

Abstract
To gain better understanding of the distributions of the culturable Lactobacillus species in the chicken intestinal tract, we collected ceca, and distal ileum from 10 3-weeks-old broiler chickens. Lactobacillus strains from cecal lumen contents (M-CL), and those associated with mucosa of ceca (M-CM) and ileum (M-IM) were recovered on de Man, Rogosa and Sharpe (MRS) agar plates, and used for microbiota analysis. The total cecal content (T-CL) was also used directly for microbiota analysis. We purposefully focused on MRS-recovered populations to gain understanding of the culturable subpopulations of Lactobacillus, since the culturability is an important phenotype in order to exploit the chicken gut microbiota as a resource for development of probiotics. The V1–V3 regions of 16S rRNA gene was amplified from genomic DNA samples, and the pooled amplicons were analyzed by MiSeq sequencing with paired-end read 300 cycle option. Among MRS groups, Firmicutes were significantly higher in M-IM and M-CL as compared to M-CM, whereas Proteobacteria were significantly higher in M-CM as compared to M-IM and M-CL at p < 0.05. Among Lactobacillus, L. salivarius (36%) and L. johnsonii (21%) were higher in M-IM as compared to M-CL (L. salivarius, 28%; L. johnsonii, 15%), and M-CM (L. salivarius, 20%; L. johnsonii, 11%). L. crispatus was found significantly higher in M-CL as compared to M-IM (p < 0.01) whereas L. gasseri was found significantly higher in M-IM as compared to M-CM (p < 0.05). L. aviarius, and L. fornicalis were only observed in T-CL. In summary, Lactobacillus populations recovered on MRS vary with different regions and locations in chicken GIT, which might indicate their distinct functional roles in different gastrointestinal tract (GIT) niches, and some species of Lactobacillus are not culturable on MRS agar media. This study is the first attempt to define culturable Lactobacillus subpopulations in the chicken intestinal tract comprehensively using 16S rRNA gene profiling, and the findings of this study will be used as a platform to develop a new strategy for isolation of effective Lactobacillus probiotic candidates based on comparative analyses of chicken gut microbiota.
Keywords: broiler, gastrointestinal tract, Lactobacillus, microbiota, probiotics
Introduction
Due to the increased risk associated with the development of antibiotic resistance in bacteria, the use of antibiotic growth promoters (AGPs) in animal industry has been completely banned in Europe since January 1, 2006 and has been in the process of reduction or complete elimination in several countries, including the United States (Dibner and Richards, 2005; Huyghebaert et al., 2011). The use of probiotics as an alternative to AGP has been rapidly increasing in recent years (Ahasan et al., 2015). Microbes that are commonly used as probiotics include various species of the genera Lactobacillus, Bifidobacterium, and Enterococcus (Moreira et al., 2005). Although the microbial communities are distributed throughout the GIT, their composition was found heterogeneous along the different regions of GIT in chicken (Yeoman et al., 2012; Choi et al., 2014; Ranjitkar et al., 2016), pigs (Looft et al., 2014), and cattle (Mao et al., 2015). The variations in microbial composition can occur not only in different segments along GIT, but can also at different locations (lumen vs. mucosa) in the same region (Gong et al., 2002; Looft et al., 2014). Diverse groups of microbes reside in various regions and locations of the GIT and this might indicate differential functional roles they play in maintaining host health. Thus, in this study we characterized the bacterial communities across the different regions and locations of the GIT of chickens with a focus on the genus Lactobacillus, which have been most commonly considered for probiotics, through microbiota analysis of the bacterial cells recovered on MRS agar plates. By characterizing bacterial cells recovered on MRS agar plates, we eliminate unculturable Lactobacillus strains from the downstream analysis, retaining only culturable strains. If necessary, this step can be followed by identification and isolation of the species that demonstrate promising utility as probiotics based on comparative metagenomic analysis (16S rRNA gene profiling, and/or shot-gun metagenomics). For example, when a comparative microbiota/microbiome analysis indicates particular species (or strains) as effector species (or strains), the culturability of the corresponding species can be first confirmed by the presence of corresponding DNA signatures in culture-recovered bacterial populations before any attempt can be made to isolate the target species (strains) for further evaluation as promising probiotics. It is important to note that current method for 16S rRNA gene profiling using Illumina sequencing has a limited resolution and often cannot differentiate even at a species-level, while a strain-level analysis is impossible. It is mainly due to short lengths of the target regions in 16S rRNA gene that are sequenced, and inevitable sequencing errors from PCR and sequencing step. However, with the increasing interest in exploring intra-species variations, novel methods have been developed to overcome the current limitations enabling microbiota analysis at a strain-level (Ellegaard and Engel, 2016).
Lactobacillus strains were found to enhance tight junctions, and thereby reducing intestinal permeability in both in vitro studies with Caco-2 cells (Anderson et al., 2010; Miyauchi et al., 2012) and in vivo study with mice (Xu et al., 2016). However their distribution at species level, and functional activity may differ in different regions and locations of the GIT. Lactobacillus strains that are tightly associated with mucosa might possess better properties as probiotics than those found in lumen, and detailed characterization of Lactobacillus populations in both lumen and mucosa of different regions may be very helpful in the quest for isolating good probiotic candidates. Although MRS agar is the most commonly used medium for isolation of Lactobacillus strains, the scope of the culturability on MRS agar for diverse Lactobacillus species has not been systematically evaluated. In addition, since the use of candidate Lactobacillus strains for probiotic applications would require the culturability of the strains, in this study we adopted the approach of characterizing Lactobacillus strains recovered on MRS agar plates.
