

Abstract
Single-cell flow cytometric techniques have been indispensable to improving our understanding of the phenotype and function of immune cell subsets that are important in both rejection and tolerance post-transplant. Mass cytometry, or Cytometry by Time-of-Flight (CyTOF), is a single cell–based platform that utilizes antibodies conjugated to rare heavy metal ions for analysis of cellular proteins by a time-of-flight mass spectrometer. This new technology allows for the evaluation of over 40 simultaneous cellular parameters in a single sample because the limitation of spectral overlap, seen in conventional flow cytometry, is eliminated. In this review we discuss the current state of mass cytometry, describe the advantages and disadvantages over multi-parameter flow cytometry, introduce novel methods of high-dimensional data analysis and visualization, and review some recent studies using mass cytometry to profile the immune system of healthy people, and transplant recipients.
Introduction
Fluorescence-based flow cytometry has clearly become the standard choice for phenotypic and functional analysis of single cells and is crucial to our understanding of the immune response post-transplantation. Indeed, fluorescence-labeled antibodies and flow cytometry have been essential tools for both immune monitoring and basic science research in immunology, and indispensable to the field of transplantation. Advances in technology, including new fluorophores and tools to analyze and display data now allow for the quantitation of 15–20 parameters on a single cell [1]. However, spectral overlap or spillover between fluorescent signals, limits the expansion of the flow cytometry platform to additional parameters. An alternative technique, mass cytometry, also known as cytometry by time-of-flight or commercially known as CyTOF, has been developed [2]. In contrast to flow cytometry where the fluorescently-tagged cellular proteins are excited by lasers and quantitated via optical filters and photomultiplier tubes, mass cytometry utilizes antibodies conjugated to rare heavy-metal isotopes. The isotopes are covalently linked to antibodies or other probes via a chelator protein. The workflow of labeling cells for mass cytometry is very similar to that of flow cytometry (Figure 1). After labeling with antibodies, the cells as a single-cell suspension are introduced into the nebulizer where the cells are deposited in a fine spray of droplets. Cells then travel through an argon plasma, covalent bonds are broken to produce free atoms which become charged [3]. The resulting ion cloud is filtered through a quadropole, selecting for heavy-metal reporter ions of mass range 80–200 which are the separated by their mass-to-charge ratio in a time-of-flight detector [1]. Ion counts are then converted to electrical signals and integrated into single-cell events [3]. Since the readout of atomic masses is very discrete, the potential for higher multiplexing is substantially enhanced.
Figure 1. Workflow of a Typical Mass Cytometry Experiment.
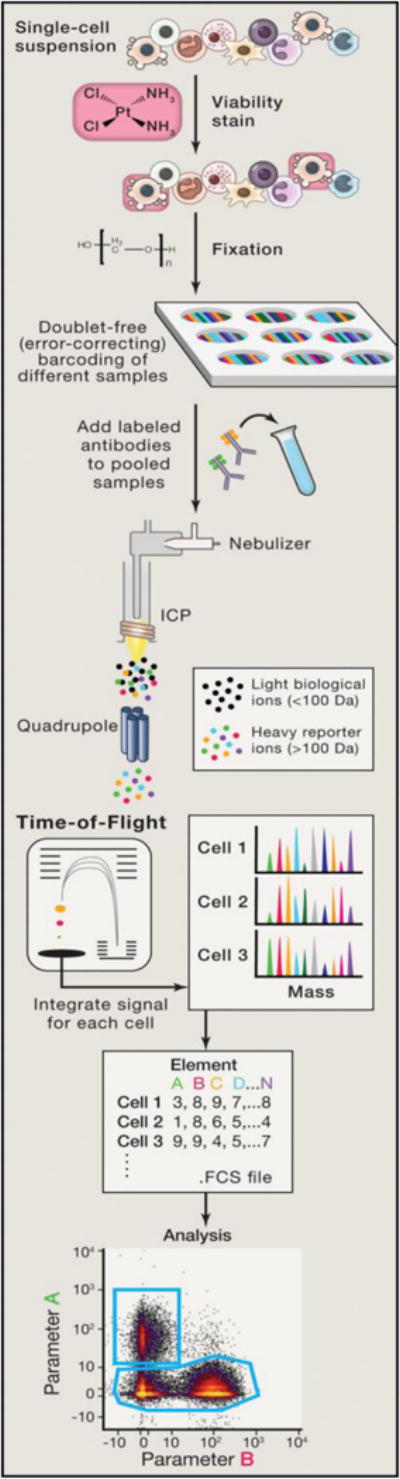
Single cells are acquired, and a viability stain is applied to mark dead cells for exclusion from analyses. Fixation can optionally be applied at this point to preserve the cell state. Multiple samples can be barcoded with unique combinations of heavy metal tags, enabling them to be pooled together prior to staining to minimize technical variability at this step. After pooling samples into one tube, cells are then incubated with antibodies targeted against proteins of interest. Cell permeabilization can be performed if intracellular targets are to be measured. Cells are nebulized into droplets as they are introduced into the mass cytometer. They then travel into an inductively coupled argon plasma (ICP), in which covalent bonds are broken and ions are liberated. The ion cloud is filtered by a quadrupole to remove common biological elements and enrich the heavy metal reporter ions to be quantified by time-ˇof-ˇflight mass spectrometry. Ion signals are integrated on a per-ˇcell basis, resulting in single-ˇcell measurements for downstream analysis. Data are compiled in an FCS file that can then be parsed and plotted in a variety of ways (from Spitzer and Nolan, Cell 165:780-790;2016). FCS, Flow Cytometry Standard.
