is visible with simple maneuvers such as finger tapping, where slowing of rapid movements, progressive slowing with repetition, and movement amplitude decrements are demonstrated readily in the clinic. The failure of DRT to normalize motor function implies that some important aspect(s) of normal striatal dopaminergic signaling is irretrievably impaired in PD.
DRT Has Two Major Components
DRT effects in PD are complex. As noted in the seminal papers of Cotzias et al., there are both rapid and longer term therapeutic effects of L-dopa.3,4 While Cotzias and colleagues noted rapid onset of L-dopa effects, they also found that some PD subjects experienced continued improvement for several days after reaching stable daily L-dopa doses. Similarly, some of their PD subjects experienced a slow decline in motor function over days to weeks after stopping L-dopa treatment. Muenter and Tyce subsequently characterized these 2 primary L-dopa effects as the short duration response (SDR) and the long duration response (LDR).5,6 In both responses, bradykinesia, rigidity, and (usually) tremor improve with therapy. The SDR begins rapidly, sometimes within minutes of L-dopa administration, lasts minutes to hours, and then declines, with clinical improvement roughly parallel to plasma L-dopa levels. The LDR is sustained improvement that builds up over days of repeated L-dopa treatment and decays over similar intervals after treatment cessation. The SDR is usually explained by correlating it with L-dopa pharmacokinetics, which begs the question of what dopamine actions are normalized during the SDR. The LDR is generally treated as an unexplained pharmacodynamic phenomenon. Nutt and Holford interpreted the existence of the SDR and LDR as implying more than one mechanism of L-dopa action, a proposal consistent with the concept that striatal dopaminergic signaling has diverse actions.7
SDR & LDR Features
Cotzias et al. described the LDR qualitatively in their original clinical observations of successful L-dopa treatment of PD. Muenter and Tyce used a clinical disability rating scale to evaluate effects of L-dopa treatment. They evaluated clinically stable, treated PD subjects after overnight withdrawal of L-dopa (~10 hours; the “practical off” state) and then after administration of their customary oral L-dopa dose. Several subjects exhibited significant differences between these “off state” disability scale measurements and their pre-treatment disability scores, indicating improved baseline function in the “practical off” state after prolonged treatment. Muenter and Tyce reported also that some of their subjects exhibited functional decline 3–5 days after stopping L-dopa.
LDR kinetics are incompletely understood. Because patients often undergo repeated dose adjustments after initiation of therapy, and only rarely in controlled settings, it is difficult to measure the magnitude of the LDR after therapy initiation. Modeling of data accumulated in the DATATOP trial, which followed initially treatment naïve subjects, indicated that maximum benefits of L-dopa may not be achieved for months.8 LDR decline after therapy cessation is better studied, particularly with rigorous paradigms developed by Nutt and colleagues in which motor performance is measured regularly after L-dopa discontinuation.9.10.11 This is an operational definition of the LDR as gradually declining motor function after therapy cessation, and assumes that this decline is a reversal of the same phenomenon underlying gradual improvement after therapy initiation. Dopaminergic therapy is withdrawn from patients with consistent responses to stable treatment regimens in controlled settings. Some standard motor task, such as finger tapping rates, is used to assess motor performance. Motor performance off medication can be compared with baseline (treated) performance and also with subjects’ pre-treatment performances (Figure 1). SDR effects can be estimated by measuring the effects of acutely administered L-dopa, including intravenous L-dopa administration to sidestep the pharmacokinetic complexities of oral L-dopa administration.
Figure 1.
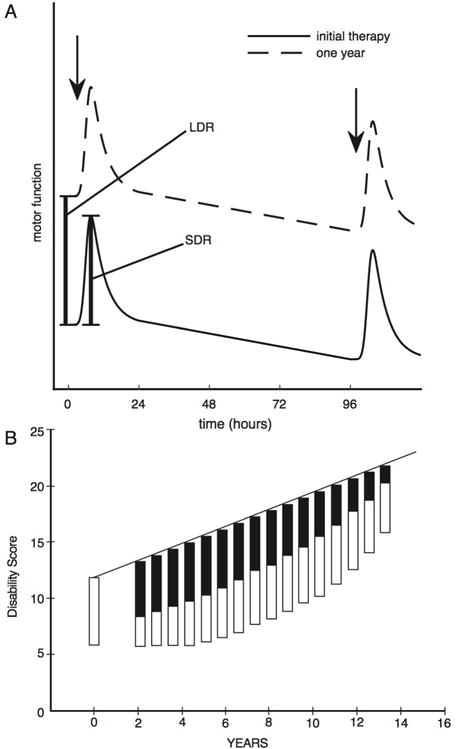
Clinical characteristics of the SDR and LDR effects. (A) Schematic representation of the SDR and LDR effects (adapted from Nutt et al., Short and long-duration responses to levodopa during the first year of therapy, Ann Neurology, 1997, Figure 1).9 Drug-naïve PD patients were given IV levodopa infusions, and motor function assessed with a finger-tapping task. After 4 days, a second infusion was given. Patients returned after one year and underwent the same protocol after PD medications were held overnight. The LDR is visible as the upward migration of motor performance immediately prior to the first IV levodopa infusion after one year of treatment. The SDR immediately follows IV levodopa infusions. (B) From Clissold et al., Longitudinal study of the motor response to levodopa in Parkinson’s disease, Mov Disord, 2006, Figure 5.20 Schematic of the progression of the magnitude of the short- (open boxes; SDR) and long- (solid boxes; LDR) duration responses to levodopa. The solid line represents disability in the untreated state and is a partial function of the magnitude of the LDR. With disease progression, the LDR wanes and the SDR becomes a relatively larger component of the levodopa response. Higher scores indicate increasing disability.
These studies suggest that the LDR declines over days to weeks. Pharmacodynamic modeling of declining motor performance after treatment cessation estimates the half-life of L-dopa and bromocriptine motor effects, predominantly the LDR, at approximately 8 days.12 This inference is consistent with results of the ELLDOPA trial, in which the L-dopa treated participants had better UPDRS scores than placebo treated participants after a 2 week washout period.2 The ELLDOPA experience highlights an important aspect of the LDR; it complicates interpretation of disease-modifying trial outcomes.
By various measures, the LDR accounts for 30% to 50% of the total (SDR + LDR) response to L-dopa and is responsible for much of the sustained-uniform response to L-dopa in patients with early PD.6.13 The LDR is present in more advanced PD but diminishes with disease progression, with the SDR becoming a more important treatment component.9.11,14,15,16 In a longitudinal study following PD subjects over a 4 year interval, Nutt et al. used their rigorous LDR evaluation protocol and documented more rapidly declining LDRs with disease progression.11 The SDR persisted and increased in magnitude with advancing disease, perhaps because more rapidly declining LDRs resulted in lower baseline levels of motor function at the times of acute L-dopa administrations. The declining LDR and increase of the SDR accounts partly for the emergence of motor fluctuations. The SDR but not the LDR is associated with dyskinesias.
Analogous results were found in Kempster’s careful longitudinal study of a small group (N=34) of PD subjects followed for over 2 decades from treatment inception (Figure 1B).17–20 The SDR was preserved in some subjects with PD of many years duration. Those advanced PD subjects with declining SDRs tended to exhibit overt dementia, suggesting that pathologies outside the basal ganglia are responsible for the loss of the SDR (Figure 2). Alternatively, patients may have developed dose-limiting side-effects as non-motor features became more prominent.
Figure 2.
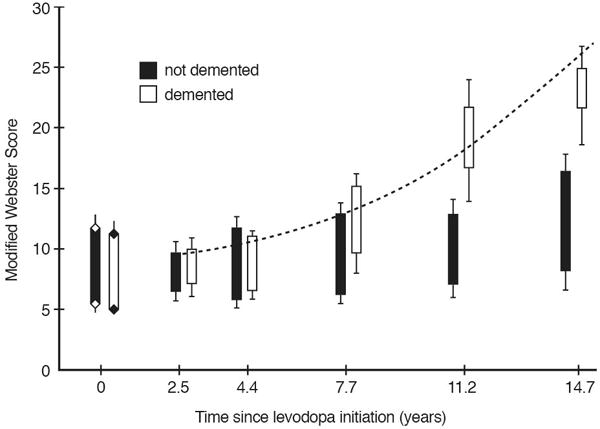
The natural history of LDR and SDR with disease progression in Kempster’s cohort. From Alty et al., Longitudinal study of the levodopa motor response in Parkinson’s disease: relationship between cognitive decline and motor function, Mov Disord, 2009, Figure 5.19 Modified Webster Scores of PD patients in the practical “off” state (tops of boxes) and 60–90 minutes after their usual levodopa dose (bottom of boxes) as a function of disease duration for non-demented (black boxes) and demented (MMSE < 24, white boxes) patients. The Modified Webster Score is a standardized assessment of motor function, with higher scores indicating worse function. Note the diminished magnitude of the SDR in demented vs non-demented patients.