The precise identification of Lactobacillus isolates by phenotypic method is difficult, because phenotypic properties beyond the common fermentation tests are often required, and around 17 phenotypic tests are required to identify Lactobacillus at species level (Moreira et al., 2005). Only around 30% of the total vaginal and intestinal lactobacilli from humans were identified correctly at the species level by the most commonly used commercially available biochemical kit (Song et al., 1999). Alternatively, taxonomic identification of the strains belonging to genus Lactobacillus can be performed at species level with high accuracy based on DNA sequencing of the variable regions in 16S ribosomal RNA (16S rRNA) gene (Woo et al., 2002; Piotrowska et al., 2016).
Hence, the main aim of this study is to analyze bacterial populations recovered on MRS agar media via deep sequencing of the V1–V3 region of 16S rRNA gene in order to better understand the structure and distribution of the culturable subpopulations of Lactobacillus in different regions and locations of the GIT of broiler chickens.
Materials and Methods
Sample Collection and Processing
Cobb 500 broiler chickens were provided ad libitum access to water and an antibiotic-free corn-soybean meal diet. At the age of 3 weeks, 10 birds were humanely sacrificed, and ceca and distal end of ileum (5 cm) were aseptically collected according to the animal use protocol (No. 16047) approved by the IACUC committee at the University of Arkansas. The age of 3 weeks was chosen because the gut microbiota are established stably around this age (Ranjitkar et al., 2016). Cecal lumen contents were serially diluted and plated on MRS agar plates. To isolate bacteria associated with cecal mucosa or ileal mucosa, each mucosa sample was washed in sterile PBS buffer (pH 7.4) after removing luminal contents for four times, and homogenized in 20 ml PBS using Bullet Blender® (Next Advance). The supernatant was collected, serially diluted, and plated on MRS agar plates. The MRS agar plates were incubated overnight at 37°C under microaerophilic condition. Bacterial pellets were recovered from MRS plates with lowest dilutions (1 plate per sample) by resuspending all colonies in 5 ml PBS followed by centrifugation. The lowest dilutions were used to maximize the number of colonies collected for each sample: 10-fold dilution was used for M-CL and the supernatant without dilution was for M-CM and M-IM. The average log10CFUs per sample (mean ± standard error) was 6.02 ± 0.18, 3.71 ± 0.18, and 3.23 ± 0.21 for M-CL, M-CM, and M-IM samples, respectively.
DNA Extraction and PCR
Genomic DNA was extracted from each pellet (equal amount) by using DNeasy Blood and Tissue Kit (Qiagen). Genomic DNA of total bacteria in cecal lumen was also extracted directly without culturing on MRS plates using QIAamp Fast DNA Stool Minikit (Qiagen). Thus, we had altogether 40 genomic DNA samples: 10 MRS-recovered cells from each of cecal lumen (M-CL), cecal mucosa (M-CM), and ileal mucosa (M-IM), and 10 total bacterial cells from cecal lumen (T-CL). The V1–V3 region of the 16S rRNA gene was amplified from the genomic DNA samples using barcode-tagged universal primers; 27F (5′-AGRGTTYGATYMTGGCTCAG-3′) and 533R (5′-TTACCGCGGCTGCTGGCAC-3′) with attached Illumina adapters. Details regarding primers, enzymes, and PCR conditions were previously described (Mandal et al., 2016). The amplicons were purified from 0.7% agarose gel electrophoresis after verifying the length of amplicons. After the concentration of each amplicon sample was measured using Qubit dsDNA broad range assay kit (Life Technologies, United States), the amplicons were pooled at an equal amount. The pooled sample was gel-purified from 6% TBE gel (Invitrogen, United States), and sent for Illumina sequencing at the University of California (Riverside, CA, United States) using MiSeq paired-end reads with 300 cycles.
Data Analysis
All MiSeq paired-end sequence reads were analyzed by Quantitative Insights into Microbial Ecology, QIIME version 1.9.1 (available at http://qiime.sourceforge.net/; Caporaso et al., 2010). General pipelines for data analysis was previously described in details (Mandal et al., 2016). Forward and reverse ends sequences were joined together by using join_paired_ends.py command followed by formatting barcodes using customized Perl script, before extracting barcodes using extract_barcodes.py option. Demultiplexing and quality filtering were performed by split_libraries_fastq.py with default options. OTU picking was performed by using reference sequences from NCBI RefSeq 16S RNA database (O’Leary et al., 2016) and Swarm algorithm (Mahé et al., 2014). Taxonomic classification was performed by using reference taxonomy file from NCBI RefSeq 16S RNA sequences and SortMeRNA algorithm (Kopylova et al., 2012). NCBI RefSeq 16S RNA sequences are curated, non-redundant and quality controlled (Pruitt et al., 2007; O’Leary et al., 2016). We used this database instead of greengenes database for better taxonomic assignment at species level. Cumulative sum scaling (CSS) method with QIIME was used to normalize the OTU BIOM (biological observation matrix) before taxonomic assignment and alpha diversity calculation. Beta diversity estimates were calculated by using beta_diversity_through_plots.py options of QIIME with even sampling depth of 8000. Analysis of similarities (ANOSIM) between groups were performed using unweighted UniFrac distance metric (compare_categories.py, QIIME). Statistical significance in alpha diversity indices and different taxa among various groups were measured using one-way analysis of variance (ANOVA) followed by post hoc Student’s t-test using JMP Genomics 7 software.