Comparison of Mass Cytometry and Flow Cytometry
Since the first report of single-cell mass cytometry in 2009 from a group at the University of Toronto [2], substantial developments and improvements have occurred. In terms of the hardware, the commercially available Helios system (Fluidigm) has 135 detection channels, allowing flexibility as more metal tags are developed and is barcode enabled.
In the initial mass cytometry reports, in was necessary for investigators to conjugate the heavy-metal isotopes to the antibodies for use in experiments. This has been simplified recently by the commercial availability of metal labeling kits and pre-conjugated antibodies (Fluidigm) although only a few hundred unique pre-conjugated antibodies are currently available for either mouse or human. A challenge in all cytometry experiments is the development of the antibody panel. In flow cytometry, especially for multiparameter experiments (>12 colors) it is critical that the spectral overlap between fluorescent dyes is considered in the assignment of antibodies to fluorochome channels [4]. Although overlap between fluorochromes is not a concern for mass cytometry, the mass of the metal when paired to a specific antibody must be considered as the high and low end of the mass window (80–200) have somewhat lower intensities [5]. Thus markers with higher expression could be used in dimmer channels while markers with lower expression should be used in the channels with maximum sensitivity, which are centered just higher than the middle of the range. It is important to validate each antibody by testing cell subsets known to express and those that do not express the specific marker and in some cases to compare the staining results to those obtained with the same antibody used in the flow cytometry configuration. There is debate as to whether the controls typically used for flow cytometry, such as isotype-matched control antibodies or fluorescence-minus-one-like controls, are useful for mass cytometry [6]. Optimization of the antibody panel, including shuffling antibody and metal pairs, is necessary for best results. Pre-configured screening panels of 5–17 markers to determine basic mouse or human phenotyping or functions are commercially available (Fluidigm) and are a good starting point for analyses.
Similar to flow cytometry the use of barcodes for multiplexing mass cytometry has been reported by several groups [7–9]. One example uses seven metals in a binary fashion to generate 128 barcodes to uniquely label each well of a 96-well plate. Individual wells are then combined, into one tube, for further sample processing and later debarcoded for analysis [7]. A commercially available barcoding kit (Fluidigm) enables unique barcoding of 20 samples which are subsequently stained and acquired as one multiplexed sample. Clearly there are many advantages to cellular barcoding including the requirement for smaller antibody volumes and reduced time on the machine for data acquisition. Importantly, data quality is improved by barcoding as variability between samples is minimized. Further, immune cell subsets could be detected by mass cytometry from only 10,000 cells, potentially expanding analysis to patient samples that were previously deemed too limited in cell numbers to analyze [10]. Since mass cytometry processes only a fraction of the events/second that a flow cytometer can, there is drift of signal intensity over time that could introduce channel bias. Further, “batch effects” are seen between runs thus a good quality control sample should be used to account for system variations.
