

Abstract
Chorea-Acanthocytosis (ChAc), a neurodegenerative disorder, results from loss-of-function-mutations of chorein-encoding gene VPS13A. In tumour cells chorein up-regulates ORAI1, a Ca2+-channel accomplishing store operated Ca2+-entry (SOCE) upon stimulation by STIM1. Furthermore SOCE could be up-regulated by lithium. The present study explored whether SOCE impacts on neuron apoptosis. Cortical neurons were differentiated from induced pluripotent stem cells generated from fibroblasts of ChAc patients and healthy volunteers. ORAI1 and STIM1 transcript levels and protein abundance were estimated from qRT-PCR and Western blotting, respectively, cytosolic Ca2+-activity ([Ca2+]i) from Fura-2-fluorescence, as well as apoptosis from annexin-V-binding and propidium-iodide uptake determined by flow cytometry. As a result, ORAI1 and STIM1 transcript levels and protein abundance and SOCE were significantly smaller and the percentage apoptotic cells significantly higher in ChAc neurons than in control neurons. Lithium treatment (2 mM, 24 hours) increased significantly ORAI1 and STIM1 transcript levels and protein abundance, an effect reversed by inhibition of Serum & Glucocorticoid inducible Kinase 1. ORAI1 blocker 2-APB (50 µM, 24 hours) significantly decreased SOCE, markedly increased apoptosis and abrogated the anti-apoptotic effect of lithium. In conclusion, enhanced neuronal apoptosis in ChAc at least partially results from decreased ORAI1 expression and SOCE, which could be reversed by lithium treatment.
Introduction
Chorein promotes activation of phosphoinositide-3-kinase (PI3K)-p85-subunit and thus participates in the regulation of actin polymerization and cell survival1–3. Loss-of-function-mutations of the chorein encoding gene VPS13A (vacuolar protein sorting-associated protein 13A) lead to chorea-acanthocytosis (ChAc)4, 5, a progressive autosomal recessive neurodegenerative disease with hyperkinetic movements, impaired cognitive functions, myopathy with increased plasma levels of creatine kinase, and erythrocyte acanthocytosis4, 6–8. The movement and motor disorders of chorea-acanthocytosis include limb chorea, parkinsonism, dystonia, tongue protrusion, dysarthria, dysphagia, tongue and lip biting as well as progressive distal muscle wasting and weakness8.The derangements of motor function are paralleled by cognitive impairment, behavioral changes and epileptic seizures8. The progressive neurodegeneration eventually leads to severe disability and early death8. Gene targeted mice lacking functional chorein display erythrocyte shape changes9, neuronal apoptosis10 and altered behaviour10.
Chorein is expressed in a wide variety of tissues11–13 and participates in the regulation of diverse functions including dopamine release14, platelet activation13, cytoskeletal architecture15, endothelial cell stiffness12, and tumour cell survival3. Most importantly, chorein is decisive for the survival of neurons and skeletal muscle cells4, 16.
Regulators of cell survival and cell death include alterations of cytosolic Ca2+ activity ([Ca2+]i)17, 18. [Ca2+]i could be increased by Ca2+ release from intracellular stores with subsequent store-operated Ca2+ entry (SOCE) through the pore-forming Ca2+ channel subunits ORAI1, ORAI2 and/or ORAI319. Following store depletion, the ORAI isoforms are activated by the Ca2+ sensing proteins STIM1 and/or STIM220–22. ORAI1 and SOCE are up-regulated by the PI3K pathway and thus at least in some cell types sensitive to chorein23. Moreover, ORAI1 and SOCE could be up-regulated by lithium24, which has been shown to counteract neurodegeneration25–27.
The present study explored whether chorein deficiency and lithium impact on neuronal ORAI1 expression, SOCE and/or cell survival. To this end skin fibroblasts from ChAc patients and age-matched healthy individuals were reprogrammed and differentiated to neurons and ORAI transcript levels, ORAI protein abundance, SOCE and apoptosis determined without or with prior lithium treatment.
Results
In order to test whether the pathophysiology of chorea-acanthocytosis (ChAc) involves deranged neuronal regulation of the Ca2+ release activated Ca2+ channel ORAI1 and/or its regulator STIM1, experiments were performed with neurons generated from induced pluripotent stem cells (iPSCs). The iPSCs were generated from skin fibroblasts of healthy individuals (control neurons) and patients with chorea-acanthocytosis (ChAc neurons).
ORAI1 and STIM1 transcript levels and protein abundance
qRT-PCR was utilized for the quantification of the transcript levels encoding ORAI1 or STIM1. As apparent from Fig. 1a,b, the neurons from both, healthy individuals and ChAc patients did express ORAI1 and STIM1. The ORAI1 and STIM1 transcript levels were significantly lower in neurons derived from ChAc patients than in neurons derived from healthy individuals (Complete gel picture in Supplementary Figure S1).