Results
After demultiplexing and quality filtering, there was 1,350,414 assembled sequence reads ranging from 444 to 574 bp with median sequence length 546 bp. Summarizing raw vs. CSS normalized otu biom table resulted in mean sample depth of 33,760.35 ± 3,311.22 and 1,488 ± 11.72 reads per sample, respectively. CSS normalized otu biom table was used further for taxonomy assignment and alpha diversity analysis.
Taxonomy Assignment
Phylum Level
Taxonomic analysis among MRS groups revealed Firmicutes (83.83%) as the predominant phylum followed by Proteobacteria (13.83%). Firmicutes were found significantly higher in cecal lumen (M-CL) and ileal mucosa (M-IM) as compared to cecal mucosa (M-CM) at p < 0.05 (Figure 1), but there was no significant difference between M-CL and M-IM. On the contrary, Proteobacteria were found significantly higher in M-CM as compared to M-IM and M-CL at p < 0.05 (Figure 1).
FIGURE 1.

Relative abundance of different phyla. Different letters indicate significance at p < 0.05. Total bacterial cells from cecal lumen (T-CL). MRS-recovered cells from cecal lumen (M-CL), cecal mucosa (M-CM), and ileal mucosa (M-IM).
Genus Level
Relative abundance of different genera recovered from MRS groups (≥1% of all MRS groups) is shown in Figure 2. Lactobacillus, Enterococcus, and Citrobacter were the major predominant genera recovered from MRS groups. Lactobacillus was observed significantly higher in M-IM and M-CL as compared to M-CM (p < 0.01), whereas Citrobacter was significantly higher in M-CM as compared to M-IM (p < 0.05). Although Lactobacillus was predominant genus in each MRS group, recovery of other genera demonstrated that MRS agar medium also supports the growth of the strains belonging to Enterococcus and Citrobacter.
FIGURE 2.

Relative abundance of different genera. MRS-recovered cells from cecal lumen (M-CL), cecal mucosa (M-CM), and ileal mucosa (M-IM).
Species Level
Among the major Lactobacillus species identified, relative abundance of L. salivarius was highest in all three groups followed by L. johnsonii. Both L. salivarius (36%) and L. johnsonii (21%) were higher in M-IM as compared to M-CL (L. salivarius, 28%; L. johnsonii, 15%) and M-CM (L. salivarius, 20%; L. johnsonii, 11%) as shown in Figure 3. L. crispatus was found higher in M-CL as compared to M-CM and M-IM, but significant difference was found only between M-CL and M-IM (p < 0.01). Similarly, L. gasseri was found significantly higher in M-IM as compared to M-CM (p < 0.05).
FIGURE 3.
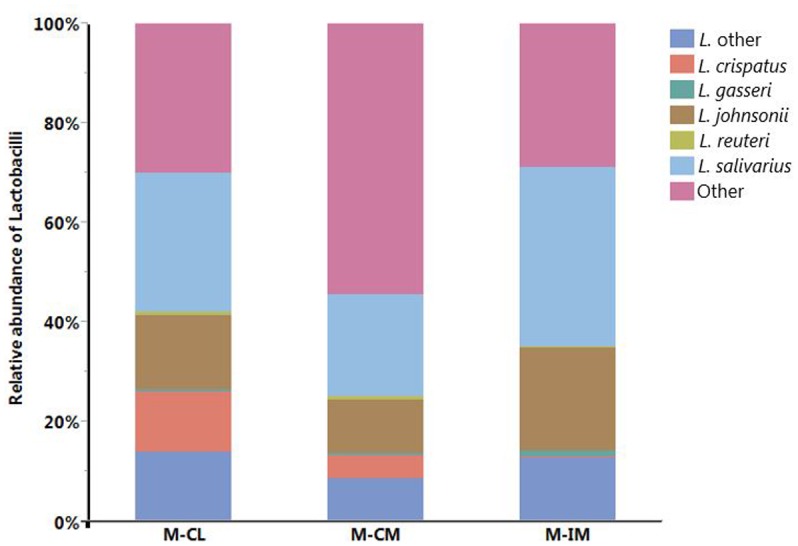
Relative abundance of different Lactobacillus species. MRS-recovered cells from cecal lumen (M-CL), cecal mucosa (M-CM), and ileal mucosa (M-IM).