Mass cytometry does have some technical limitations when compared to flow cytometry (Table 1). Flow cytometry can be performed on live cells with recovery of sorted cells for subsequent functional analysis. In contrast, the cells must be fixed for mass cytometry, and the cells are ultimately vaporized and thus not recoverable. Further, unlike flow cytometry where virtually all of the cells in the sample can be analyzed, in mass cytometry a large proportion of cells in the sample are lost in the instrument resulting in less than 50% of the injected cell sample being analyzed [11]. Forward and side scatter can’t currently be measured on a mass cytometer thus the ability to easily discriminate size and granularity to distinguish between granulocytes, lymphocytes, cell doublets and cellular debris, is lost. To compensate for the lack of light scattering properties, and to assure that all cells are counted (cells will only be identified if they are bound to a metal within the mass range of the mass cytometer), cells containing DNA are labeled with iridium-containing intercalators. Further, live-dead stains such as cisplatin are used to determine viability. [5]. Moreover, the sensitivity of tagged antibodies used in mass cytometry is lower than the same antibodies used for flow cytometry since the chelating polymer that is generally used allows a maximum of 100 metal reporter ions to be attached to an antibody molecule [1]. Thus, the molecules/cell detected by mass cytometry is ten-fold less than can be detected by flow cytometry, consequently if a cellular protein is in low abundance, it may not be detected by mass cytometry. Currently, mass cytometry is in its infancy and thus it can be expected that improvements in technology will minimize the limitations and enhance utility.
Table 1.
Comparison of features of flow cytometry and mass cytometry
| Fluorescence Flow Cytometry | Mass Cytometry | |
|---|---|---|
| Probes | fluorescent | stable mass isotope |
| Max number of measurements (current) | 19 | 37 |
| Compensation | Compensation necessary to avoid signal overlap and spillover | discrete signals, minimal overlap |
| Sensitivity (molecules/cell) | 40 | 400–500 |
| Sampling efficiency | >95% | <50% |
| Acquisition rate/s | > 25000 | 500 |
| Light scatter properties | forward and side scatter measured | not measured (DNA stains used to distinguish cells) |
| Cellular proteins measured | surface markers, intracellular cytokines, signaling proteins | surface markers, intracellular cytokines, signaling proteins |
| Cell preparation | live or fixed cells | fixed cells resuspended in pure water |
| Cell sorting | cells can be sorted for functional assays | cells are destroyed, no sorting possible |
| Cellular Barcoding and Sample Multiplexing | yes | yes |
| Data files | Flow Cytometry Standard (FCS) | Flow Cytometry Standard (FCS) |
| Data analysis | generally user-guided | requires newer complex analysis techniques |
| robe cost (per test) | $2.00–$5.00 | $2.00–$5.00 |
Analysis and Visualization of Mass Cytometry Data
The development of mass cytometry has ushered in a new era in analysis and visualization of the immune system. While traditional flow cytometers struggle to distinguish around 18 different markers per cell due to fluorescence spectra overlap, mass cytometers routinely examine around 40 or more markers per cell simultaneously. What this means for immunologists studying the human immune response is that many new combinations of markers can be examined, allowing researchers to discover and define new, more specific subsets of cells with potentially important and unique functions. Additionally, rather than focusing on certain branches of the immune system (e.g., CD4+ T cells) for particular experiments or analyses, a mass cytometry experiment can gain a much more comprehensive, systems view of the state of the immune system in patients by examining all branches at once.
While traditional techniques involving manual gating can still be used, the very strengths gained by using so many markers (i.e., finding new populations of biologically-relevant cells defined by unique combinations of makers) won’t be realized in this manner. One of the first steps in analyzing high dimensional data should be to visualize it. When working with flow cytometry data, researchers routinely plot cells based on two markers in bi-axial plots, allowing them to identify key cell populations. The human brain has a remarkable ability to discover patterns, however it is not suited to finding these patterns when dimensions exceed three or four. With high dimensional data, we can use dimensionality reduction techniques to bring the dimensionality of the data down to a level that is suited for plotting. These techniques attempt to preserve the relationships between points in high dimensional space (in our case, cells defined by the expression of ~40 markers) in a lower dimensional setting (typically two or three). One of the most commonly used techniques in this area is principal components analysis (PCA) (Figure 2).