A crucial point is that LDR induction is seen only with repetitive treatments, as it is not reinstated after a single intravenous dose of L-dopa during drug holidays.21 Relatively infrequent treatment may be sufficient to induce the LDR since daily, relatively high (250 mg) L-dopa doses are reported to elicit the LDR in early PD.22
Pharmacokinetic vs Pharmacodynamic LDR Mechanisms
Two broad categories of LDR mechanisms were considered: pharmacokinetic and pharmacodynamic.6.13 The pharmacokinetic hypothesis postulated the existence of a brain reservoir that accumulates L-dopa and slowly releases dopamine. A plausible hypothesis was that surviving nigrostriatal terminals were a central reservoir of dopamine synthesized from exogenous L-dopa and the decline of the LDR with disease progression could be explained by gradual loss of residual nigrostriatal terminals. A critical prediction is that the LDR should be specific for L-dopa and not occur with dopamine agonists. This prediction was falsified by several studies.12,23,24,25 The LDR is sustained by intravenous infusion of the dopamine agonist apomorphine and is induced by treatment with D2-receptor selective dopamine agonists.12,23,24,25 The decay rate of agonist induced LDR was essentially identical to the decay rate of the L-dopa induced LDR.12,24.25 The LDR is also documented in Dopa Responsive Dystonia (DRD).26 The LDR in DRD has similar decay kinetics to those found in PD, but DRD has essentially normal presynaptic dopamine storage capacity. If the LDR is based on presynaptic storage of dopamine, the LDR of DRD should exceed that of PD.26 The decline of the LDR with disease progression does not clearly parallel the decline in putaminal nigrostriatal terminal density. In Kempster’s cohort, off-state disability, which is a partially a function of LDR magnitude, declined linearly over 2 decades.17–20 In contrast, a prospective, longitudinal study of putaminal nigrostriatal dopaminergic terminal loss in PD with [11C]dihydrotetrabenazine positron emission tomography indicates exponential decline in terminal density, approaching a plateau in more advanced disease.27 Kordower et al. studied nigrostriatal terminal integrity as a function of disease duration in a set of well-characterized post-mortem PD specimens and described a similar non-linear trajectory of putaminal nigrostriatal terminal loss.28 In more advanced PD subjects, Kordower et al. document an almost complete absence of nigrostriatal terminals in the dorsal striatum. These results point away from a pharmacokinetic explanation and implicate a pharmacodynamic effect of dopamine signaling.
Mapping Clinical Pharmacology onto Known Dopamine Functions
DRT clinical pharmacology has 3 major features to correlate with normal striatal dopaminergic actions – the ceiling effects of treatment, the SDR, and the LDR. A correlate of the ceiling effect would have to be an aspect of striatal dopamine action that is irretrievably disrupted with nigrostriatal terminal degeneration in PD. The temporal and other features of the SDR and LDR offer criteria for plausible mapping of these phenomena onto known striatal dopaminergic functions. Mechanisms responsible for the SDR should act with time courses in the minutes to hours range. The persistence of the SDR in advanced PD indicates that the SDR does not require many nigrostriatal terminals. In contrast, changes in mechanisms responsible for the LDR should occur over days to weeks and require multiple and/or chronic exposures to dopaminergic stimulation. The LDR is elicited by dopamine agonists with relatively long half-lives, suggesting that it is a function of tonic dopamine action.
Organization of the Nigrostriatal Projection
Substantia nigra (SN) dopaminergic neurons constitute a tiny fraction of human brain neurons with an estimated total of ~1.2 million (~600,000 per side) neurons.29 Each neuron of this small population gives rise to large axonal arborizations with particularly dense innervation of dorsal striatal projection neurons. Projections from the most medial portion of the SN complex, the ventral tegmental area, target other forebrain targets such as ventral striatum, frontal cortex, and amygdala. Within the striatum, a single SN neuron may contact as many as 75,000 striatal neurons.30 This “broadcast” connectional anatomy suggests that striatal dopaminergic neurotransmission conveys general signals.
SN dopaminergic neurons project topographically to striatal subregions with reciprocal striatonigral afferents from their projection target regions. This architecture is not entirely closed as there is some overlap in nigrostriatal neuron projections.31 Different striatal regions exhibit functional specialization as they are nodes in roughly parallel and functionally differentiated circuits that course through the whole cortico-basal ganglia-thalamic-cortical loop.32 In crude terms, more anterior regions such as the caudate are specialized for cognitive functions, the more ventral regions are specialized for limbic (motivational) functions, and the dorsal putamen for motor functions. It is likely that there is complex subregional functional specialization of cortico-basal ganglia-thalamic-cortical loops. The regional specialization of striatal regions and corresponding functional specialization of cortico-basal ganglia-thalamic-cortical loops suggests that striatal dopaminergic signaling performs uniform operations across the striatum with functional specificity residing at the level of striatal circuitry.
The “broadcast” concept is consistent also with the ultrastructure of nigrostriatal terminals. Striatal projection neuron dendrites exhibit prominent spines which are the termination sites of both cortical (and thalamic) and SN dopaminergic projections. The canonical microcircuit of a striatal projection neuron spine consists of a glutamatergic excitatory cortical (or thalamic) neuron terminal synapsing on the spine “head” and a dopaminergic terminal synapsing on the spine “neck” (Figure 3). This triadic arrangement is consistent with dopamine action regulating striatal projection neuron function via modulation of corticostriate synapse function.
Figure 3.
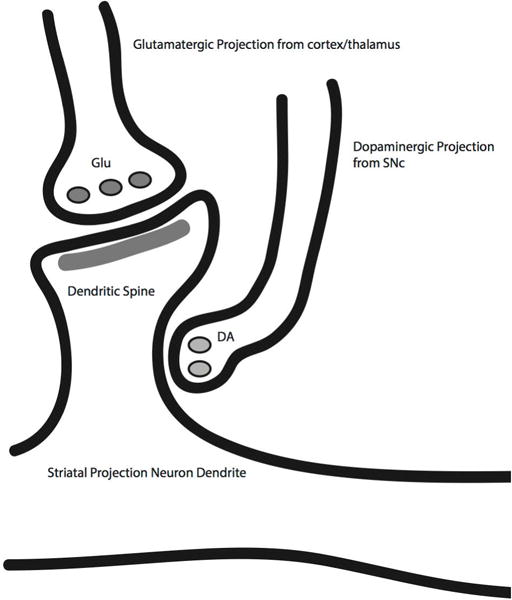
Schematic diagram of the striatal “synaptic triad.” Cortical (and thalamic afferents) synapse on striatal projection neuron spine heads with dopaminergic terminals on spine necks. Dopaminergic synapses are well positioned to both modulate glutamatergic signaling onto medium spiny projection neurons and plasticity of corticostriate synapses.
Phasic Striatal Dopamine Signaling
Dopaminergic nigrostriatal neurons exhibit tonic regular firing punctuated by phasic bursts of action potentials that trigger bolus release of dopamine within the striatum. These bursts result in brief (100–300 milliseconds) and sharply contoured pulses of striatal dopamine release with substantial, transient increases in extracellular dopamine. Most of the physiologic literature on striatal dopaminergic signaling is devoted to explaining the role(s) of phasic activity. Convergent theoretical and experimental results over the past couple of decades suggests that phasic nigrostriatal dopaminergic signaling mediates reinforcement learning (for concise reviews, see Glimcher33 or Kerflin and Janak34; for detailed review, see Schultz35). An important concept is that phasic nigrostriatal dopaminergic neurotransmission mediates reward prediction error (RPE) signals for reinforcement learning. In the best-validated models of reinforcement learning, temporal difference models, the value of an organism’s current state is estimated as the net value of future expected rewards. High value rewards with a high probability of attainment in the near future contribute greatly to the current state value. Conversely, potential future rewards with lower intrinsic value, a lower probability of attainment, and/or longer latency to attainment contribute less. At each time point, the organism compares their current state value estimate to their previous prediction; the difference is the RPE. These RPEs are then used to update ensembles of cached sets of estimated values of environmental stimuli and actions. The concept that nigrostriatal dopaminergic signaling mediates reinforcement learning dovetails nicely with the concept that nigrostriatal dopaminergic function is important for habit learning.36
In seminal experiments by Schultz and colleagues, phasic nigrostriatal dopaminergic neuron activity exhibits RPE signal properties. Schultz’s group studied substantia nigra (SN) dopaminergic neuron activity as monkeys learned associations between visual cues and rewards. In untrained animals, reward presentation was followed rapidly by a burst of SN neuron activity. Phasic dopaminergic neuron activity and the inferred striatal dopamine bolus elicited by this unexpected reward constitute a positive RPE signal. As animals learned the stimulus reward associations, the burst of SN activity following the reward subsides, while the visual cue is followed by a burst of SN activity (and presumed striatal dopamine bolus). Migration of the positive RPE signal from a previously unexpected reward to the predicting cue is an explicit prediction of temporal difference models (Figure 4). Also consistent with temporal difference models, omission of an expected reward results in reduced nigrostriatal neuron activity. Temporal difference models result in other predictions that can be evaluated experimentally. One example is predictions about the magnitude of positive reward prediction error signals and the history of rewards. In clever experiments, Bayer and Glimcher demonstrated that firing rates of dopaminergic SN neurons under conditions of varying rewards follow the predicted relationships.37
Figure 4.