OTU Heatmap at Species Level
The OTU heatmap that consists of only Lactobacillus species, constructed with QIIME, revealed that L. aviarius and L. fornicalis were detected only from the total bacterial group (T-CL) as shown in Figure 4. Although these species were found only in a subset of T-CL samples, their relative abundance was significantly high as indicated by the green colors. Some other Lactobacillus species such as L. aviarius, L. equigenerosi, L. agilis, L. gallinarum, L. satsumensis, and L. capillatus were also detected in negligible amounts, in only one or two samples of the total bacterial group or MRS groups, which may be due to the errors during PCR or Illumina sequencing step.
FIGURE 4.

Heatmap of normalized OTU table consisting of Lactobacillus species only. Heatmap was constructed with make_otu_heatmap.py option of QIIME with log transformation where all zeros were set to a small value (1/2 the smallest non-zero entry), and data was translated to non-negative after log transformation, and num_otu_hits was set to 0. The abundance of Lactobacillus species decreases as the intensity of color decreases from green to yellow. Total bacterial cells from cecal lumen (T-CL). MRS-recovered cells from cecal lumen (M-CL), cecal mucosa (M-CM), and ileal mucosa (M-IM).
Alpha Diversity
The observed OTUs ranged from 20 to 71 for all samples together. The alpha diversity measured with observed OTU metric was not significantly different among M-CL, M-CM, and M-IM. But as expected, the alpha diversity was significantly higher in the samples for which genomic DNA was directly isolated from total bacteria (T-CL) as compared to the samples recovered from MRS medium at p < 0.01 as shown in Figure 5.
FIGURE 5.
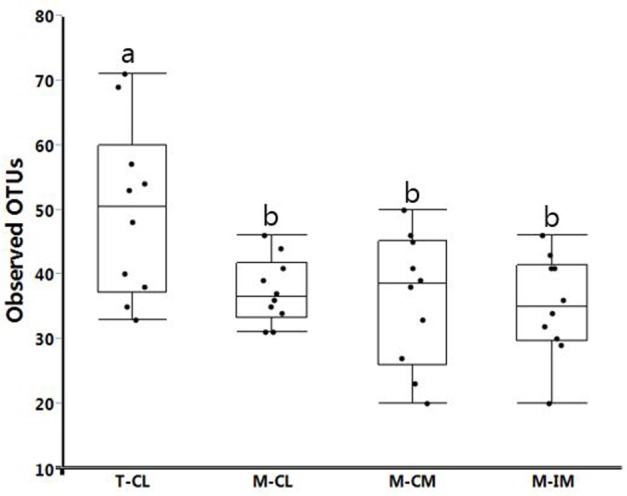
Alpha diversity in different groups measured with Observed_otus metric. Bars with different letters represent statistical significance at p < 0.01. Total bacterial cells from cecal lumen (T-CL). MRS-recovered cells from cecal lumen (M-CL), cecal mucosa (M-CM), and ileal mucosa (M-IM).
Beta Diversity
Unweighted unifrac distance metric was used to calculate ANOSIM. ANOSIM results showed that there were significant differences in bacterial community structure among different groups (M-CL, M-CM, M-IM, and T-CL; R = 0.67, p = 0.001) as illustrated in PCoA plot in Figure 6A. Similarly, the difference in bacterial community structure was observed among the groups of samples isolated from MRS medium (M-CL, M-CM, and M-IM; R = 0.13, p = 0.01) as shown in Figure 6B, and also between cecal and ileum mucosal samples (R = 0.18, p = 0.02) as shown in Figure 6C.
FIGURE 6.

PCoA plots showing significant difference in bacterial community structure. (A) Among all groups analyzed; MRS-recovered cells from cecal lumen (M-CL), cecal mucosa (M-CM) and ileal mucosa (M-IM), and total bacterial cells from cecal lumen (T-CL) (R = 0.67, p = 0.001). (B) Among MRS groups; M-CL, M-CM, and M-IM (R = 0.13, p = 0.01). (C) Between two different regions of gut; M-CM and M-IM (R = 0.18, p = 0.02).
Discussion
Although the use of different species of Lactobacillus as probiotics in chickens has shown beneficial effects (Zhang et al., 2007; Mappley et al., 2013; Saint-Cyr et al., 2017), there is still a lack of solid scientific basis for probiotic actions, and thus effective strategies to isolate promising probiotic strains. Comprehensive investigation of Lactobacillus populations in chicken GIT might provide important insights for better understanding of their roles in host function, and therefore for development of better screening strategies to identify more effective probiotic strains. Comprehensive characterization of chicken gut microbiota through the use of high throughput next generation sequencing (HT-NGS) has been limited as compared to human gut microbiota (Shaufi et al., 2015). It has already been reported that the relative abundance of Lactobacillus varies among different segments of the GIT in chickens (Gong et al., 2007; Ranjitkar et al., 2016) using culture independent method. Only one study reported the analysis of mucosa associated microbiota in chicken GIT via high-throughput sequencing of 16S rRNA gene sequences (Gong et al., 2007). Thus, there is very limited information available regarding topological differences of Lactobacillus population found in chicken GIT.