Figure 2. Mass cytometry (CyTOF) data on PBMCs from a pediatric liver transplant recipient [31] is depicted using three different algorithms: PCA, tSNE, and SPADE.
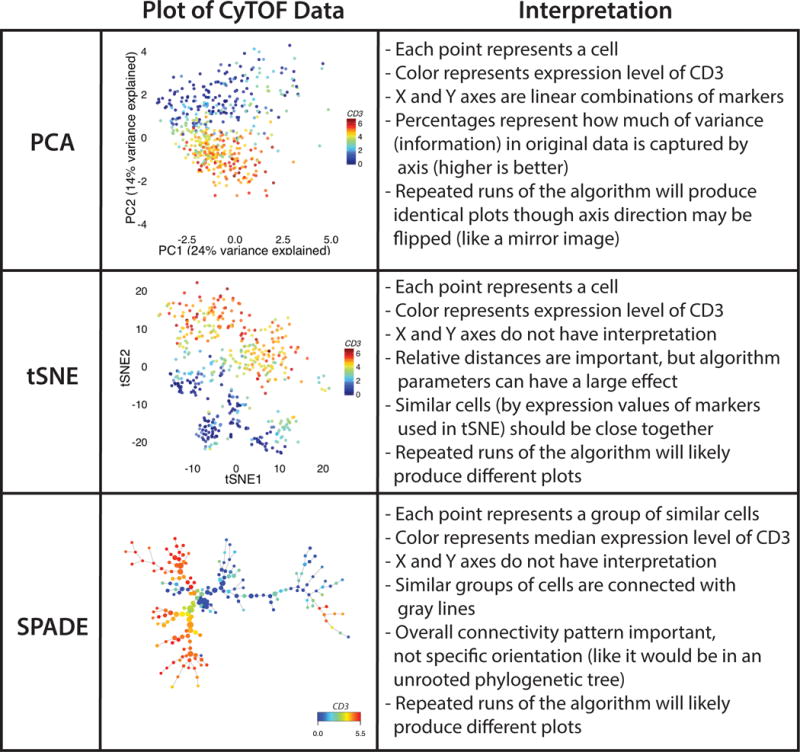
In each plot, the color corresponds to the level of CD3 on each cell (or group, in the case of SPADE). With PCA, one can see general separation of two different branches of the immune system. T cells are in the lower half of the plot and non-T cells are in the upper half. Replotting the same data using tSNE reveals a similar separation, but one can now see two groups of CD3+ cells in the upper half of the plot (which represent CD4 and CD8 T cells) and other groups of cells as well. Using SPADE similarly reveals the major branches of the immune system, including the separation of CD4 and CD8 T cells (the two branches on the left). PBMC, peripheral blood mononuclear cell; PCA, principal component analysis; SPADE, spanning tree progression analysis of density normalized events.
To understand PCA without getting into the linear algebra behind it, it’s best to think about how to represent a 3D object in two dimensions (and then extend that notion into higher dimensions). If you have data points in 3D that are laid out to form a cylinder, PCA would choose a side view as a vantage point since most of the variation in the data lies along that axis, rather than a head-on view where most of the data points would be on top of each other. Of course, this is a simplification of what is actually going on, but the underlying intuition with PCA should be that it rotates your high dimensional data so that your vantage point (in a lower dimension) preserves the most information about the data. The new axes (principal components) which define your vantage point are simple linear combinations of the original variables, making the axes interpretable. In practice, PCA will tell you how much of the cumulative variance is explained by the number of principal components chosen (typically the first two or three). This will give you an idea about how much information is being lost while doing this dimensionality reduction. Many researchers aim for at least 75% of the total variance in the data to be explained by the components chosen.
The strengths of PCA are that is well-established, interpretable (the PCA axes are just weighted combinations of the original variables), simple, and consistent (there is only one best rotation). The chief drawback of PCA is its linear nature. To understand that, imagine your data were a spiral in 2D. What would the best representation of that data be in one dimension? Well, the key insight is that a spiral is effectively one dimensional, so you would like your data points to be plotted in one-dimension corresponding to their position along the curve of the spiral. How would PCA deal with this problem? The answer: not very well, since it would end up clumping points from different parts of the spiral together in the one dimensional representation. Despite this, PCA is still a very powerful first step in analyzing high dimensional data, but it is best to keep in mind its pitfalls.