Figure 1.
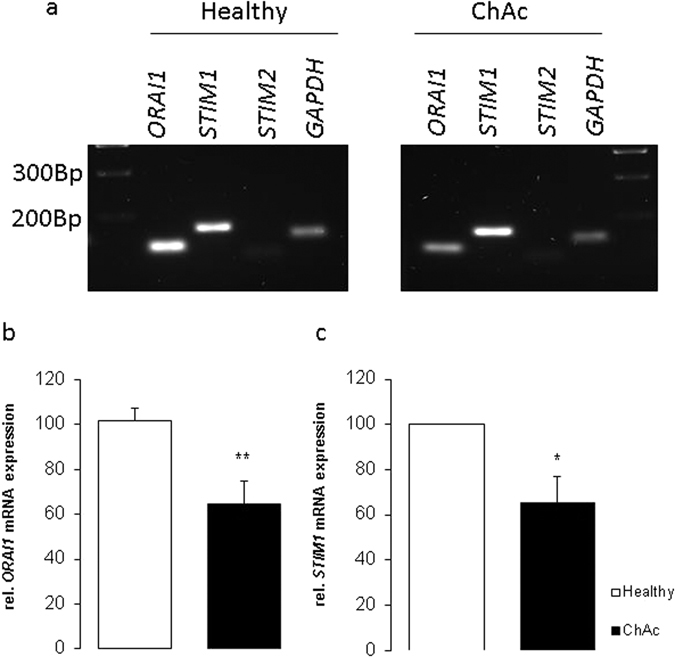
ORAI1 and STIM1 transcript levels in neurons generated from ChAc patients and healthy volunteers. (a) Original gels illustrating specificity of PCR products. (b,c) Arithmetic means (±SEM, n = 4) of (b) ORAI1 and (c) STIM1 transcript levels in neurons isolated from healthy volunteers (white bars) or from ChAc patients (black bars). *(p < 0.05), **(p < 0.01) indicates statistically significant difference to respective value from healthy volunteers.
In order to test whether transcription of ORAI1 and STIM1 in ChAc neurons is sensitive to lithium, ChAc neurons were incubated for 24 hours in the absence or presence of 2 mM lithium. The ORAI1 and STIM1 mRNA abundance was subsequently determined using qRT-PCR. As displayed in Fig. 2, ORAI1 and STIM1 transcript levels were in ChAc neurons significantly increased by treatment with 2 mM LiCl. The ORAI1 and STIM1 transcript levels were similar in lithium treated ChAc neurons and in untreated neurons from healthy volunteers. The effect of lithium on ORAI1 and STIM1 transcript levels was reversed by additional treatment with the serum & glucocorticoid inducible kinase (SGK1) inhibitor GSK650394 (10 µM). In the presence of lithium and GSK650394, the ORAI1 and STIM1 transcript levels were even significantly lower than the transcript levels of untreated ChAc neurons (Fig. 2) (Complete gel picture in Supplementary Figure S2).
Figure 2.
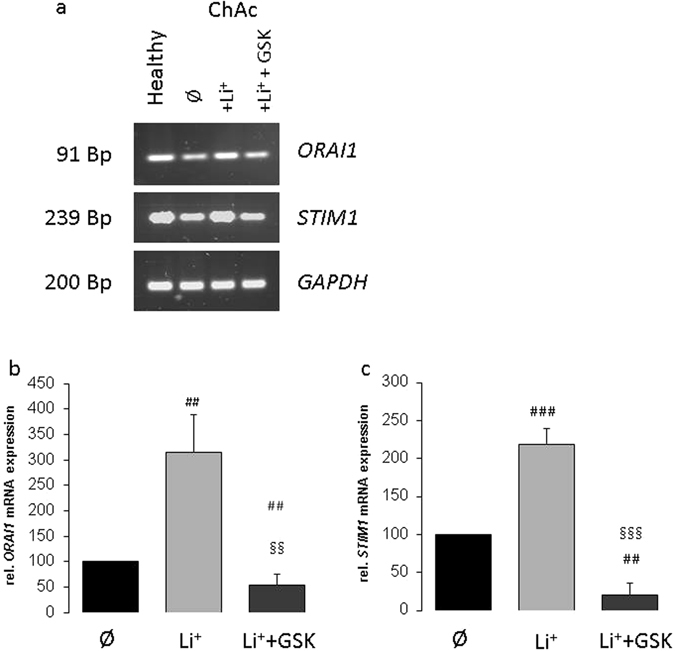
Effect of lithium treatment without and with SGK1 inhibitor GSK650394 on ORAI1 and STIM1 transcript levels in ChAc neurons. (a) Original gels illustrating specificity of PCR products. (b,c) Arithmetic means (±SEM, n = 5) of (b) ORAI1 and (c) STIM1 transcript levels in neurons generated from ChAc patients without (black bars) or with lithium (24 h, 2 mM) treatment without (light grey bar) and with (dark grey bar) presence of SGK1 inhibitor GSK650394 (10 µM). ##(p < 0.01), ###(p < 0.001) indicates statistically significant difference to respective value in the absence of lithium treatment. §§(p < 0.01), §§§(p < 0.001) indicates statistically significant difference to respective value in the absence of GSK650394.