Gong et al. (2002) reported differences in bacterial populations between lumen and mucosa of chicken caeca through terminal restriction fragment length polymorphism (T-RFLP). The 16S rRNA gene-based analysis of mucosa-associated bacterial populations in chicken GIT revealed Lactobacillus as a predominant genera in upper GIT where L. salivarius and L. aviarius were predominant species in genus Lactobacillus (Gong et al., 2007). Similarly previous studies reported Lactobacillus species higher in ileum than cecum (Ranjitkar et al., 2016; Wang et al., 2016). We also noticed higher percentage of L. salivarius and L. johnsonii in ileal mucosa as compared to cecal lumen and cecal mucosa, albeit there was no significant differences among them. This is in agreement with our findings at phylum level where Firmicutes were higher in ileal mucosa as compared to cecal lumen and cecal mucosa, but significant difference was observed only between ileal mucosa and cecal mucosa. Observation of other genera that do not belong to lactic acid bacteria (LAB), such as Citrobacter and Bacillus, among the MRS groups suggests the limited selectivity of MRS agar for LAB strains as demonstrated earlier (Hartemink and Rombouts, 1999; Quartieri et al., 2016). Our report on the limited selectivity of MRS agar should be considered carefully when MRS agar is used as a means to estimate CFUs of LAB strains present in animal GIT samples. We reported L. salivarius to be a predominant species in all regions and locations of the GIT, which is in agreement with recent studies in chickens that reported higher percentage of L. salivarius in both cecum and ileum at the age of 36 (Ranjitkar et al., 2016), and at ileal mucosa at the age of 35 (Wang et al., 2016). These recent findings are in agreement with the previous reports that L. salivarius are consistently detected in older birds (Knarreborg et al., 2002; Guan et al., 2003). In this study, L. crispatus was found significantly higher in cecal lumen than ileal mucosa whereas L. gasseri was found significantly higher in ileal mucosa as compared to cecal mucosa. L. crispatus can be found in vertebrate GIT and is a Lactobacillus species frequently isolated from human vaginal tract (Witkin et al., 2007; El Aila et al., 2009). However, we should consider different factors including age, diet, litter type, horizontal gene transfer, chicken type, geography, climate, environment, feed additive, etc. before direct comparison of the present study with other findings, since these factors can affect chicken GIT microbiota (Qu et al., 2008; Danzeisen et al., 2011; Wang et al., 2016).
We observed L. aviarius and L. fornicalis only in total bacterial group. Failure to recover these species from MRS agar may be due to the followings reasons; these species either (i) require strictly anaerobic condition (L. aviarius), or (ii) grow well under anaerobic condition although being facultative anaerobic (L. fornicalis) as compared to microaerophilic condition at 37°C, which was used in this study (Fujisawa et al., 1984; Dicks et al., 2000; Baele et al., 2003). Alternatively, some of these species may have unique metabolic requirements that are not provided in MRS media. Observation of significantly higher alpha and beta diversity in total bacterial group (T-CL) as compared to MRS groups is obvious. Among the MRS groups (M-CL, M-CM, and M-IM), alpha diversity was observed higher in cecal lumen followed by cecal mucosa and ileal mucosa, although there was no significant difference. This was in agreement with ANOSIM results which showed differences in bacterial community structure among different MRS groups. Thus results from both alpha and beta diversity revealed difference in bacterial diversity between cecum and ileum, which is similar with the previous findings (Shaufi et al., 2015; Ranjitkar et al., 2016).
In summary, L. salivarius was found as a dominant species in all three regions of the GIT. Relative abundance of Lactobacillus not only varied with different regions of the GIT but also varied between lumen and mucosa of the same region. All the Lactobacillus species present in chicken GIT samples may not be cultured on MRS agar media. Analysis of alpha diversity and beta diversity revealed differences in the structure of MRS-recovered bacterial communities among different regions and locations of the GIT.
To our knowledge, in most studies to isolate effective probiotics in poultry as well as in other food-producing animals, the first step is isolation of strains that belong to the target taxonomic group (e.g., Lactobacillus genus), followed by a screening of the strains for various desirable phenotypes, including resistance to acidic pH or bile acid, ability to inhibit the growth of pathogenic bacteria in vitro, and particular enzymatic properties among others (Asghar et al., 2016; Kizerwetter-Świda and Binek, 2016). However, this approach has the following inherent limitations: (1) the screening is conducted with randomly picked strains from a large pool of bacterial strains, (2) the number of strains screened is critically limited due to the labor and time required for the process, and (3) the suitability of the screening criteria for in vivo efficacy remains questionable (Morelli, 2000). For these reasons, this current approach remains ineffective, limiting our ability to exploit the gut microbiota as a rich resource for development of more effective probiotics.
On the other hand, the use of culture-independent approaches (16S rRNA gene profiling, and shot-gun metagenome analysis) have provided new insights on the function of gut microbiota in overall body functions (Singh et al., 2014; Choi et al., 2015; Yan et al., 2017), and are expected to reveal some core members of gut microbiota that play crucial roles in promoting gut health and thus growth performance in poultry. For example, Stanley et al. (2016) attempted to identify probiotic candidates for broilers based on their association with desirable productivity outcomes using microbiota analyses. Although the lack of consistency in the microbial shifts across the three animal trials was shown as a major challenge for this effort, this new approach demonstrated in Stanley et al. (2016) has a great potential for identification of effective probiotics. On the other hand, Buffie et al. (2015) identified Clostridium scindens as a species associated with resistance to C. difficile gut colonization in both mice and humans using comparative microbiota analysis and mathematical modeling, and experimentally demonstrated that oral administration of C. scindens significantly enhanced resistance to C. difficile colonization in mice.