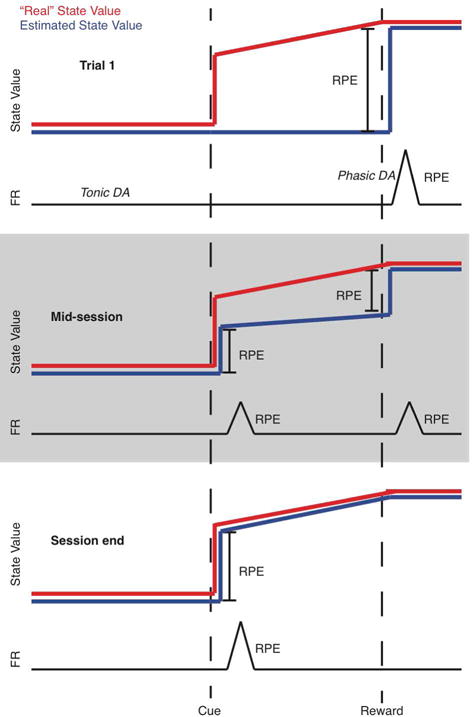
Temporal difference model reward prediction error signaling. The red trace indicates actual state value; the blue trace indicates estimated state value; the black trace represents dopamine neuron firing rates (FR). In a hypothetical task, a cue predicts a reward and reward timing with 100% certainty. Note that the state value gradually increases after the predictive cue as the rewarding event moves closer. On trial 1 (top panel), the agent is unaware of the cue-reward association, so the state value estimate is low until the reward is delivered. This is reflected as a phasic increase in DA neuron firing at reward delivery. After many trials (“mid-session”), the agent associates the cue with reward according to the rules of temporal difference models, but is not yet certain of the 100% correspondence between cue and reward. Therefore, the state value estimate jumps twice, reflected in two smaller phasic DA firing increases. With more experience, the agent understands that the cue predicts reward with 100% certainty, and the phasic DA signal migrates entirely to the cue.
While a large body of work supports the embodiment of RPE signals in phasic nigrostriatal dopaminergic neuron activity, recent results suggest somewhat different roles in other aspects of behavior. Dopaminergic neurotransmission and reinforcement learning can be dissociated under specific conditions.38,39.40 Palmiter and colleagues, for example, used mice with ablated aromatic acid decarboxylase genes to demonstrate learning in typical reinforcement learning paradigms in the absence of dopaminergic signaling.39
Recent technical advances make it possible to measure calcium transients in striatal dopaminergic terminals and axons in awake, behaving mice. These transients likely reflect phasic nigrostriatal neuron bursting. Parker et al. recorded rapid calcium transients in dorsomedial (analogous to caudate) and ventral (limbic) striata of mice performing a reversal learning task.41 Consistent with the concept of phasic dopaminergic signaling embodying RPEs, dorsomedial and ventral striatal rapid calcium transients were associated with rewards and reward predicting cues. Dorsomedial striatal calcium transients, however, were also associated with movement direction selection. This result is consistent with involvement of dopaminergic signaling in movement execution independent of learning or motivation, but is also consistent with more complex reinforcement learning models. These results indicate also that functional specificity of dopaminergic signaling resides at the level of striatal subregions.
In a technical tour de force, Howe and Dombeck recorded these calcium transients in dorsal striatal dopaminergic terminals and axons in awake, locomoting mice.42 Rapid calcium transients were associated with locomotion accelerations and not with unpredicted rewards. Optogenetic stimulation mimicking dorsal striatal phasic dopamine release produced accelerations. In strong control experiments, Howe and Dombeck demonstrated reward associated calcium transients in ventral striatal nigrostriatal axons-terminals. This work associates dorsal striatal phasic dopamine signaling with specific kinematic features of movement, consistent with other reports of movement-related phasic changes in nigrostriatal dopamine neuron firing.43,44,45
Phasic dorsal nigrostriatal dopaminergic signaling likely plays a critical role in important aspects of fine motor performance distinct from its well established role in reinforcement learning based on primary rewards. Some conceptual models of nigrostriatal phasic signaling in motor performance suggest that dopaminergic signaling is involved in feedback control of motor performance by matching sensory feedback about motor performance to internal representations of desired actions.46,47,48 These concepts bear a general family resemblance to temporal difference models of reinforcement learning in that they incorporate updating internal representations of action outcomes. In an ingenious experiment, Gadagkar et al. evaluated the role of phasic nigrostriatal signaling in fine coordination of a complex motor act, zebra finch singing.49 By manipulating auditory feedback, Gadagkar et al. were able to correlate dopaminergic neuron behavior with perceived song performance. Phasic activation and suppression of dopaminergic neurons correlated well with perceived better than expected and worse than expected song performance, respectively. These results are consistent with phasic nigrostriatal signaling participating in evaluation of motor performance relative to some internal benchmark.
The Ceiling Effect of Treatment and Loss of Phasic Signaling
Given the impressive degree of posterior putaminal striatal nigrostriatal terminal loss in early PD, likely in excess of 60% of terminals, it is hard to imagine that normal phasic dopaminergic signaling is preserved in motor specialized striatal regions in PD patients.50 The likely disruption of normal phasic signaling probably has both anatomic and functional components. Many posterior putamen neurons undoubtedly lose much of their dopaminergic innervation. While the remaining nigrostriatal neurons probably continue to exhibit phasic firing, it is very likely that magnitude of phasic dopamine release diminishes.51,52 The dopamine that is released is also likely to be cleared more slowly because extracellular dopamine is normally removed rapidly by dopamine transporters on nigrostriatal terminals. Loss of nigrostriatal terminals, with consequent loss of dopamine transporters, results in extended residence of dopamine in the extracellular space.51 Loss of many dopaminergic terminals and reduced capacity to rapidly clear extracellular dopamine markedly degrade the pulsatile character of normal phasic nigrostriatal signaling. This is not likely to be corrected by L-dopa supplementation and will not be mimicked by dopamine agonists.
It is likely that loss of normal phasic signaling accounts for persistent motor deficits in well-treated, early PD patients, suggesting an interesting conclusion. Given the impressive functional improvements in early PD patients, phasic dopamine signaling represents only a fraction of relevant dopamine actions. In terms of movement control, phasic striatal dopamine signaling may be a “fine-tuning” mechanism superimposed on other dopamine actions that likely account for the SDR and LDR.
Dopaminergic Maintenance of Corticostriate Synaptic Plasticity – A Mechanism for the SDR?
Key features of the SDR - its close relationship to plasma L-dopa levels, relatively rapid onset and offset, and persistence in the presence of substantial nigrostriatal terminal loss - indicate a direct effect of L-dopa derived dopamine on striatal neurons via a hormone-like effect. This is also consistent with the SDR-like effects of dopamine agonists, as initially documented by Cotzias in experiments with apomorphine.53 As dopamine acts via modulatory G-protein coupled receptors, this cannot be a conventional fast inhibitory or excitatory neurotransmitter effect. The canonical triadic arrangement of striatal projection neuron spines, corticostriate neuron terminals, and closely adjacent nigrostriatal terminals suggests a particularly important role for dopaminergic signaling in modulating corticostriate neurotransmission. A strong candidate SDR mechanism is dopaminergic maintenance of corticostriate synaptic plasticity.
Yttri and Dudman recently evaluated the role of corticostriate synaptic plasticity in a methodologically sophisticated study of movement control.54 They demonstrated that brief closed loop optogenetic photostimulation of striatal projection neurons during limb movements, mimicking physiologic striatal projection neuron bursting, produced increasing changes in limb movement velocity. Selectively stimulating direct or indirect pathway neurons during fast movements increased or decreased velocity, respectively. Importantly, the opposite occurred with photostimulation only during slow movements. That is, direct pathway activation now slowed movements, while indirect pathway activation sped them up. Furthermore, increased and decreased limb movement velocities were present during non-stimulated trials, suggesting persistent changes in corticostriatal networks, inferred to be secondary to changes in corticostriate synapse plasticity. The effects of optogenetic stimulation manifested over minutes, and with cessation of optogenetic stimulation, limb movement velocities returned gradually to their pre-stimulation states. In a complementary analysis, Yttri & Dudman developed a computational model of corticostriate synaptic plasticity governing striatal projection neuron bursting and movement kinematics that nicely reproduced the results of their experiments. This sophisticated analysis and set of experiments links modulation of corticostriate synapse plasticity to specific kinematic features of movement.
The optogenetic stimulation approach utilized by Yttri & Dudman did not directly alter striatal dopaminergic neurotransmission. The inferred changes in corticostriate synapse plasticity took place in the absence of changes in nigrostriatal dopaminergic signaling and likely against the background of tonic ambient dopamine levels. Yttri & Dudman showed also that behavioral effects of optogenetic stimulation were blocked by systemically administered dopamine antagonists. A large literature demonstrates that normal modulation of corticostriate synaptic plasticity requires dopamine receptor activation (see reviews by Calabresi and colleagues).55,56 This is true for both long term potentiation (LTP) and long term depression (LTD), including the spike timing dependent plasticity (STDP) thought to be critical for fine modulation of synaptic strength.57,58,59.60,61,62 The great majority of experiments studying dopaminergic modulation of corticostriate synaptic plasticity utilize ex vivo slice preparations in which phasic dopaminergic signaling is absent. This strongly suggests that tonic dopamine receptor activation has a permissive effect on normal corticostriate synaptic plasticity independent of phasic dopamine release. This conclusion is supported by computational modeling of dopamine effects on STDP.63
A permissive-paracrine effect of dopamine on corticostriate synaptic plasticity is consistent with SDR features. The time course of minutes to hours for the effects documented by Yttri and Dudman, as well as in ex vivo slice preparations, is consistent with the SDR. The requirement for some degree of striatal dopamine receptor activation, though not necessarily precise modulation of extracellular striatal dopamine levels, is also consistent with the incomplete response to DRT. This is not to argue that phasic changes in striatal dopamine levels have no consequence - the magnitude and timing of striatal dopamine receptor activation likely modulate corticostriate synaptic plasticity. We suggest that this fine modulation is subtle, however, at least in terms of motor control. The concept of a permissive-paracrine tonic dopamine effect on corticostriate synaptic plasticity as the basis for the SDR is consistent with in vivo data in treated PD patients. Positron emission tomography studies of extracellular striatal dopamine levels indicate that PD patients with motor fluctuations and dyskinesias exhibit larger, briefer changes in extracellular dopamine levels with L-dopa treatment than patients with stable responses.64 These effects likely reflect progressive loss of nigrostriatal terminals, and consequently a reduced “buffer” compartment to maintain striatal extracellular dopamine levels within a broadly physiologic limit.65 With disease progression, striatal extracellular dopamine levels increasingly depend on the unregulated synthesis and release of dopamine from non-dopaminergic neurons after L-dopa treatment. Serotoninergic neurons are believed to be especially important in this respect.66
The waning SDR in advanced PD may reflect loss or dysfunction of corticostriate synapses. Braak and Del Tredici note the frequent presence of α-synuclein inclusions in cortical layer V, the site of corticostriate projection neuron perikarya, in advanced PD. They suggest that this cortical pathology leads to corticostriate synaptic dysfunction and loss, with consequent loss of the DRT response.67 This hypothesis is consistent with Kempster’s description of waning SDR in overtly demented patients.19
The LDR and Learning
The gradual onset and decline of the LDR is consistent with some form of neuronal learning as its basis. Therefore, in contrast to our proposal that corticostriate synapse plasticity mediates the SDR, it has also been suggested that corticostriate synapse plasticity underlies the LDR.68 Interesting experiments with rodent models of parkinsonism explored this concept.