Western blotting was employed to test whether the differences of ORAI1 and STIM1 transcript levels between healthy individuals and ChAc patients were paralleled by similar differences in protein abundance. As illustrated in Fig. 3a,b, the ORAI1 and STIM1 protein abundance was significantly lower in ChAc neurons than in neurons from healthy volunteers. In line with the qRT-PCR results, ORAI1 and STIM1 protein abundance was significantly higher in ChAc neurons following a 24 hours incubation in the presence than in the absence of 2 mM lithium (Fig. 3c,d). Accordingly, lithium up-regulates ORAI1 and STIM1 protein expression. The ORAI1 and STIM1 protein abundance was in lithium treated ChAc neurons similarly high as in untreated neurons from healthy volunteers. The additional treatment with the SGK1 inhibitor GSK650394 (10 µM) decreased the ORAI1 and STIM1 protein abundance. In the presence of both, lithium and GSK650394, the ORAI1 and STIM1 protein abundance were not significantly different from the ORAI1 and STIM1 protein abundance in the absence of lithium and GSK650394 (Fig. 3) (Complete Western blot in Supplementary Figure S3 for ORAI1 and Figure S4 for STIM1).
Figure 3.
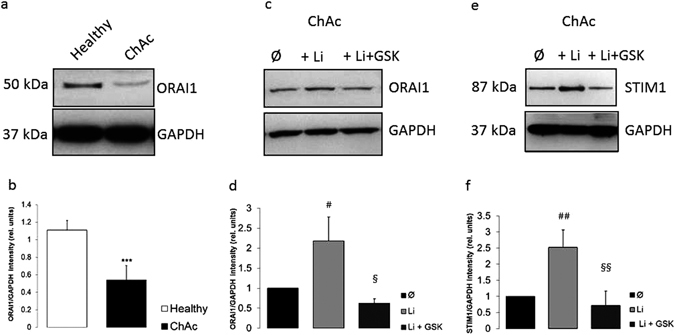
ORAI1 and STIM1 protein abundance in neurons generated from healthy volunteers and from ChAc patients without or with lithium treatment without and with SGK1 inhibitor GSK650394. (a) Original Western blot of ORAI1 protein abundance in neurons differentiated from healthy volunteers (healthy) and in neurons from ChAc patients (ChAc). (b) Arithmetic means (±SEM, n = 4) of ORAI1 protein levels in neurons generated from healthy volunteers (white bar) and in neurons from ChAc patients (black bar). (c) Original Western blot of ORAI1 protein abundance in neurons from ChAc patients without (Ø) and with prior lithium (24 h, 2 mM) treatment without (+Li) and with (+Li + GSK) additional presence of SGK1 inhibitor GSK650394 (10 µM). (d) Arithmetic means (±SEM, n = 4) of ORAI1 protein levels in neurons differentiated from ChAc patients without (black bar) or with lithium (24 h, 2 mM) treatment without (light grey bar) and with (dark grey bar) presence of SGK1 inhibitor GSK650394 (10 µM). (e) Original Western blot of STIM1 protein abundance in neurons from ChAc patients without (Ø) and with prior lithium (24 h, 2 mM) treatment without (+Li) and with (+Li + GSK) additional presence of SGK1 inhibitor GSK650394 (10 µM). (f) Arithmetic means (±SEM, n = 3) of STIM1 protein abundance in neurons differentiated from ChAc patients without (black bar) or with lithium (24 h, 2 mM) treatment without (light grey bar) and with (dark grey bar) presence of SGK1 inhibitor GSK650394 (10 µM). ***(p < 0.001) indicates statistically significant difference to respective value in neurons from healthy volunteers, #(p < 0.05), ##(p < 0.01) indicates statistically significant difference to respective value in absence of lithium. §(p < 0.05), §§(p < 0.01) indicates statistically significant difference to respective value in the absence of GSK650394.
Store operated Ca2+ entry
Decreased expression of ORAI1 and STIM1 were expected to be paralleled by impaired store operated Ca2+ entry (SOCE). Fura2 fluorescence was thus employed to quantify the cytosolic Ca2+ concentration ([Ca2+]i). For determination of SOCE, the intracellular stores were emptied by exposure of the cells to the sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin (1 µM) in the absence of extracellular Ca2+. Re-addition of extracellular Ca2+ in the continued presence of thapsigargin resulted in a sharp increase of [Ca2+]i reflecting SOCE. As shown in Fig. 4a–c, emptying the intracellular Ca2+ stores with thapsigargin was followed by a transient increase in [Ca2+]i to similar values in control neurons and ChAc neurons. The increase of [Ca2+]i following re-addition of extracellular Ca2+ in the continued presence of thapsigargin was significantly blunted in ChAc neurons as compared to control neurons (Fig. 4d,e). Both, slope and peak of [Ca2+]i increase following re-addition of extracellular Ca2+ were significantly lower in ChAc neurons than in control neurons. SOCE was virtually abrogated by ORAI1 inhibitor 2-APB (50 µM) (data not shown).