In the study by Buffie et al. (2015) the use of the C. scindens strain originated from different source was successful in demonstrating the probiotic efficacy, suggesting that the genetic capacity conferring resistance is probably well-conserved within the C. scindens species. However, an increasing body of studies are pointing to the fact that intra-species variations on genetic capacity is quite common (Greenblum et al., 2015). In some cases, different strains from the same species can act in an opposite manner as previously reported by Fåk and Bäckhed (2012) that L. reuteri ATCC PTA 4659 was linked to weight loss while L. reuteri L6798 was linked to weight gain in mice. These findings suggests that the probiotic candidates identified by comparative microbiota analysis should be strain-specific in some cases and thus need to be isolated from appropriate samples used for the microbiota analysis.
However, when the target species or strains are identified, the next step to isolate the strains represented by the identified signature DNA sequences (e.g., specific 16S rRNA gene sequences) would encounter multiple challenges to overcome, primarily due to the complex microbiota background from which the target strains are to be isolated. One major challenge can be the culturability of the target strains, because DNA sequence data do not provide information regarding culturability of each member of a microbiota. However, a comparative microbiota analysis between culture-recovered bacteria such as shown in our study (e.g., M-CL) and direct microbiota (e.g., T-CL) can identify the culturable members in the microbiota as illustrated in Figure 4. This information would ensure that the efforts to retrieve target strains is an achievable goal, although the practical strategies to isolate the strains based on DNA signatures still remains to be developed.
We reason that the conventional approach to isolate probiotics should move toward this new direction to fully exploit gut microbiota in poultry as a valuable resource to develop probiotics that would be more effective in positively modulating gut microbiota, thereby preventing diseases, and promoting health and growth performance in poultry. Our study is conducted on a small scale, but it is the first attempt to define MRS-recovered Lactobacillus subpopulations in GIT of chickens with the long-term goal of developing more effective Lactobacillus probiotic candidates based on system-wide comparative microbiota analyses.
Author Contributions
BA and YK designed the experiment. BA conducted the study, analyzed the data and wrote the manuscript. BA and YK revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful for the funding support from Arkansas Biosciences Institute for this study. We are also thankful for Rabindra K. Mandal for his suggestions during data analysis with QIIME.
Footnotes
Funding. This study was conducted by the funding support from Arkansas Biosciences Institute.
References
- Ahasan A., Agazzi A., Invernizzi G., Bontempo V., Savoini G. (2015). The beneficial role of probiotics in monogastric animal nutrition and health. J. Dairy Vet. Anim. Res. 2:00041 10.15406/jdvar.2015.02.00041 [DOI] [Google Scholar]
- Anderson R. C., Cookson A. L., McNabb W. C., Park Z., McCann M. J., Kelly W. J., et al. (2010). Lactobacillus plantarum MB452 enhances the function of the intestinal barrier by increasing the expression levels of genes involved in tight junction formation. BMC Microbiol. 10:316 10.1186/1471-2180-10-316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghar S., Arif M., Nawaz M., Muhammad K., Ali M. A., Ahmad M. D., et al. (2016). Selection, characterisation and evaluation of potential probiotic Lactobacillus spp. isolated from poultry droppings. Benef. Microbes 7 35–44. 10.3920/BM2015.0020 [DOI] [PubMed] [Google Scholar]
- Baele M., Vancanneyt M., Devriese L. A., Lefebvre K., Swings J., Haesebrouck F. (2003). Lactobacillus ingluviei sp. nov., isolated from the intestinal tract of pigeons. Int. J. Syst. Evol. Microbiol. 53 133–136. 10.1099/ijs.0.02206-0 [DOI] [PubMed] [Google Scholar]
- Buffie C. G., Bucci V., Stein R. R., McKenney P. T., Ling L., Gobourne A., et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517 205–208. 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H., Kim G. B., Cha C. J. (2014). Spatial heterogeneity and stability of bacterial community in the gastrointestinal tracts of broiler chickens. Poult. Sci. 93 1942–1950. 10.3382/ps.2014-03974 [DOI] [PubMed] [Google Scholar]
- Choi K. Y., Lee T. K., Sul W. J. (2015). Metagenomic analysis of chicken gut microbiota for improving metabolism and health of chickens - a review. Asian Australas. J. Anim. Sci. 28 1217–1225. 10.5713/ajas.15.0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzeisen J. L., Kim H. B., Isaacson R. E., Tu Z. J., Johnson T. J. (2011). Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS ONE 6:e27949 10.1371/journal.pone.0027949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibner J. J., Richards J. D. (2005). Antibiotic growth promoters in agriculture: history and mode of action. Poult. Sci. 84 634–643. [DOI] [PubMed] [Google Scholar]