Beeler and colleagues studied acquisition and extinction of motor performance in homozygous Pitx3 knockout mice.69 In these mutants, nigrostriatal neurons degenerate gradually with marked (~90%) loss of dorsal striatal dopamine. These mice exhibit grossly normal motor function but are impaired on some learning tasks, including a rotarod task. In a possible LDR analogue, L-dopa administered once daily prior to testing sessions restored the ability of Pitx3 knockout mice to improve performance with trial repetition. L-dopa administration outside trial sessions had no effect. With L-dopa treatment discontinuation, performance did not deteriorate immediately but decayed over several days with repeated task performance. Critically, rotarod performance of rats trained on a different task (treadmill running) in the absence of levodopa did not decay. This indicates that the “learning” effects of levodopa loss were task-specific. Measurement of striatal dopamine levels confirmed that pharmacokinetic factors could not account for the slow decline in motor performance after L-dopa discontinuation. These results are consistent with the concept that some form of dopamine mediated learning underlies the LDR.
One potential problem with invoking this form of motor learning as the basis for the LDR is that the LDR manifests as a general improvement in motor performance, as opposed to specific deficits in learning new tasks. This fact may be reconciled with a learning conception of the LDR by dopamine replacement preventing unlearning (extinction) of previously learned motor behaviors. In a complementary experiment, Dowd and Dunnett used the rat unilateral 6-hydroxydopamine lesion model to study an analogue of declining motor function after DRT discontinuation.70,71 Rats were trained in a lateralized reaction time task and rewarded for selecting a target either to the right or left of midline after presentation of an instructive cue. Unilateral 6-hydroxydopamine lesions were performed after training to a high level of accuracy. After a period of weeks without training, rats were retested. Task performance was initially almost normal, but performance contralateral to the lesioned side declined over several days. As with the Pitx3 mice experiments, the initially normal performance excludes a motor deficit per se. The decline in motor performance contralateral to the 6-hydroxydopamine lesion closely paralleled the decline in performance of unlesioned rats after unidirectional reward omission, and similar results were observed using intrastriatal injections of dopamine antagonists.72 Striatal dopaminergic denervation mimicked extinction of a learned motor act.
A learning conception of the LDR can be reconciled with diffuse effects of DRT in maintaining the ability to perform previously learned motor behaviors. However, it does not obviously account for the general effect of DRT in improving bradykinesia. An interesting literature on striatal dopaminergic modulation of speed or strength of movement may cast light on the nature of the LDR.
Vigor and Striatal Dopamine Signaling
Another important concept of nigrostriatal dopamine action is that it plays an important role in regulating “vigor;” the speed or strength of actions. Salamone and others argued persuasively that a great deal of experimental data are best understood in the context of a role for striatal dopamine in efficiently allocating effort by scaling actions to motivational states.73,74,75 Interfering, for example, with striatal dopamine signaling reduces the effort an animal is willing to put into obtaining a food reward. All movements incur costs, if only some energy costs, and more vigorous (faster or more sustained) actions incur higher costs. In the view of Salamone et al., striatal dopaminergic signaling is crucial for estimating the context dependent cost/benefit tradeoffs of actions. This idea fits well with a number of observations about PD. An obvious potential clinical correlate is bradykinesia. There is considerable literature on deficient movement amplitude scaling in PD, and decrementing amplitude of simple movements and phenomena like micrographia are common clinical observations in PD. More speculatively, deficient vigor could form part of the substrate of the apathy found commonly in PD patients.
Mazzoni et al tested the hypothesis that bradykinesia results from abnormal effort allocation in PD using a speed-accuracy trade-off task.76 Subjects were asked to move their hand, within a specific velocity range, into a target area. The task was repeated until 20 movements were executed with an appropriate velocity. PD patients and control subjects exhibited similar accuracy on velocity-matched movements, but it took more trials for PD patients to generate movements in the target velocity range. This was interpreted as PD patients having a different perception of the cost/benefit ratio of movements compared to control subjects.76 Baraduc et al. obtained analogous results in DBS treated PD subjects performing a reaching task, and other recent studies of reward/effort trade-offs in PD subjects are consistent with dopaminergic modulation of vigor.77,78,79,80 Electrophysiologic studies of MPTP-treated non-human primates are also consistent with dopaminergic modulation of vigor.81
Panigraphi et al. examined the role of nigrostriatal dopaminergic signaling in “vigor” using a mouse model of progressive nigrostriatal degeneration.82 MitoPark mice have a selective deletion of a crucial mitochondrial transcription factor restricted to midbrain dopaminergic neurons. These mice exhibit gradual post-natal death of midbrain dopaminergic neurons and slowly progressive bradykinesia over months. Panigraphi et al. trained MitoPark mice to perform a joystick task with varying thresholds of limb movement velocities needed to obtain rewards. Young MitoPark mice readily learned the task and performed well. Task performance declined, primarily due to impaired velocity of limb movements, in parallel with nigrostriatal neuron degeneration. During testing sessions, the limb movement velocity thresholds for reward were varied in blocs. Bradykinetic MitoPark mice appropriately modulated limb velocity with bloc changes, even though limb velocities were often inadequate to obtain rewards. These analyses indicated that MitoPark mice learned the appropriate reward contingencies but were not able to scale limb movement velocity appropriately. These results parallel the human experiments of Mazzoni et al.76 and Baraduc et al.77, and suggest that striatal dopamine deficiency causes a defect in the regulation of movement vigor, not a learning deficit per se.
A complementary experiment by Cagniard et al. also supports a role for tonic dopamine signaling in modulating vigor.83 These workers used mice with an inducible knockdown of the dopamine transporter (IDATKD) that results in a chronic, moderately hyperdopaminergic state. After DAT knockdown, these mice exhibit increased tonic dopaminergic midbrain neuron activity with normal phasic activity. Cagniard et al. trained IDATKD mice in a standard task in which increasing effort is required to obtain rewards. To eliminate the possibility that DAT knockdown would affect task learning, animals were trained to a high performance level prior to induced DAT knockdown. After DAT knockdown, these animals are willing to work harder than wild-type mice for equivalent rewards. In an appropriate control experiment, Cagniard et al. showed that IDATKD mice had normal learning on a control task after induced KD knockdown.
Reconciling Learning and Vigor
In an effort to reconcile these two apparently disparate concepts of dopaminergic signaling, Niv et al. elaborated a temporal difference model of reinforcement learning in which subjects make choices about both action selection and action vigor.84 In any given setting, differing choices of actions and degree of vigor will incur differing benefits and costs. The goal is to optimize the net value of rewards per unit of time, which is the value of the chosen action minus the value of costs incurred by the action (Figure 5). The costs include the vigor with which the action is performed. An important element of this calculation is to estimate the cost not only of one alternative action versus another but also the cost of doing nothing. In economic terms, the real cost of a decision – the opportunity cost – is the relative value of the alternative(s) not chosen. An important point made by Niv et al. is that less vigorous actions not only delay the immediate rewards associated with the chosen actions but also delay all future rewards. How, then, to estimate the opportunity cost of inaction? In the Niv et al. model, the opportunity cost of inaction is the preceding average rate of rewards. If the average rate of rewards is high, then doing nothing is costly and more vigorous performance is incented. If the average rate of rewards is low, the penalty of inaction is lower and there is less incentive to act vigorously. Niv et al. show that outputs from their model simulations duplicate results of typical animal experiments examining response vigor.
Figure 5.