Figure 4.
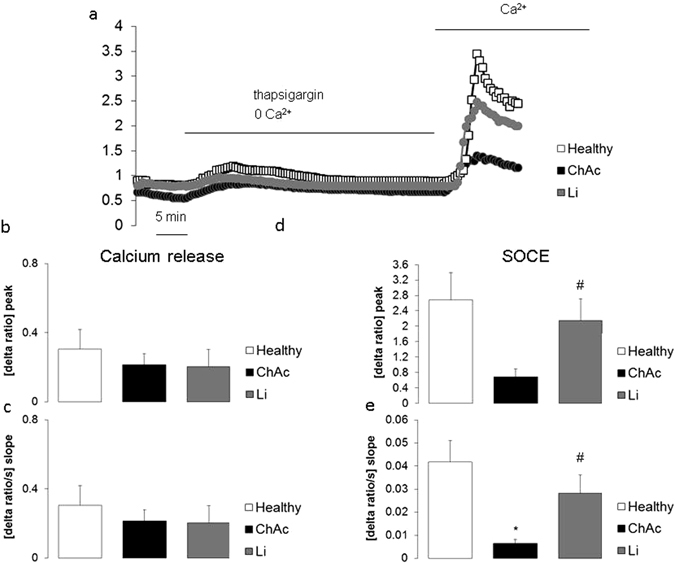
Intracellular Ca2+ release and store-operated Ca2+ entry (SOCE) in neurons from healthy volunteers and from ChAc patients without or with lithium treatment. (a) Representative tracings of Fura-2 fluorescence-ratio in fluorescence spectrometry before and following extracellular Ca2+ removal and addition of thapsigargin (1 µM), as well as re-addition of extracellular Ca2+ in neurons generated from healthy volunteers (white squares) and from ChAc patients without (black circles) and with (grey circles) lithium (24 h, 2 mM) treatment. (b,c). Arithmetic means (±SEM, n = 37–74 cells from 4 individuals) of slope (b) and peak (c) increase of fura-2-fluorescence-ratio following addition of thapsigargin (1 µM) in control neurons (white bar) and in ChAc neurons without (black bar) and with (grey bar) lithium (24 h, 2 mM) treatment. (d,e) Arithmetic means (±SEM, n = 37–74 cells from 4 individuals) of slope (d) and peak (e) increase of fura-2-fluorescence-ratio following re-addition of extracellular Ca2+ in neurons from healthy volunteers (white bars) and in neurons from ChAc patients without (black bar) and with (grey bar) lithium (24 h, 2 mM) treatment. *(p < 0.05) indicates statistically significant difference to respective value in neurons from healthy volunteers, #(p < 0.05) indicates statistically significant difference to respective value in absence of lithium.
Further experiments explored the effect of lithium on SOCE. To this end, ChAc neurons were incubated for 24 hours in the presence or absence of 2 mM lithium. As a result, without triggering SOCE, [Ca2+]i was in ChAc neurons similar in the absence or presence of lithium (data not shown). Both, peak and slope of SOCE were, however, significantly higher in lithium treated than in untreated ChAc neurons (Fig. 4d,e). Following lithium treatment, SOCE was in ChAc neurons of similar magnitude as in untreated neurons from healthy volunteers.
Apoptosis
Flow cytometry was employed to explore whether the differences in SOCE were paralleled by similar differences in apoptotic neuronal death. Apoptotic neurons were identified by measurement of annexin-V-binding and propidium iodide uptake. As apparent from Fig. 5a,b, the percentage of propidium iodide harboring and annexin-V-binding cells was significantly higher in ChAc neurons than in control neurons.
Figure 5.