- Dicks L. M., Silvester M., Lawson P. A., Collins M. D. (2000). Lactobacillus fornicalis sp. nov., isolated from the posterior fornix of the human vagina. Int. J. Syst. Evol. Microbiol. 50 1253–1258. 10.1099/00207713-50-3-1253 [DOI] [PubMed] [Google Scholar]
- El Aila N. A., Tency I., Claeys G., Verstraelen H., Saerens B., Santiago G. L., et al. (2009). Identification and genotyping of bacteria from paired vaginal and rectal samples from pregnant women indicates similarity between vaginal and rectal microflora. BMC Infect. Dis. 9:167 10.1186/1471-2334-9-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegaard K. M., Engel P. (2016). Beyond 16S rRNA community profiling: intra-species diversity in the gut microbiota. Front. Microbiol. 7:1475 10.3389/fmicb.2016.01475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fåk F., Bäckhed F. (2012). Lactobacillus reuteri prevents diet-induced obesity, but not atherosclerosis, in a strain dependent fashion in Apoe-/- mice. PLoS ONE 7:e46837 10.1371/journal.pone.0046837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T., Shirasaka S., Watabe J., Mitsuoka T. (1984). Lactobacillus aviarius sp. nov.: a new species isolated from the intestine of chickens. Syst. Appl. Microbiol. 5 414–420. [Google Scholar]
- Gong J., Forster R. J., Yu H., Chambers J. R., Sabour P. M., Wheatcroft R., et al. (2002). Diversity and phylogenetic analysis of bacteria in the mucosa of chicken ceca and comparison with bacteria in the cecal lumen. FEMS Microbiol. Lett. 208 1–7. 10.1111/j.1574-6968.2002.tb11051.x [DOI] [PubMed] [Google Scholar]
- Gong J., Si W., Forster R. J., Huang R., Yu H., Yin Y., et al. (2007). 16S rRNA gene-based analysis of mucosa-associated bacterial community and phylogeny in the chicken gastrointestinal tracts: from crops to ceca. FEMS Microbiol. Ecol. 59 147–157. 10.1111/j.1574-6941.2006.00193.x [DOI] [PubMed] [Google Scholar]
- Greenblum S., Carr R., Borenstein E. (2015). Extensive strain-level copy-number variation across human gut microbiome species. Cell 160 583–594. 10.1016/j.cell.2014.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan L. L., Hagen K. E., Tannock G. W., Korver D. R., Fasenko G. M., Allison G. E. (2003). Detection and identification of Lactobacillus species in crops of broilers of different ages by using PCR-denaturing gradient gel electrophoresis and amplified ribosomal DNA restriction analysis. Appl. Environ. Microbiol. 69 6750–6757. 10.1128/AEM.69.11.6750-6757.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartemink R., Rombouts F. M. (1999). Comparison of media for the detection of bifidobacteria, lactobacilli and total anaerobes from faecal samples. J. Microbiol. Methods 36 181–192. [DOI] [PubMed] [Google Scholar]
- Huyghebaert G., Ducatelle R., Van Immerseel F. (2011). An update on alternatives to antimicrobial growth promoters for broilers. Vet. J. 187 182–188. 10.1016/j.tvjl.2010.03.003 [DOI] [PubMed] [Google Scholar]
- Kizerwetter-Świda M., Binek M. (2016). Assessment of potentially probiotic properties of Lactobacillus strains isolated from chickens. Pol. J. Vet. Sci. 19 15–20. 10.1515/pjvs-2016-0003 [DOI] [PubMed] [Google Scholar]
- Knarreborg A., Simon M. A., Engberg R. M., Jensen B. B., Tannock G. W. (2002). Effects of dietary fat source and subtherapeutic levels of antibiotic on the bacterial community in the ileum of broiler chickens at various ages. Appl. Environ. Microbiol. 68 5918–5924. 10.1128/AEM.68.12.5918-5924.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopylova E., Noe L., Touzet H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28 3211–3217. 10.1093/bioinformatics/bts611 [DOI] [PubMed] [Google Scholar]
- Looft T., Allen H. K., Cantarel B. L., Levine U. Y., Bayles D. O., Alt D. P., et al. (2014). Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 8 1566–1576. 10.1038/ismej.2014.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahé F., Rognes T., Quince C., De Vargas C., Dunthorn M. (2014). Swarm: robust and fast clustering method for amplicon-based studies. PeerJ. 2:e593 10.7717/peerj.593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal R. K., Jiang T., Al-Rubaye A. A., Rhoads D. D., Wideman R. F., Zhao J., et al. (2016). An investigation into blood microbiota and its potential association with Bacterial Chondronecrosis with Osteomyelitis (BCO) in Broilers. Sci. Rep. 6:25882 10.1038/srep25882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao S., Zhang M., Liu J., Zhu W. (2015). Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci. Rep. 5:16116 10.1038/srep16116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mappley L. J., Tchórzewska M. A., Nunez A., Woodward M. J., Bramley P. M., La Ragione R. M. (2013). Oral treatment of chickens with Lactobacillus reuteri LM1 reduces Brachyspira pilosicoli-induced pathology. J. Med. Microbiol. 62 287–296. 10.1099/jmm.0.051862-0 [DOI] [PubMed] [Google Scholar]