In response to this problem, several techniques in non-linear dimensionality reduction have been developed. Of particular note, given its recent popularity in visualizing high dimensional flow data, is t-distributed stochastic neighbor embedding (t-SNE) [12]. t-SNE has been used in several recent papers to visualize mass cytometry data [13–15]. The goal of t-SNE is to create a faithful lower dimensional representation of high dimensional data that preserves the overall relationships between data points. When t-SNE begins, each data point in high dimensional space has a corresponding data point in two-dimensional space (on the plot). These points in 2D are laid out randomly at the start. For each data point, the distances to other points in 2D and in the original, higher dimensional space are used to calculate probabilities with the Gaussian distribution being used for the high dimensional distances and the t-distribution (from which the algorithm gets its name) being used in 2D. The algorithm then iterates in an attempt to make these probability distributions similar, which has the effect of moving similar data points in high dimensional space closer to each other. You can think of each point in 2D as being acted on by attractive and repulsive forces. These forces are determined by how its probability distribution in 2D space corresponds to its probability distribution in high dimensional space [12]. The points in 2D continue to move at each iteration until they stabilize. The end result is that points close together in 2D should be close together in high dimensional space. This pairwise distance calculation and movement for all points means that t-SNE is computationally intensive and may require a long time to complete with a large number of data points. Borrowing techniques from physics to solve N-body problems efficiently has speeded up the process, but it will still typically be much slower than something like PCA.
The strength of t-SNE is its non-linear nature, giving it the ability to represent complex non-linear relationships (e.g. spirals). This makes t-SNE particularly attractive for plotting immunological data where populations of interest may be contorted in many different ways. The drawbacks of t-SNE include its difficulty of interpretation (t-SNE axes do not have interpretation in and of themselves) and its stochastic nature (the problem does not always converge on the same solution, meaning subsequent t-SNE plots of the same data will vary). Nonetheless, t-SNE has now become a mainstream technique for the visualization of mass cytometry data. It can also be used in combination with manual gating (of the t-SNE plot) to identify populations of interest.
To make t-SNE plots more interpretable, Cheng et al. have developed a method, One-SENSE, which allows the researcher to create two or three groups of markers (e.g., trafficking markers and differentiation markers) and then plot cells in a t-SNE-like plot based on these groups. For each grouping, it performs t-SNE with a one-dimensional output using the markers in that group. Cells can then be plotted based on the output of these one dimensional t-SNEs creating 2 or 3D plots [13]. Each axis would be the one-dimensional t-SNE output using the markers in that user-defined group. This provides a little more insight into the clusters of cells in the resulting plots since the axes have more biological meaning.
One of the strengths of using such high dimensional data is the ability to identify new cell populations that would not have been found with a smaller number of markers. Several techniques have been developed which can identify populations automatically in high dimensional flow data. Many of these techniques are combined with a visualization component.
SPADE (spanning tree progression analysis of density normalized events) is probably the most well-known mass cytometry analysis technique as it was published in one of examples for the application of the mass cytometer to human samples [16]. The SPADE technique downsamples highly dense regions and then performs agglomerative hierarchical clustering on cells in high dimensional space [17]. The resulting clusters can then be visualized in a minimum spanning tree (a tree with the minimum total edge weight). In a SPADE tree, clusters of cells that are neighbors are similar to each other. It is important to note that since this is a tree structure, the x and y axes do not have intrinsic meaning (and branches can be freely rotated as in a mobile without changing the structure of the tree). A drawback of SPADE is the necessity to specify the number of clusters one expects to find in the data beforehand and the lack of single-cell resolution. However, SPADE remains an extremely useful tool for mass cytometry analysis.
t-SNE and other dimensionality reduction techniques can be used as a first step for downstream automated detection of populations. One method which uses this idea with mass cytometry data is ACCENSE (automatic classification of cellular expression by nonlinear stochastic embedding). The discovery of populations can occur directly in high dimensional space, but ACCENSE performs t-SNE on mass cytometry data first, distilling the relationships between cells down to just two dimensions. It then finds the populations within the resulting low dimensional representation [14]. The resulting populations can then be further examined for markers of interest. A strength of ACCENSE is that it does not require the user to specify the number of populations she or he expects to find in the data and it allows the researcher to examine each cell individually if needed.