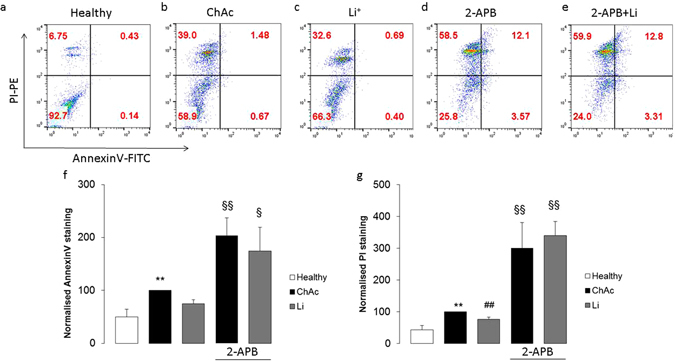
Phosphatidylserine translocation and propidium iodide uptake in neurons from healthy volunteers and from ChAc patients without and with treatment with lithium and/or 2-APB. (a–e) Representative dot blots of propidium iodide staining versus annexin-V-binding of neurons generated from (a) healthy volunteers and from (b–e) ChAc patients (b) without treatment and with lithium treatment (2 mM) alone (c), with 50 µM 2-APB alone (d), and with lithium and 2-APB together (e). (f,g) Arithmetic means (±SEM, n = 4–8 from 4 individuals) of the normalised (f) annexin-V-binding or (g) propidium iodide stained neurons from healthy volunteers (white bars) and from ChAc patients without (black bars) or with (grey bars) 2 mM lithium treatment without (left bars) and with (right bars) presence of 50 µM 2-APB (2-APB). **(p < 0.01) indicates statistically significant difference to respective value in neurons from healthy volunteers, ##(p < 0.01) indicates statistically significant difference to respective value in absence of lithium, §(p < 0.05), §§(p < 0.01) indicates statistically significant difference to respective value in absence of 2-APB (ANOVA).
The percentage of propidium iodide harboring and annexin-V-binding ChAc neurons was significantly lower following a 24 hours incubation in the presence than in the absence of lithium (Fig. 5b,c). Exposure of ChAc neurons with ORAI1 blocker 2-APB (50 µM) dramatically and significantly increased the percentage of propidium iodide harboring and annexin-V-binding cells and virtually abrogated the effect of lithium on the percentage of propidium iodide harboring and annexin-V-binding ChAc neurons (Fig. 5d,e).
Discussion
The present study uncovers a novel pathophysiological mechanism in chorea-acanthocytosis, i.e. deranged regulation of ORAI1 and STIM1 expression with subsequent impairment of store operated Ca2+ entry (SOCE). SOCE is significantly and markedly down-regulated in neurons lacking functional chorein.
SOCE may trigger oscillations of cytosolic Ca2+ activity ([Ca2+]i)28, resulting from rapid Ca2+ entry due to activation of SOCE followed by inhibition of ORAI1 and rapid decrease of [Ca2+]i due to subsequent Ca2+ extrusion29. The repetitive short pulses of [Ca2+]i activate Ca2+ dependent transcription factors and reorganize the actin filament network without triggering the detrimental consequences of sustained increases of [Ca2+]i 30, 31.
The Ca2+ oscillations influence diverse cellular functions32, 33 including entry into the S and the M phase of the cell cycle34 and cell survival35, 36. Along those lines the ORAI isoforms19, and their regulators STIM 1 or 220 contribute to the orchestration of survival, proliferation, and migration of tumor cells37–40 and neural stem/progenitor cells41. As shown here, pharmacological inhibition of ORAI1 triggers apoptosis and disrupts the supporting effect of lithium on neuronal cell survival.
In contrast to Ca2+ oscillations, sustained increases of cytosolic Ca2+ activity have been shown to stimulate apoptosis in a variety of cell types42–44. Thus, cell survival or death depends on a delicate balance between stimulators and inhibitors of Ca2+ entry.
The present study sheds some light on the signalling linking chorein deficiency and ORAI1 protein abundance. Previous observations in other cell types revealed that ORAI1 expression is up-regulated by the PI3K dependent45 serum & glucocorticoid inducible kinase SGK146, 47. Along those lines, the effect of lithium is reversed in the presence of SGK1 inhibitor GSK650394. SGK1 is effective by NFκB dependent up-regulation of ORAI1 expression and by inhibition of Nedd4-2 triggered degradation of ORAI1 protein46, 47. Chorein deficiency impairs activation of PI3K1–3 and is thus expected to disrupt PI3K/SGK1/ NFκB dependent ORAI1 upregulation. The observed signalling may thus well contribute to the anti-apoptotic effect of PI3K which confers survival of a wide variety of cells including cancer cells48–51 and neurons52–55.
The present observations further uncover an anti-apoptotic effect of lithium in neurons from chorea-acanthocytosis. The effect of lithium apparently requires up-regulation of SOCE. Lithium has previously been shown to counteract neurodegeneration in several diseases such as Huntington’s chorea, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis as well as spinocerebellar ataxias type 1 and type 325–27, 56, 57. Mechanisms involved in the neuroprotective effect of lithium include direct or Akt-mediated inhibition of glycogen synthase kinase GSK-3β, Akt-mediated inhibition of the proapoptotic forkhead box class O transcription factor Foxo3a and murine double minute (MDM), stimulation of production and activity of neuroprotective brain derived neurotrophic factor BDNF, up-regulation of antiapoptotic protein Bcl-2, as well as down-regulation of proapoptotic transcription factor p53, of the proapoptotic proteins Bad and Bax, of glutamate excitotoxicity, of calpain and of oxidative stress27, 58. We show here that the protective effect of lithium is abrogated by Orai1 inhibition. In view of the present observations, it is tempting to speculate that lithium may modify the course of neurodegeneration in part by up-regulating neuronal ORAI1 expression and SOCE thus stimulating proliferation of neuronal progenitor cells and inhibiting neuronal apoptosis.