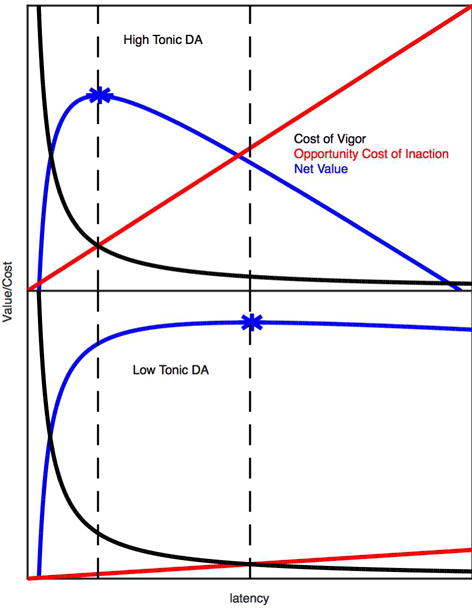
Schematic representation of varying opportunity cost of inaction (tonic dopamine signaling) on vigor. Optimal latency to initiate movement (as a surrogate for response vigor). Top – high tonic dopamine, bottom – low tonic dopamine. Black curves – cost of movement vigor, which is very high at short latencies and independent of tonic dopamine levels. Red lines – opportunity cost of the action relative to inaction, which has a higher slope at higher tonic dopamine levels. Blue curves – net action value as a function of latency (expected action value minus the cost of vigor and opportunity cost). The maximum net action value (optimum latency) is indicated with an asterisk. Note the optimal latency to maximize future rewards shifts to the right with decreasing tonic dopamine levels.
Accounting of rewards and reward magnitudes over long intervals would be necessary to accurately estimate the prior average rate of rewards. Niv et al. suggest that this signal is tonic striatal dopamine signaling. In their simulations, manipulating the average rate of reward has an identical effect to manipulating striatal dopamine in typical experiments. Niv et al. demonstrate that increasing the average rate of reward has a generally “energizing” effect on actions, with action vigor increasing not only for the specific actions in a learned task but also for actions generally. This is a potential correlate of DRT relief of bradykinesia.
Niv et al. point out another implication of their model.84 In a particularly interesting set of simulations, Niv et al. report that the slower responding resulting from simulating reduced tonic dopamine signaling - reduced prior average rate of reward - results in less switching between different actions. Less vigorous actions are energetically less costly, which raises the opportunity cost of switching to a different action. This simulated outcome may be analogue of phenomena such as freezing of gait when attempting turns and pallilalia.
The Niv et al. model is consistent with some recent experimental results.85,86 Hamid et al. examined striatal extracellular dopamine across multiple time scales in rats performing a complex adaptive decision making task. Minute to minute changes in dopamine concentration correlated with task reward rate and a measure of task vigor.85 Beeler et al. employed dopamine transporter knockdown mice (DATKD) with moderate, chronic elevations in striatal dopamine and a clever behavioral paradigm that required mice to both learn new responses for rewards and to adjust the amount of effort needed to maintain their body weight.86 DATKD mice learned as well as control mice but worked harder to obtain equivalent rewards. DATKD mice acted with greater vigor, exhibiting distorted coupling between the magnitudes of reward and effort, presumably because their chronically elevated striatal dopamine levels leads to misperception of the prior average rate of rewards.
Vigor and the LDR
Invigorating movement via increased striatal tonic dopaminergic signaling as a mimic of increasing the background rate of rewards is a good hypothesis to explain the LDR. While based largely on correlation with preclinical experimental and theoretical literature, this hypothesis is consistent with the work of Mazzoni et al. and others who have examined vigor in PD subjects (see above).76,77,78,79,80 In an interesting experimental correlate, Panigraphi et al. treated bradykinetic MitoPark mice with daily L-dopa, which improved both locomotion and limb movement velocities.82 In a result strongly reminiscent of the LDR, bradykinesia improved gradually over several weeks (Figure 6). This non-physiologic but clinically relevant DRT improved estimation of appropriate movement vigor. This result is consistent with a gradual effect of dopamine replacement in correcting dysregulated matching of motivational state to action. The equation of tonic dopaminergic modulation of vigor and the LDR, however, raises some interesting questions and opens the door to potentially interesting experiments.
Figure 6.
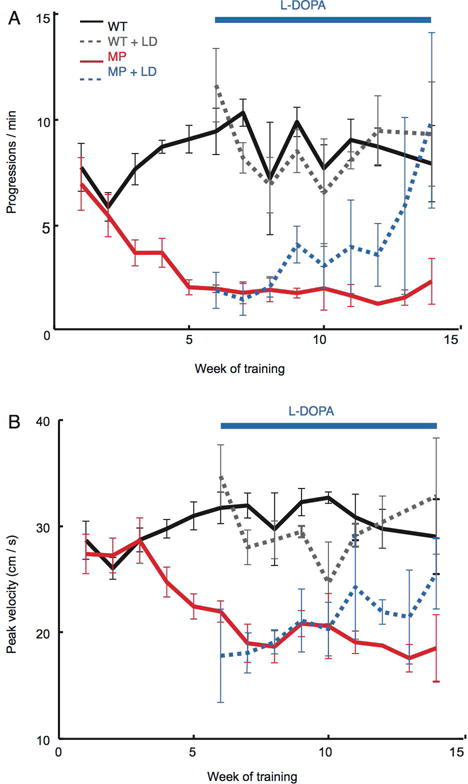
LDR-like effect of levodopa in MitoPark mice. Number of locomotor bout initiations (“progressions”, panel A) and maximal locomotor velocity (panel B) in MitoPark (MP) and wildtype (WT) mice moving freely in an open field. MitoPark mice initiated fewer locomotor bouts with lower peak velocities as their midbrain dopamine neurons degenerated (red lines). Once treated with levodopa, both measures of motor function returned to near baseline levels, but only with repeated dosing (dashed blue lines). Modified for formatting from Panigrahi et al, Dopamine is Required for the Neural Representation and Control of Movement Vigor, Cell, 2015, Figure 6).80
If the dopaminergic signal provided by L-dopa and dopamine agonists is the basis for computing the average rate of reward, how is that signal analyzed and translated into invigorated action? The LDR phenomenon clearly builds up (and declines) over days to weeks. This implies some kind of relatively long duration plasticity. To date, neither human nor preclinical experiments have explored this aspect of the LDR. In the Hamid et al. experiments, which demonstrated correlations between movement vigor, striatal extracellular dopamine concentrations, and average rate of rewards, the duration of measurements was over hours, not days. The experiment of Mazzoni et al. examined treated PD treated subjects. Other human experiments examining the relationship between movement vigor and energy costs in PD studied chronically treated subjects in the “practical off” state and after acute treatment.78,79,80
To test the concept that the LDR results from chronic treatment restoring a more normal relationship between movement vigor and perceived costs of movement, experiments could be performed in treatment naïve PD subjects, in the same subjects immediately after treatment initiation, and weeks after achievement of stable treatment regimens. The LDR = Increased Vigor induced by DRT hypothesis predicts that untreated patients should have markedly abnormal reward/effort trade-offs in behavioral paradigms of the type used by Mazzoni et al., that these abnormalities would not improve immediately after treatment initiation, and that the vigor-perceived cost relationship would improve gradually over days to weeks. Improvements in the vigor-perceived cost relationship should correlate with measures of overall clinical improvement, particularly bradykinesia. Falsification of these predictions would suggest strongly that normalizing vigor modulation is not the basis of the LDR.
The MitoPark mice utilized by Panigraphi et al. might be a useful platform to explore mechanisms underlying this type of relatively long duration plasticity. This model appears to exhibit an analogue of the LDR (see above) and temporal correlations between the gradual improvement in motor performance following L-dopa treatment and potential mechanisms would provide logical points of departure in the search for specific mechanisms underlying the LDR.
Potential Neuronal Mechanisms of the LDR
While mimicry of tonic striatal dopaminergic modulation of vigor is an attractive hypothesis to explain the LDR, this is a psychophysical construct that does not speak to the neuronal mechanisms by which it is implemented. If corticostriate plasticity underlies the SDR, where do circuit changes occur that mediate the LDR? If tonic dopamine signaling represents the average rate of reward, there must be an “accounting” or “integrating” mechanism that stores the information conveyed by tonic dopamine and adjusts responses accordingly. Recent rodent and non-human primate lesion experiments indicate that the striatum mediates longer term estimates of prior reward histories, influencing both action selection and action vigor.87,88
Zhuang et al. put forward the interesting hypothesis that the LDR results from chronic DRT normalization of corticostriate synaptic plasticity.68 As discussed above, a permissive-paracrine effect of DA in maintaining corticostriate synaptic plasticity is a more plausible mechanism for the SDR. The computational model of corticostriate synaptic plasticity of Yttri & Dudman, however, has an interesting feature that could be the basis for a corticostriate synaptic plasticity based explanation of the LDR.54 In the Yttri & Dudman model, the range of movement velocities and underlying corticostriate synaptic plasticity changes are assumed to exist in the form of Gaussian distributions of potential states. Rewarded movements shift the means of the distributions. To model maintenance of a physiological range of movement velocities, this model incorporates a “restorative set point” that tends to pull movement velocities back towards the original mean. Extending this model to explain both the SDR and the LDR, a permissive-paracrine effect of DA maintains relatively normal corticostriate synaptic plasticity, accounting for the SDR. The magnitude of tonic DA action (average background rate of reward) influences the “set point,” determining the mean of the distribution of permissible movement velocities. This hypothesis provides a mechanistic basis for our psychophysical explanation of the LDR and may be testable in rodent parkinsonism models.
Another possibility is that the LDR is based on plasticity that is an emergent property of basal ganglia circuits under tonic striatal dopaminergic stimulation. This concept derives from the clinical observation that subthalamic or pallidal deep brain stimulation (DBS) in PD patients does not reach peak effects (at least for bradykinesia) for days to weeks after stimulation parameter changes. In an analogous experiment, Wang et al. observed persistent beneficial effects, lasting days after cessation of stimulation, of a novel subthalamic DBS protocol in MPTP-treated non-human primates.89 The physiologic basis of these effects is unknown but the fact they are elicited by STN DBS suggests extra-striatal mechanisms unrelated directly to striatal dopamine actions.