Researchers analyzing mass cytometry data may also want to discover which populations are significantly different between treatment groups. CITRUS (cluster identification, characterization, and regression) from Bruggner et al. combines automated population discovery with regression [18]. In CITRUS, data from all samples are combined and hierarchical clustering is performed. The cluster proportions are then determined per sample and then these are fed into a regression model (e.g., LASSO logistic regression) along with the group to which the sample belongs. For typical CyTOF experiments, at least 8 samples per experimental group should be used in order to have the necessary statistical power and robustness. The utility of CITRUS is being able to find significantly different populations between experimental groups and the downstream analysis of these populations (which can be graphed for different markers to get an idea of what types of cells they are).
This section only provided a brief overview of the myriad of techniques being developed for use on mass cytometry data. Aghaeepour et al., have provided an excellent evaluation of many of these techniques based on the FlowCAP dataset for readers interested in how they compare to manual gating and how well they classify samples [19]. Thus far, most algorithms have been adapted from standard machine learning algorithms and applied to flow data, but few have begun to incorporate the expert knowledge of the experimentalist using the technique. What has become apparent is that this burgeoning field will require more intense collaboration between algorithm developers and immunologists in order to find lasting solutions that are useful and interpretable.
Application of Mass Cytometry to Immunology and Transplantation
Mass cytometry allows for the deep profiling of cellular subpopulations along with an unprecedented ability to define relationships between phenotype and function in both healthy people and during disease [13, 15, 20–28]. In one of the first comprehensive studies using mass cytometry, Bendall and colleagues dissected the functional complexity of hematopoiesis [16]. Single-cell analysis of 34 parameters performed on healthy bone-marrow cells. In addition to surface markers, the signaling behavior of specific cell subsets was examined after ex vivo stimulation. This unique systems-level view of hematopoiesis demonstrated novel and unexpected signaling responses during hematopoietic development. More recently, the same group used a similar strategy to comprehensively analyze normal B cell lymphopoiesis and identify a novel early B cell population [21].
Mass cytometry has been applied to several studies that examine T cell phenotypes and function in healthy people and during viral infections [13, 15, 24, 25, 29]. Newell and colleagues analyzed CD8+ T cell diversity including functional parameters such as cytokines, cytotoxic mediators and antigen-specific T cells [25]. They observed that there were more than 200 functional phenotypes represented by distinct CD8+ T cell subsets and that virus-specific cells have both shared and unique phenotypic and functional attributes depending on the virus. Recently the multiplexing capacity of mass cytometry, cellular barcoding, and high-dimensional analysis, was leveraged to simultaneously probe T cell trafficking and functional markers across eight different human tissues [15]. In addition to demonstrating that T cells exhibit diverse patterns of cytokine secretion in different tissue environments, mass cytometry also highlighted that combinatorial expression of trafficking receptors and cytokines better defines tissue specificity and suggesting the presence of tissue-specific signature. Similarly, the multiplexing power of mass cytometry was used to characterize human NK cell subsets. Human NK cells express a multitude of both activating and inhibitory receptors which regulate cell function Interestingly, the findings demonstrated the extraordinary phenotypic diversity of NK cell receptors and suggests that a given individual may produce up to 30,000 distinct subsets of NK cells [23].
One of the major advantages of mass cytometry is the ability to multiplex over 40 cellular measurements from samples where there is a limited sample volume, as is often the case in clinical samples [10]. In a recent study, the phenotypical and functional immune response to surgical trauma was examined [22]. Serial blood samples from 32 patients undergoing hip replacement were analyzed, by mass cytometry, for 35 cell-surface proteins and intracellular phospho-specific epitopes at 1 h, 24 h, 72 h, and 6 weeks after surgery. The results indicated that there is indeed an immune signature of surgical trauma and that clinical recovery is associated with expansion of subsets of CD14+ monocytes.