In view of the present observations clinical studies appear warranted exploring the effect of lithium treatment on the clinical course of chorea-acanthocytosis. To the extent that neuronal apoptosis is the decisive pathophysiological mechanism leading to this devastating disease, lithium treatment may delay or even halt the deterioration of neuronal function in affected patients.
In conclusion, lack of Chorein in chorea-acanthocytosis downregulates ORAI1 expression and store operated Ca2+ entry leading to compromised neuronal cell survival. Conversely, lithium up-regulates store operated Ca2+ entry and attenuates neuronal apoptosis, an effect abrogated by pharmacological inhibition of ORAI1.
Methods
Generation of iPSCs
The study has been approved by the Ethical Commission of the University of Tübingen (598/2011). Informed consent was obtained from all participants and/or their legal guardian/s. Human dermal fibroblasts were isolated from ChAc patients (n = 2) and healthy volunteers (n = 3). We confirm that all methods performed, including obtaining of consent, were performed in accordance with the relevant guidelines and regulations as approved by the ethics committee. Dermal fibroblasts were cultivated in fibroblast medium, consisting of DMEM (Biochrom, Berlin, Germany) supplemented with 10% fetal calf serum (FCS, Life technologies, Thermo Fisher Scientific, Waltham, Massachusetts) and 1% L-Glutamine (Biochrom). Induced pluripotent stem cells (iPSCs) were generated following a protocol published previously59, with minor modifications. In brief, 1 × 105 fibroblasts were electroporated (Nucleofector 2D, Lonza) with a total of 1 μg per plasmid carrying the sequences for hOCT4, hSOX2, hKLF4, hL-MYC and hLIN28. After cultivation in fibroblast medium for 1 day, 2 ng/ml FGF-2 (Peprotech) was supplemented to the medium. From day 3 on, cells were cultivated in Essential 8 (E8) medium containing 100 µM NaB (Sigma-Aldrich). iPSC colonies were picked manually after 3–4 weeks and further expanded in Matrigel coated 6-well plates. At passage 7–10, iPSCs were characterized and frozen in E8 medium supplemented with 40% KOSR (Thermo Fisher Scientific), 10% DMSO (Sigma-Aldrich), and 1 µM Y-27632 (Selleckchem, Munich, Germany). Characterization of generated iPSCs included genomic validation via exclusion of plasmid-integration, SNParray analysis for genetic integrity, and resequencing of mutation site, as well as functional validation via confirmation of expression of pluripotency marker, and verification of the in vitro differentiation potential as described previously60.
Differentiation of iPSCs to neurons
Cortical neurons were generated as described previously61. Briefly, neural induction of iPSCs was achieved by addition of dual SMAD inhibitors (10 µM SB431542 (Sigma-Aldrich) and 500 nM LDN-193189 (Sigma-Aldrich)) to 3N medium. Cells were collected at day 10 and further expanded by cultivation in 3N medium supplemented with 20 ng/ml FGF-2 for 2 days. From day 12 on, cells were cultivated in 3N medium with medium change every other day. Cell cultures were passaged at day 27 and replated appropriately for the specific assay (RNA/Protein isolation: 5 × 105 cells per cm2; FACS analysis: 2.5 × 105 per cm2; Ca2+ measurements: 5 × 104 per cm2). Where indicated, 2 mM lithium (Sigma-Aldrich) was added to the medium 24 hours prior to measurements. Analysis was performed between day 37 and 41.
Quantitative PCR
Transcript levels of ORAI1, STIM1, STIM2, and house keeping Gapdh were determined by RT-PCR as described previously23, 62. Total RNA was extracted in TriFast (Peqlab, Erlangen, Germany) according to the manufacturer’s instructions. After DNAse digestion reverse transcription of 2 µg RNA was performed using random hexamers (Roche Diagnostics, Penzberg, Germany) and SuperScriptIII reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Real-time polymerase chain reaction (RT-PCR) amplification of the respective genes were set up in a total volume of 20 µl using 40 ng of cDNA, 500 nM forward and reverse primer and 2x GoTaq® qPCR Master Mix (Promega, Hilden, Germany) according to the manufacturer’s protocol. Cycling conditions were as follows: initial denaturation at 95 °C for 2 minutes, followed by 40 cycles of 95 °C for 15 seconds, 55 °C for 15 seconds and 68 °C for 20 seconds. For amplification the following primers were used (5′->3′orientation):
GAPDH:
fw: TGAGTACGTCGTGGAGTCCAC;
rev: GTGCTAAGCAGTTGGTGGTG
ORAI11:
fw: CGTATCTAGAATGCATCCGGAGCC;
rev: CAGCCACTATGCCTAGGTCGACTAGC
STIM1:
fow: CCTCGGTACCATCCATGTTGTAGCA
rev: GCGAAAGCTTACGCTAAAATGGTGTCT
STIM2:
for: CAAGTTGCCCTGCGCTTTAT
rev: ATTCACTTTTGCACGCACCG
Specificity of PCR products was confirmed by analysis of a melting curve. Real-time PCR amplifications were performed on a CFX96 Real-Time System (Bio-Rad, Munich, Germany) and all experiments were done in duplicate. The house-keeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was amplified to standardize the amount of sample RNA.