Another possible class of explanations for the LDR is some form of structural plasticity within the basal ganglia. Experimental nigrostriatal neuron degeneration or administration of dopamine receptor antagonists are described by several groups as producing structural changes in in striatal projection neuron dendrites, striatal interneuron connectivity, and corticostriate synapses.90,91,92,93,94,95,96 Of note, dendritic spine loss after 6-OHDA lesioning is confined to indirect pathway SPNs, and D2 receptor agonists are capable of inducing the LDR.92 Administration of L-dopa to mice with 6-hydroxydopamine striatal lesions produces dendritic alterations in striatal projection neurons.95 These kinds of plastic changes are not restricted to the striatum. Fan et al. demonstrated a relatively rapid, within weeks, increase in the density of external globus pallidus – subthalamic neuron synapses following 6-hydroxydopamine striatal lesions in mice.90
As pointed out some years ago by Nutt and Holford, understanding mechanisms underlying the LDR might allow development of interventions to prolong the period in which the LDR plays a major role in response to dopaminergic agents.7 This is usually the period of maximum function, and even modest extensions of that period could have a major impact on patient quality of life.
Striatal Dopamine Actions Through the Lens of DRT Clinical Pharmacology
Correlation of DRT clinical pharmacology with some features of striatal dopamine actions suggests a tripartite model of striatal dopamine modulation of motor performance (Figure 7a). Relatively rapid, over seconds to minutes, “fine tuning” of motor acts is secondary to phasic dopamine signaling. Slower, over the course of minutes to hours, permissive-paracrine effects of dopaminergic support of corticostriate synaptic plasticity accounts for the SDR. Finally, tonic striatal dopamine signaling provides a measure of the prior rate of reward and the motivational “set point” for action vigor, which broadly influences motor performance. This set point provides the foundation for the modulatory activities of corticostriate synapse plasticity. Each one of these putative components is incompletely understood, and what may be the most quantitatively important, the LDR, is likely the least understood.
Figure 7.
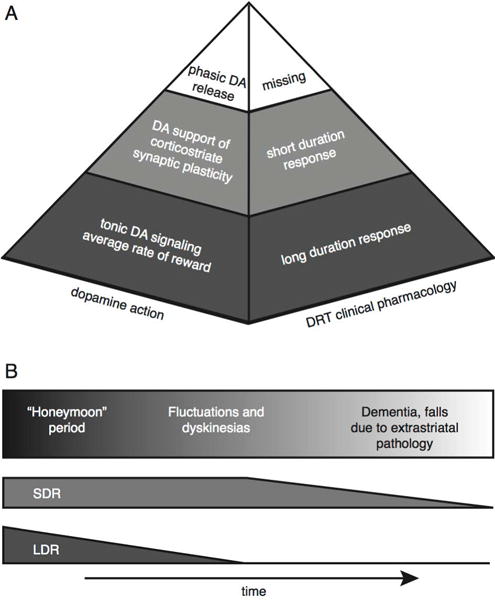
(A) Proposed dopamine action “pyramid” as related to the clinical pharmacology of DRT. Phasic dopamine (DA) release acts on a very short time-scale and is responsible for “fine-tuning.” It is lost in early PD, explaining why dopamine replacement therapy cannot fully restore motor function. Dopamine supports normal corticostriatal synaptic plasticity, which operates on intermediate time scales (minutes to hours) and is responsible for the SDR. Tonic dopamine signaling indicates the average rate of reward over hours to weeks, and is responsible for the LDR. (B) Proposed changes in dopamine actions during progression of Parkinson Disease. In the “honeymoon period,” the LDR allows infrequent dosing of dopamine replacement therapy to provide stable motor function. As the LDR wanes, motor fluctuations and dyskinesias emerge, but the preserved SDR allows at least temporary restoration of motor function with DRT. With continued disease progression, pathology spreads to cortex and cortical afferents become dysfunctional or degenerate, reducing the SDR. In many patients, SDR decline is paralleled by other extrastriatal pathologies that contribute to disability.
An important conceptual limitation of this model is that it is based on phenomena occurring in a multisystem neurodegenerative disorder. Analogies between clinical phenomena and normal actions may be confounded by compensatory mechanisms in the disease state. Pathologies outside the basal ganglia may also be salient (as discussed briefly in the context of the SDR). Nonetheless, this model appears to identify important areas for future investigation and suggests some testable predictions.
This model also suggests an alternative way of looking at PD progression (Figure 7b). Important milestones for PD patients include the emergence of motor fluctuations and DRT non-responsive symptoms like cognitive and postural-gait deficits.97
The intervals between these milestones can be characterized in terms of changes in the 3 different striatal dopamine signaling actions. The early “honeymoon” period of treated PD is characterized by loss of phasic striatal dopamine signaling, but DRT partially restores corticostriate synaptic plasticity and partially normalizes reward/effort scaling (i.e, vigor). A second phase is characterized by decline of the mechanism(s) underpinning estimation of appropriate levels of vigor. In the final phase, extra-basal ganglia pathologies lead to non-DRT responsive features and decline of the SDR because of cortical pathologies impairing corticostriate plasticity.
Potential Clinical Implications
Our analysis may have some implications for contemporary clinical practice, clinical experiments, and novel therapeutic interventions. If the LDR is a function of tonic, stable, striatal dopamine signaling, then interventions to restore or maintain tonic signaling might be beneficial. A potentially relevant existing clinical intervention is continuous L-dopa delivery via carbidopa-levodopa intestinal gel (CLIG). A recent PET imaging experiment indicates that CLIG produces sustained, increased striatal dopamine levels.98 It would be potentially interesting to determine if CLIG improves or reinstates the LDR in more advanced PD subjects.
As dopamine agonists can induce and sustain the LDR, this could be a rationale for relatively early use of dopamine agonists. This would be a somewhat different rationale than prior, and largely unsupported, suggestions that early agonist treatment retards disease progression or delays the emergence of dyskinesias.99 Agonist treatment, however, comes with increased risk of side-effects, notably impulse control disorders, compared to L-dopa preparations. A plausible explanation for the lower therapeutic index of dopamine agonists is a variant of the “over-dose” hypothesis.100 Dopamine agonists indiscriminately activate dopamine receptors in all parts of the striatal complex, including the ventral striatum. With relative preservation of nigrostriatal innervation in the ventral striatum, treatment with dopamine agonists likely causes pathological activation of ventral striatal dopamine receptors, distorting the motivational functions of this part of the striatal complex.
An intervention that improved tonic dopamine signaling in the dorsal striatum selectively might sustain the LDR without the risk of dopamine agonist associated impulse control disorders. Novel gene therapy methods may allow sub-regionally targeted and modulated increases in tonic dopamine signaling in the PD striatum.101,102 Clinical trials are underway with some of these approaches and it might be possible to assess LDR effects of these interventions.
As discussed above (Potential Neuronal Mechanisms of the LDR), it is also plausible that the LDR results from basal ganglia circuit changes and that STN DBS might induce LDR-like effects. If correct, this would be an additional rationale for earlier use of STN DBS. STN DBS induction of enhanced LDR or LDR-like effects might partly explain the reported benefits of relatively early use of STN DBS.103 Evaluation of potential LDR effects should be feasible in this patient population and might cast light on LDR mechanisms.
Further study of the LDR in the context of existing and novel clinical interventions has the potential to both improve understanding of this important phenomenon and improve clinical practice.
Acknowledgments
We thank two anonymous reviewers for helpful criticisms. We thank Bill Dauer and Kent Berridge for useful criticisms, Josh Berke for sharing an unpublished manuscript, and Josh Dudman and Peter Kempster for supplying part of a figure. Supported by R21 NS088302, R56 NS082941, P50 NS091856, K08 NS072183, and the Michael J. Fox Foundation.
Footnotes
Author Contributions: Design and conception – RLA & DKL. Drafting and revision of manuscript – RLA & DKL. Design and revision of figures – DKL & RLA.
Potential Conflicts of Interest:
Neither Dr. Albin nor Dr. Leventhal have any financial relationships that could be perceived as a conflict of interest.