It is evident that mass cytometry has enormous potential to define biologically important phenotypic and functional, including cytokines and phospho-signaling, changes in the immune system after transplantation. This technology will enable researchers to study mechanistic differences in defined populations of cells to yield a system-wide view of responses to specific immunosuppressive regimes. Moreover, mass cytometry is particularly amenable to biomarker discovery since multiple parameters can simultaneously be examined at the single-cell level from clinical samples. To date, however there have been just two studies published that applied this technology specifically to transplantation. Yabu and colleagues [30] used mass cytometry to profile the peripheral immune system of 20 highly sensitized kidney transplant recipients who underwent desensitization therapy (10 responders and 10 non-responders) to lower HLA antibodies and enable transplantation. PBMC were obtained prior to the initiation of therapy and analyzed for 33 parameters by mass cytometry. They utilized a multivariate decision tree model and demonstrated that kidney transplant candidates with low numbers of transitional B cells and high numbers of Tregs were less likely to respond to desensitization treatment. In this study, all of the responders, and only three of the non-responders received a transplant and had similar borderline rejection episodes and graft outcomes at one year. Additional studies are clearly necessary to validate these findings and determine if the numbers of transitional B cells and Treg can be used to predict response to desensitization therapy.
Our group has utilized single-cell mass cytometry to characterize the immune cell populations in a group of operationally-tolerant pediatric liver transplant recipients. PBMC from seven operational tolerant recipients, eight recipients on low dose single agent tacrolimus and five age-matched controls were examined for 25 parameters (22 immune markers and 191Iridium, 193Iridium and 195cisplatin to distinguish single live cells) by mass cytometry [31]. PCA demonstrated that tolerant patients exhibit a differential pattern of immune cell frequencies compared to patients on immunosuppression and normal controls. Further, by Citrus analysis a distinct population (CD3+, CD4+, CD5+, CD25+, CD38−/lo, CD45RA−) was specifically and significantly increased in tolerant liver transplant recipients. Analysis by both mass and flow cytometry identified a T cell subset of Operational Tolerance (TOT) that correlates with tolerance in pediatric liver transplant recipients.
Mass cytometry is a powerful new addition for single-cell analysis and will likely co-exist with flow cytometry for the foreseeable future as each has unique advantages and disadvantages (Table 1). Clearly the establishment of the mass cytometry technology, in a lab or facility, requires a significant investment in infrastructure and training, especially in data analysis. Although, individual antibodies for mass cytometry are priced similarly to those for flow cytometry, most laboratories have substantial stocks of flourophore-labeled antibodies, making the start-up for an individual mass cytometry experiment costly. Further the purchase price of a mass cytometer, operation (argon gas), and maintenance contracts for the instrument are more costly that a state-of-the art flow cytometer. Currently, mass cytometry is not high-throughput as the average time on the instrument, per sample, is increased as compared to flow cytometry. The true power of mass cytometry is discovery, by casting a wider net one can identify novel markers that can then be translated to a flow cytometry platform for diagnostics and basic science research.
Acknowledgments
SMK and OMM are funded in part by NIH RO1 AI113130, R21 AI115313, R21 AI119686, UO1 AI104342 and the Transplant and Tissue Engineering Center of Excellence at Lucile Packard Children’s Hospital. SS is funded by the NLM postdoctoral trainee fellowship (T-15 LM007033-32). AHL was supported by F32 DK94548 and a Thrasher Research Fund Early Career Award.