Western Blotting
Protein abundance of ORAI1, STIM1 and GAPDH was determined by Western blotting as described previously23, 62. To this end, cells were centrifuged for 5 minutes at 240 g and 4 °C. The pellet was washed twice with ice cold PBS and suspended in 200 μl ice-cold RIPA lysis buffer (Thermo Fisher Scientific, USA) containing Halt Protease and Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, USA). Protein concentration was determined using the Bradford assay (BioRad, München, Germany). 100 µg of protein were solubilized in sample buffer at 95 °C for 5 min. The proteins were separated by a 10% SDS-PAGE in a Glycine-Tris buffer and electro-transferred onto nitrocellulose membranes for 70 min. After blocking with 5% milk in TBST at room temperature for 1 h, the membranes were incubated with primary anti-ORAI1 antibody (1:1000, Proteintec), anti STIM1 antibody (1:2000, Cell Signaling) and anti-GAPDH antibody (1:2000, Cell Signaling) at 4 °C overnight. After washing (TBST), the blots were incubated with secondary anti-rabbit antibody conjugated with horseradish peroxidase (1:2000, Cell Signaling) for 1 h at room temperature. Protein bands were detected after additional washes (TBST) with an ECL detection reagent (Amersham, Freiburg, Germany) and quantified with Quantity One Software (BioRad, München, Germany). To assign the right protein size we used Protein-Marker VI (Peqlab, Erlangen, Germany).
Ca2+ measurements
Fura-2 fluorescence was taken as a measure of cytosolic Ca2+ concentration ([Ca2+]i), as described previously23, 62, For this purpose cells were loaded with Fura-2/AM (2 µM, Invitrogen, Goettingen, Germany) for 20 min at 37 °C. Cells were excited alternatively at 340 nm and 380 nm through an objective (Fluor 40×/1.30 oil) built in an inverted fluorescence microscope (Axiovert 100, Zeiss, Oberkochen, Germany). Emitted fluorescence intensity was recorded at 505 nm. Data were acquired using specialized computer software (Metafluor, Universal Imaging, Downingtown, USA). Cytosolic Ca2+ activity was estimated from the 340 nm/380 nm ratio. SOCE was determined by extracellular Ca2+ removal and subsequent Ca2+ re-addition in the presence of thapsigargin (1 µM, Invitrogen). For quantification of Ca2+ entry, the slope (delta ratio/s) and peak (delta ratio) were calculated following re-addition of Ca2+.
Experiments were performed with Ringer solution containing (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 2 CaCl2, 2 Na2HPO4, 32 HEPES, 5 glucose, pH 7.4. To reach nominally Ca2+-free conditions, experiments were performed using Ca2+-free Ringer solution containing (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 2 Na2HPO4, 32 HEPES, 0.5 EGTA, 5 glucose, pH 7.4.
Analysis of apoptosis
Propidium iodide (PI, Sigma-Aldrich) uptake was taken as a measure of cell membrane permeability and annexin V-FITC (Immunotools, Friesoythe, Germany) binding as a measure of cell membrane scrambling with phosphatidylserine translocation at the cell surface, as described previously63, 64. To this end, cells were incubated in 100 µl Ringer solution containing 5 mM Ca2+ and 1 µl annexin V-FITC After incubation, the cells were centrifuged at 1000 r.p.m. for 5 minutes. Then, cells were stained with annexin V-FITC (Immunotools, Friesoythe, Germany) to assess phosphatidylserine exposure and propidium iodide (PI) to estimate cell membrane integrity. To this end, cells were incubated in 100 µl Ringer solution containing 5 mM Ca2+ and 1 µl annexin V-FITC (Immunotools, Friesoythe, Germany) and 0.075 µl PI stock solution (25 mg/ml PI in PBS) for 15 min at 37 °C. Then, 100 µl of Ringer solution were added and annexin V-FITC as well as PI fluorescence were determined by flow cytometry using a FACS calibur (BD, Heidelberg, Germany).