References
- 1.Schultz W. Multiple Dopamine Functions at Different Time Courses. Ann Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- 2.Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. New Eng J Med. 2004;351:2498–2508. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- 3.Cotzias GC, Van Woert MH, Schiffer LM. Aromatic amino acids and modification of parkinsonism. N Engl J Med. 1967;276:374–379. doi: 10.1056/NEJM196702162760703. [DOI] [PubMed] [Google Scholar]
- 4.Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med. 1969;280:337–345. doi: 10.1056/NEJM196902132800701. [DOI] [PubMed] [Google Scholar]
- 5.Muenter MD, Tyce GM. L-dopa therapy of Parkinson’s disease: plasma L-dopa concentration, therapeutic response, and side effects. Mayo Clin Proc. 1971;46:231–239. [PubMed] [Google Scholar]
- 6.Anderson E, Nutt J. The long-duration response to levodopa: phenomenology, potential mechanisms and clinical implications. Parkinsonism Relat Disord. 2011;17:587–592. doi: 10.1016/j.parkreldis.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 7.Nutt JG, Holford NH. The response to levodopa in Parkinson’s disease: imposing pharmacological law and order. Ann Neurol. 1996;39:561–573. doi: 10.1002/ana.410390504. [DOI] [PubMed] [Google Scholar]
- 8.Holford NH, Chan PL, Nutt JG, et al. Disease progression and pharmacodynamics in Parkinson disease - evidence for functional protection with levodopa and other treatments. J Pharmacokinet Pharmacodyn. 2006;33:281–311. doi: 10.1007/s10928-006-9012-6. [DOI] [PubMed] [Google Scholar]
- 9.Nutt JG, Carter JH, Van Houten L, Woodward WR. Short- and long-duration responses to levodopa during the first year of levodopa therapy. Ann Neurol. 1997;42:349–355. doi: 10.1002/ana.410420311. [DOI] [PubMed] [Google Scholar]
- 10.Nutt JG, Woodward WR, Carter JH, Gancher ST. Effect of long-term therapy on the pharmacodynamics of levodopa. Relation to on-off phenomenon. Arch Neurol. 1992;49:1123–1130. doi: 10.1001/archneur.1992.00530350037016. [DOI] [PubMed] [Google Scholar]
- 11.Nutt JG, Carter JH, Lea ES, Sexton GJ. Evolution of the response to levodopa during the first 4 years of therapy. Ann Neurol. 2002;51:686–693. doi: 10.1002/ana.10189. [DOI] [PubMed] [Google Scholar]
- 12.Hauser RA, Holford NH. Quantitative description of loss of clinical benefit following withdrawal of levodopa-carbidopa and bromocriptine in early Parkinson’s disease. Mov Disord. 2002;17:961–968. doi: 10.1002/mds.10226. [DOI] [PubMed] [Google Scholar]
- 13.Zappia M, Nicoletti A. The role of the long-duration response to levodopa in Parkinson’s disease. J Neurol. 2010;257:S284–S287. doi: 10.1007/s00415-010-5731-0. [DOI] [PubMed] [Google Scholar]
- 14.Zappia M, Oliveri RL, Montesanti R, et al. Loss of long-duration response to levodopa over time in PD: implications for wearing-off. Neurology. 1999;52:763–767. doi: 10.1212/wnl.52.4.763. [DOI] [PubMed] [Google Scholar]
- 15.Stocchi F, Berardelli A, Vacca L, et al. Apomorphine infusion and the long-duration response to levodopa in advanced Parkinson’s disease. Clin Neuropharmacol. 2003;26:151–155. doi: 10.1097/00002826-200305000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Wider C, Russmann H, Villemure JG, et al. Long-duration response to levodopa in patients with advanced Parkinson disease treated with subthalamic deep brain stimulation. Arch Neurol. 2006;63:951–955. doi: 10.1001/archneur.63.7.951. [DOI] [PubMed] [Google Scholar]
- 17.Ding C, Ganesvaran G, Alty JE, et al. Study of levodopa response in Parkinson’s disease: Observations on rates of motor progression. Mov Disord. 2016;31:589–592. doi: 10.1002/mds.26497. [DOI] [PubMed] [Google Scholar]
- 18.Ganga G, Alty JE, Clissold BG, et al. Longitudinal study of levodopa in Parkinson’s disease: effects of the advanced disease phase. Mov Disord. 2013;28:476–481. doi: 10.1002/mds.25335. [DOI] [PubMed] [Google Scholar]
- 19.Alty JE, Clissold BG, McColl CD, et al. Longitudinal study of the levodopa motor response in Parkinson’s disease: relationship between cognitive decline and motor function. Mov Disord. 2009;24:2337–2343. doi: 10.1002/mds.22800. [DOI] [PubMed] [Google Scholar]
- 20.Clissold BG, McColl CD, Reardon KR, Shiff M, Kempster PA. Longitudinal study of the motor response to levodopa in Parkinson’s disease. Mov Disord. 2006;21:2116–2121. doi: 10.1002/mds.21126. [DOI] [PubMed] [Google Scholar]
- 21.Nutt JG, Carter JH, Woodward WR. Long-duration response to levodopa. Neurology. 1995;45:1613–1616. doi: 10.1212/wnl.45.8.1613. [DOI] [PubMed] [Google Scholar]
- 22.Zappia M, Oliveri RL, Bosco D, et al. The long-duration response to L-dopa in the treatment of early PD. Neurology. 2000;54:1910–1915. doi: 10.1212/wnl.54.10.1910. [DOI] [PubMed] [Google Scholar]
- 23.Nutt JG, Carter JH. Apomorphine can sustain the long-duration response to L-DOPA in fluctuating PD. Neurology. 2000;54:247–250. doi: 10.1212/wnl.54.1.247. [DOI] [PubMed] [Google Scholar]
- 24.Barbato L, Stocchi F, Monge A, et al. The long-duration action of levodopa may be due to a postsynaptic effect. Clin Neuropharmacol. 1997;20:394–401. doi: 10.1097/00002826-199710000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Stocchi F, Vacca L, Berardelli A, et al. Long-duration effect and the postsynaptic compartment: study using a dopamine agonist with a short half-life. Mov Disord. 2001;16:301–305. doi: 10.1002/mds.1070. [DOI] [PubMed] [Google Scholar]
- 26.Nutt JG, Nygaard TG. Response to levodopa treatment in dopa-responsive dystonia. Arch Neurol. 2001;58:905–910. doi: 10.1001/archneur.58.6.905. [DOI] [PubMed] [Google Scholar]
- 27.de la Fuente-Fernández R, Schulzer M, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol. 2011;69:803–810. doi: 10.1002/ana.22284. [DOI] [PubMed] [Google Scholar]
- 28.Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Lorenzo Alho AT, Suemoto CK, et al. Three-dimensional and stereological characterization of the human substantia nigra during aging. Brain Struct Funct. 2016;221:3393–3403. doi: 10.1007/s00429-015-1108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuda W, Furuta T, Nakamura KC, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haber SN. The place of dopamine in the cortico-basal ganglia circuit. Neuroscience. 2014;282:248–257. doi: 10.1016/j.neuroscience.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- 33.Glimcher PW. Understanding dopamine and reinforcement learning: the dopamine reward prediction error hypothesis. Proc Natl Acad Sci USA. 2011;108:15647–15654. doi: 10.1073/pnas.1014269108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keiflin R, Janak PH. Dopamine Prediction Errors in Reward Learning and Addiction: From Theory to Neural Circuitry. Neuron. 2015;88:247–263. doi: 10.1016/j.neuron.2015.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz W. Neuronal Reward and Decision Signals: From Theories to Data. Physiol Rev. 2015;95:853–951. doi: 10.1152/physrev.00023.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- 37.Bayer HM, Glimcher PW. Midbrain dopamine neurons encode a quantitative reward prediction error signal. Neuron. 2005;47:129–141. doi: 10.1016/j.neuron.2005.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berridge KC. The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology (Berl) 2007;191:391–431. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- 39.Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Flagel SB, Clark JJ, Robinson TE, et al. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469:53–67. doi: 10.1038/nature09588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parker NF, Cameron CM, Taliaferro JP, et al. Reward and choice encoding in terminals of midbrain dopamine neurons depends on striatal target. Nat Neurosci. 2016;19:845–854. doi: 10.1038/nn.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howe MW, Dombeck DA. Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature. 2016;535:505–510. doi: 10.1038/nature18942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barter JW, Li S, Lu D, Bartholomew RA, et al. Beyond reward prediction errors: the role of dopamine in movement kinematics. Front Integr Neurosci. 2015;9:39. doi: 10.3389/fnint.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dodson PD, Dreyer JK, Jennings KA, et al. Representation of spontaneous movement by dopaminergic neurons is cell-type selective and disrupted in parkinsonism. Proc Natl Acad Sci USA. 2016;113:E2180–8. doi: 10.1073/pnas.1515941113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin X, Costa RM. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature. 2010;466:457–462. doi: 10.1038/nature09263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin HH. The basal ganglia in action. Neuroscientist. 2016 doi: 10.1177/1073858416654115. pii: 1073858416654115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fee MS. The role of efference copy in striatal learning. Curr Opin in Neurobiol. 2014;25:194–200. doi: 10.1016/j.conb.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fee MS. Oculomotor learning revisited: a model of reinforcement learning in the basal ganglia incorporating an efference copy of motor actions. Fron Neural Circ. 2012;6:1–18. doi: 10.3389/fncir.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gagagkar V, Puzerey PA, Chen R, et al. Dopamine neurons encode performance error in singing birds. Science. 2016;354:1278–1282. doi: 10.1126/science.aah6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohnen NI, Albin RL, Koeppe RA, et al. Positron emission tomography of monoaminergic vesicular binding in aging and Parkinson disease. J Cereb Blood Flow Metab. 2006;26:1198–1212. doi: 10.1038/sj.jcbfm.9600276. [DOI] [PubMed] [Google Scholar]
- 51.Garris PA, Walker QD, Wightman RM. Dopamine release and uptake rates both decrease in the partially denervated striatum in proportion to the loss of dopamine terminals. Brain Res. 1997;753:225–234. doi: 10.1016/s0006-8993(97)00003-6. [DOI] [PubMed] [Google Scholar]