Abbreviations
- CyTOF
Cytometry by Time-of-Flight
- PCA
Principal component analysis
- t-SNE
t-distributed stochastic neighbor embedding
- SPADE
spanning tree progression analysis of density normalized events)
- CITRUS
cluster identification, characterization, and regression
- ACCENSE
automatic classification of cellular expression by nonlinear stochastic embedding
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation
References
- 1.Bendall SC, et al. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33(7):323–32. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bandura DR, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–22. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 3.Spitzer MH, Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165(4):780–91. doi: 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4(8):648–55. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 5.Leipold MD, Newell EW, Maecker HT. Multiparameter Phenotyping of Human PBMCs Using Mass Cytometry. Methods Mol Biol. 2015;1343:81–95. doi: 10.1007/978-1-4939-2963-4_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newell EW, Cheng Y. Mass cytometry: blessed with the curse of dimensionality. Nat Immunol. 2016;17(8):890–5. doi: 10.1038/ni.3485. [DOI] [PubMed] [Google Scholar]
- 7.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30(9):858–67. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nassar AF, Wisnewski AV, Raddassi K. Mass cytometry moving forward in support of clinical research: advantages and considerations. Bioanalysis. 2016;8(4):255–7. doi: 10.4155/bio.15.257. [DOI] [PubMed] [Google Scholar]
- 9.Zunder ER, et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat Protoc. 2015;10(2):316–33. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Y, et al. CyTOF supports efficient detection of immune cell subsets from small samples. J Immunol Methods. 2014;415:1–5. doi: 10.1016/j.jim.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atkuri KR, Stevens JC, Neubert H. Mass cytometry: a highly multiplexed single-cell technology for advancing drug development. Drug Metab Dispos. 2015;43(2):227–33. doi: 10.1124/dmd.114.060798. [DOI] [PubMed] [Google Scholar]
- 12.van der Maaten L, Hinton G. Visualizing Data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- 13.Cheng Y, et al. Categorical Analysis of Human T Cell Heterogeneity with One-Dimensional Soli-Expression by Nonlinear Stochastic Embedding. J Immunol. 2016;196(2):924–32. doi: 10.4049/jimmunol.1501928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shekhar K, et al. Automatic Classification of Cellular Expression by Nonlinear Stochastic Embedding (ACCENSE) Proc Natl Acad Sci U S A. 2014;111(1):202–7. doi: 10.1073/pnas.1321405111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong MT, et al. A High-Dimensional Atlas of Human T Cell Diversity Reveals Tissue-Specific Trafficking and Cytokine Signatures. Immunity. 2016;45(2):442–56. doi: 10.1016/j.immuni.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–96. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29(10):886–91. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruggner RV, et al. Automated identification of stratifying signatures in cellular subpopulations. Proc Natl Acad Sci U S A. 2014;111(26):E2770–7. doi: 10.1073/pnas.1408792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aghaeepour N, et al. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10(3):228–38. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumgart S, et al. OMIP-034: Comprehensive immune phenotyping of human peripheral leukocytes by mass cytometry for monitoring immunomodulatory therapies. Cytometry A. 2016 doi: 10.1002/cyto.a.22894. [DOI] [PubMed] [Google Scholar]
- 21.Bendall SC, et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157(3):714–25. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaudilliere B, et al. Clinical recovery from surgery correlates with single-cell immune signatures. Sci Transl Med. 2014;6(255):255ra131. doi: 10.1126/scitranslmed.3009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horowitz A, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med. 2013;5(208):208ra145. doi: 10.1126/scitranslmed.3006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mason GM, et al. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J Immunol. 2015;195(5):2030–7. doi: 10.4049/jimmunol.1500703. [DOI] [PubMed] [Google Scholar]
- 25.Newell EW, et al. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36(1):142–52. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pejoski D, et al. Identification of Vaccine-Altered Circulating B Cell Phenotypes Using Mass Cytometry and a Two-Step Clustering Analysis. J Immunol. 2016;196(11):4814–31. doi: 10.4049/jimmunol.1502005. [DOI] [PubMed] [Google Scholar]
- 27.Salmon H, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44(4):924–38. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Unen V, et al. Mass Cytometry of the Human Mucosal Immune System Identifies Tissue- and Disease-Associated Immune Subsets. Immunity. 2016;44(5):1227–39. doi: 10.1016/j.immuni.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 29.Cheng Y, Newell EW. Deep Profiling Human T Cell Heterogeneity by Mass Cytometry. Adv Immunol. 2016;131:101–34. doi: 10.1016/bs.ai.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Yabu JM, Siebert JC, Maecker HT. Immune Profiles to Predict Response to Desensitization Therapy in Highly HLA-Sensitized Kidney Transplant Candidates. PLoS One. 2016;11(4):e0153355. doi: 10.1371/journal.pone.0153355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau AH, et al. Mass Cytometry Reveals a Distinct Immunoprofile of Operational Tolerance in Pediatric Liver Transplantation. Pediatric Transplantation. 2016 doi: 10.1111/petr.12795. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]