- Miyauchi E., O’Callaghan J., Butto L. F., Hurley G., Melgar S., Tanabe S., et al. (2012). Mechanism of protection of transepithelial barrier function by Lactobacillus salivarius: strain dependence and attenuation by bacteriocin production. Am. J. Physiol. Gastrointest. Liver Physiol. 303 G1029–G1041. 10.1152/ajpgi.00003.2012 [DOI] [PubMed] [Google Scholar]
- Moreira J., Mota R. M., Horta M. F., Teixeira S. M., Neumann E., Nicoli J. R., et al. (2005). Identification to the species level of Lactobacillus isolated in probiotic prospecting studies of human, animal or food origin by 16S-23S rRNA restriction profiling. BMC Microbiol. 5:15 10.1186/1471-2180-5-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli L. (2000). In vitro selection of probiotic lactobacilli: a critical appraisal. Curr. Issues Intest. Microbiol. 1 59–67. [PubMed] [Google Scholar]
- O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44D733–D745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowska A., Gosiewski T., Bulanda M., Brzychczy-Wloch M. (2016). Using of the 16S rDNA sequencing for identification of Lactobacillus species. Med. Dosw. Mikrobiol. 68 5–11. [PubMed] [Google Scholar]
- Pruitt K. D., Tatusova T., Maglott D. R. (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35 D61–D65. 10.1093/nar/gkl842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu A., Brulc J. M., Wilson M. K., Law B. F., Theoret J. R., Joens L. A., et al. (2008). Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE 3:e2945 10.1371/journal.pone.0002945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartieri A., Simone M., Gozzoli C., Popovic M., D’Auria G., Amaretti A., et al. (2016). Comparison of culture-dependent and independent approaches to characterize fecal bifidobacteria and lactobacilli. Anaerobe 38 130–137. 10.1016/j.anaerobe.2015.10.006 [DOI] [PubMed] [Google Scholar]
- Ranjitkar S., Lawley B., Tannock G., Engberg R. M. (2016). Bacterial succession in the broiler gastrointestinal tract. Appl. Environ. Microbiol. 82 2399–2410. 10.1128/AEM.02549-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Cyr M. J., Haddad N., Taminiau B., Poezevara T., Quesne S., Amelot M., et al. (2017). Use of the potential probiotic strain Lactobacillus salivarius SMXD51 to control Campylobacter jejuni in broilers. Int. J. Food Microbiol. 247 9–17. 10.1016/j.ijfoodmicro.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Shaufi M. A. M., Sieo C. C., Chong C. W., Gan H. M., Ho Y. W. (2015). Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 7 4 10.1186/s13099-015-0051-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. M., Shah T. M., Reddy B., Deshpande S., Rank D., Joshi C. (2014). Taxonomic and gene-centric metagenomics of the fecal microbiome of low and high feed conversion ratio (FCR) broilers. J. Appl. Genet. 55 145–154. 10.1007/s13353-013-0179-4 [DOI] [PubMed] [Google Scholar]
- Song Y. L., Kato N., Matsumiya Y., Liu C. X., Kato H., Watanabe K. (1999). Identification of Lactobacillus species of human origin by a commercial kit, API50CHL. Rinsho Biseibutshu Jinsoku Shindan Kenkyukai Shi 10 k77–82. [PubMed] [Google Scholar]
- Stanley D., Hughes R. J., Geier M. S., Moore R. J. (2016). Bacteria within the gastrointestinal tract microbiota correlated with improved growth and feed conversion: challenges presented for the identification of performance enhancing probiotic bacteria. Front. Microbiol. 7:187 10.3389/fmicb.2016.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Lilburn M., Yu Z. (2016). Intestinal microbiota of broiler chickens as affected by litter management regimens. Front. Microbiol. 7:593 10.3389/fmicb.2016.00593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin S. S., Linhares I. M., Giraldo P. (2007). Bacterial flora of the female genital tract: function and immune regulation. Best Pract. Res. Clin. Obstet. Gynaecol. 21 347–354. [DOI] [PubMed] [Google Scholar]
- Woo P. C., Fung A. M., Lau S. K., Yuen K. Y. (2002). Identification by 16S rRNA gene sequencing of Lactobacillus salivarius bacteremic cholecystitis. J. Clin. Microbiol. 40 265–267. 10.1128/JCM.40.1.265-267.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Li X., Wang E., He Y., Yin B., Fang D., et al. (2016). A cellular model for screening of lactobacilli that can enhance tight junctions. RSC Adv. 6 111812–111821. 10.1039/c6ra24148d [DOI] [Google Scholar]
- Yan W., Sun C., Yuan J., Yang N. (2017). Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci. Rep. 7:45308 10.1038/srep45308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoman C. J., Chia N., Jeraldo P., Sipos M., Goldenfeld N. D., White B. A. (2012). The microbiome of the chicken gastrointestinal tract. Anim. Health Res. Rev. 13 89–99. 10.1017/S1466252312000138 [DOI] [PubMed] [Google Scholar]
- Zhang G., Ma L., Doyle M. P. (2007). Salmonella reduction in poultry by competitive exclusion bacteria Lactobacillus salivarius and Streptococcus cristatus. J. Food Prot. 70 874–878. [DOI] [PubMed] [Google Scholar]