- 52.Janezic S, Threlfell S, Dodson PD, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci USA. 2013;110:E4016–25. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cotzias GC, Papavasiliou PS, Fehling C, Kaufman B, Mena I. Similarities between neurologic effects of L-dopa and of apomorphine. N Eng J Med. 1970;282:31–33. doi: 10.1056/NEJM197001012820107. [DOI] [PubMed] [Google Scholar]
- 54.Yttri EA, Dudman JT. Opponent and bidirectional control of movement velocity in the basal ganglia. Nature. 2016;533:402–406. doi: 10.1038/nature17639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calabresi P, Ghiglieri V, Mazzocchetti P, Corbelli I, Picconi B. Levodopa-induced plasticity: a double-edged sword in Parkinson’s disease? Philos Trans R Soc Lond B Biol Sci. 2015;370 doi: 10.1098/rstb.2014.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Filippo M, Picconi B, Tantucci M, et al. Short-term and long-term plasticity at corticostriatal synapses: implications for learning and memory. Behav Brain Res. 2009;199:108–118. doi: 10.1016/j.bbr.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 57.Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 58.Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 60.Pawlak V, Wickens JR, Kirkwood A, Kerr JN. Timing is not everything: Neuromodulation opens the STDP gate. Front Synaptic Neurosci. 2010;2:146. doi: 10.3389/fnsyn.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Kai L, Day M, et al. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 63.Izhikevich EM. Solving the distal reward problem through linkage of STDP and dopamine signaling. Cerebral Cortex. 2007;17:2443–2452. doi: 10.1093/cercor/bhl152. [DOI] [PubMed] [Google Scholar]
- 64.de la Fuente-Fernández R, Sossi V, Huang Z, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747–2754. doi: 10.1093/brain/awh290. [DOI] [PubMed] [Google Scholar]
- 65.de la Fuente-Fernández R, Schulzer M, Mak E, Calne DB, Stoessl AJ. Presynaptic mechanisms of motor fluctuations in Parkinson’s disease: a probabilistic model. Brain. 2004;127:888–899. doi: 10.1093/brain/awh102. [DOI] [PubMed] [Google Scholar]
- 66.Politis M, Wu K, Loane C, Brooks DJ, Kiferle L, Turkheimer FE, Bain P, Molloy S, Piccini P. Serotoninergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J Clin Invest. 2014 Mar;124(3):1340–9. doi: 10.1172/JCI71640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Braak H, Del Tredici K. Cortico-basal ganglia-cortical circuitry in Parkinson’s disease reconsidered. Exp Neurol. 2008;212:226–229. doi: 10.1016/j.expneurol.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 68.Zhuang X, Mazzoni P, Kang UJ. The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nat Rev Neurosci. 2103;9:248–256. doi: 10.1038/nrneurol.2013.57. [DOI] [PubMed] [Google Scholar]
- 69.Beeler JA, Cao ZF, Kheirbek MA, et al. Dopamine-dependent motor learning: insight into levodopa’s long-duration response. Ann Neurol. 2010;67:639–647. doi: 10.1002/ana.21947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dowd E, Dunnett SB. Movement without dopamine: striatal dopamine is required to maintain but not to perform learned actions. Biochem Soc Trans. 2007;35:428–432. doi: 10.1042/BST0350428. [DOI] [PubMed] [Google Scholar]
- 71.Dowd E, Dunnett SB. Comparison of 6-hydroxydopamine-induced medial forebrain bundle and nigrostriatal terminal lesions in a lateralised nose-poking task in rats. Behav Brain Res. 2005;159:153–161. doi: 10.1016/j.bbr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 72.Leventhal DK, Stoetzner C, Abraham R, et al. Dissociable effects of dopamine on learning and performance within sensorimotor striatum. Basal Ganglia. 2014;4:43–54. doi: 10.1016/j.baga.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salamone JD, Correa M, Farrar AM, et al. Dopamine, behavioral economics, and effort. Front Behav Neurosci. 2009;3:13. doi: 10.3389/neuro.08.013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76:470–485. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beeler JA, Frazier CR, Zhuang X. Putting desire on a budget: dopamine and energy expenditure, reconciling reward and resources. Front Integr Neurosci. 2012;6:49. doi: 10.3389/fnint.2012.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mazzoni P, Hristova A, Krakauer JW. Why don’t we move faster? Parkinson’s disease, movement vigor, and implicit motivation. J Neurosci. 2007;27:7105–7116. doi: 10.1523/JNEUROSCI.0264-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baraduc P, Thobois S, Gan J, et al. A common optimization principle for motor execution in healthy subjects and parkinsonian patients. J Neurosci. 2013;33:665–677. doi: 10.1523/JNEUROSCI.1482-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chong TT, Bonnelle V, Manohar S, et al. Dopamine enhances willingness to exert effort for reward in Parkinson’s disease. Cortex. 2015;69:40–46. doi: 10.1016/j.cortex.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gepshtein S, Li X, Snider J, Plank M, Lee D, Poizner H. Dopamine function and the efficiency of human movement. J Cogn Neurosci. 2014;26:645–65. doi: 10.1162/jocn_a_00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beierholm U, Guitart-Masip M, Economides M, et al. Dopamine modulates reward-related vigor. Neuropsychopharmacology. 2013;38:1495–1503. doi: 10.1038/npp.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Turner RS, Desmurget M. Basal ganglia contributions to motor control: a vigorous tutor. Curr Opin Neurobiol. 2010;20:704–716. doi: 10.1016/j.conb.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panigrahi B, Martin KA, Li Y, et al. Dopamine Is Required for the Neural Representation and Control of Movement Vigor. Cell. 2015;162:1418–1430. doi: 10.1016/j.cell.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 83.Cagniard B, Beeler JA, Britt JP, et al. Dopamine scales performance in the absence of new learning. Neuron. 2006;51:541–547. doi: 10.1016/j.neuron.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 84.Niv Y, Daw ND, Joel D, Dayan P. Tonic dopamine: opportunity costs and the control of response vigor. Psychopharmacology (Berl) 2007;191:507–520. doi: 10.1007/s00213-006-0502-4. [DOI] [PubMed] [Google Scholar]
- 85.Hamid AA, Pettibone JR, Mabrouk OS, et al. Melolimbic dopamine signals the value of work. Nature Neurosci. 2015;19:117–126. doi: 10.1038/nn.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beeler JA, Daw N, Frazier CR, Zhuang X. Tonic dopamine modulates exploitation of reward learning. Front Behav Neurosci. 2010;4:170. doi: 10.3389/fnbeh.2010.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muranishi M, Inokawa H, Yamada H, et al. Inactivation of the putamen selectively impairs reward history-based action selection. Exp Brain Res. 2011;209:235–246. doi: 10.1007/s00221-011-2545-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang AY, Miura K, Uchida N. The dorsomedial striatum encodes net expected return, critical for energizing performance vigor. Nat Neurosci. 2013;16:639–647. doi: 10.1038/nn.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang J, Nebeck S, Muralidharan A, et al. Coordinated reset deep brain stimulation of subthalamic nucleus produces long-lasting, dose-dependent motor improvements in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine non-human primate model of parkinsonism. Brain Stimul. 2016;9:609–617. doi: 10.1016/j.brs.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fan KY, Baufreton J, Surmeier DJ, Chan CS, Bevan MD. Proliferation of external globus pallidus-subthalamic nucleus synapses following degeneration of midbrain dopamine neurons. J Neurosci. 2012;32:13718–13728. doi: 10.1523/JNEUROSCI.5750-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sebel LE, Graves SM, Chan CS, Surmeier DJ. Haloperidol Selectively Remodels Striatal Indirect Pathway Circuits. Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Day M, Wang Z, Ding J, An X, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- 93.Fieblinger T, Graves SM, Sebel LE, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gittis A, Hang G, LaDow E, et al. Rapid Target-Specific Remodeling of Fast-Spiking Inhibitory Circuits after Loss of Dopamine. Neuron. 2011;71:858–868. doi: 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Suárez LM, Solís O, Caramés JM, et al. L-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol Psychiatry. 2014;75:711–722. doi: 10.1016/j.biopsych.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y, Meredith GE, Mendoza-Elias N, et al. Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J Neurosci. 2013;33:11655–11667. doi: 10.1523/JNEUROSCI.0288-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, Barker RA. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry. 2013;84:1258–1264. doi: 10.1136/jnnp-2013-305277. [DOI] [PubMed] [Google Scholar]
- 98.Politis M, Sauerbier A, Loane C, et al. Sustained striatal dopamine levels following intestinal levodopa infusions in Parkinson’s disease patients. Mov Dis. 2017;32:235–240. doi: 10.1002/mds.26848. [DOI] [PubMed] [Google Scholar]
- 99.Albin RL, Frey KA. Initial agonist treatment of Parkinson disease: A critique. Neurology. 2003;60:390–394. doi: 10.1212/01.wnl.0000052681.28286.52. [DOI] [PubMed] [Google Scholar]
- 100.Cools R. Dopaminergic modulation of cognitive function – implications for L-DOPA treatment in Parkinson’s disease. Neurosci Behav Rev. 2006;30:1–23. doi: 10.1016/j.neubiorev.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 101.Cederfjall E, Broom L, Kirik D. Controlled striatal DOPA production from a gene delivery system in a rodent model of Parkinson’s disease. Mol Ther. 2015;23:896–906. doi: 10.1038/mt.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lohr KM, Miller GW. VMAT2 and Parkinson’s disease: harnessing the dopamine vesicle. Expert Rev Neurother. 2014;14:1115–1117. doi: 10.1586/14737175.2014.960399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deuschl G, Rau J, Knudsen K, et al. Neurostimulation for Parkinson’s disease with early motor complications. New Eng J Med. 2013;368:610–622. doi: 10.1056/NEJMoa1205158. [DOI] [PubMed] [Google Scholar]