Statistics
Data are expressed as arithmetic means ± SEM. Statistical analysis was made by unpaired t-test or Mann-Whitney test or analysis of variance (ANOVA), as appropriate. p < 0.05 was considered as statistically significant.
Electronic supplementary material
Acknowledgements
The authors acknowledge the meticulous preparation of the manuscript by Lejla Subasic and the technical support by Yvonne Theurer. This work was supported by grants from Brigitte-Schlieben-Lange-Programm to L.P, by the Deutsche Forschungsgemeinschaft (La315-15) to F.L., by a Council of Scientific & Industrial Research (CSIR-SRF), Government of India and Deutscher Akademischer Austauschdienst (DAAD) bi-nationally supervised PhD fellowship to IS, by an EC funded Neuromics project (F5-2012-305121) to L.S. and by the Open Access Publishing Fund, University of Tuebingen. Work of R.B. is supported by the Institutional Strategy of the University of Tübingen (Deutsche Forschungsgemeinschaft, ZUK63).
Author Contributions
L.P., S.H., B.E., B.S., I.S., Y.S., P.H., R.B., M.J. performed experiments, C.S., L.S., F.L. designed the study and F.L. drafted the manuscript. All authors corrected and approved the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Ludger Schöls and Florian Lang contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06451-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.IntAct, D. I. F. I. http://www.ebi.ac.uk/intact/pages/interactions/interactions.xhtml?conversationContext=1. EMBL EBI database (2011).
- 2.Foller M, et al. Chorein-sensitive polymerization of cortical actin and suicidal cell death in chorea-acanthocytosis. FASEB J. 2012;26:1526–1534. doi: 10.1096/fj.11-198317. [DOI] [PubMed] [Google Scholar]
- 3.Honisch S, et al. Chorein addiction in VPS13A overexpressing rhabdomyosarcoma cells. Oncotarget. 2015;6:10309–10319. doi: 10.18632/oncotarget.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobson-Stone, C. et al. Chorea-Acanthocytosis. Gene Rev, updated 2010 (1993).
- 5.Ueno S, et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001;28:121–122. doi: 10.1038/88825. [DOI] [PubMed] [Google Scholar]
- 6.Dobson-Stone C, et al. Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. European journal of human genetics: EJHG. 2002;10:773–781. doi: 10.1038/sj.ejhg.5200866. [DOI] [PubMed] [Google Scholar]
- 7.Saiki S, et al. Primary skeletal muscle involvement in chorea-acanthocytosis. Movement disorders: official journal of the Movement Disorder Society. 2007;22:848–852. doi: 10.1002/mds.21437. [DOI] [PubMed] [Google Scholar]
- 8.Velayos Baeza, A. et al. In GeneReviews(R) (eds R. A. Pagon et al.) (University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved, 1993).
- 9.Walterfang M, et al. Shape alterations in the striatum in chorea-acanthocytosis. Psychiatry Res. 2011;192:29–36. doi: 10.1016/j.pscychresns.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomemori Y, et al. A gene-targeted mouse model for chorea-acanthocytosis. J Neurochem. 2005;92:759–766. doi: 10.1111/j.1471-4159.2004.02924.x. [DOI] [PubMed] [Google Scholar]
- 11.Kurano Y, et al. In vivo distribution and localization of chorein. Biochem Biophys Res Commun. 2007;353:431–435. doi: 10.1016/j.bbrc.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 12.Alesutan I, et al. Chorein sensitivity of actin polymerization, cell shape and mechanical stiffness of vascular endothelial cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2013;32:728–742. doi: 10.1159/000354475. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt EM, et al. Chorein sensitivity of cytoskeletal organization and degranulation of platelets. FASEB J. 2013;27:2799–2806. doi: 10.1096/fj.13-229286. [DOI] [PubMed] [Google Scholar]
- 14.Honisch S, et al. Chorein Sensitive Dopamine Release from Pheochromocytoma (PC12) Cells. Neurosignals. 2015;23:1–10. doi: 10.1159/000442599. [DOI] [PubMed] [Google Scholar]
- 15.Honisch S, et al. Chorein Sensitive Arrangement of Cytoskeletal Architecture. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2015;37:399–408. doi: 10.1159/000430363. [DOI] [PubMed] [Google Scholar]
- 16.Saiki S, et al. Primary skeletal muscle involvement in chorea-acanthocytosis. Mov Disord. 2007;22:848–852. doi: 10.1002/mds.21437. [DOI] [PubMed] [Google Scholar]
- 17.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 18.Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nature reviews. Neuroscience. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Putney JW., Jr. New molecular players in capacitative Ca2+ entry. J Cell Sci. 2007;120:1959–1965. doi: 10.1242/jcs.03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinelt C, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smyth JT, et al. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penna A, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu W, et al. Chorein Sensitive Orai1 Expression and Store Operated Ca2+ Entry in Rhabdomyosarcoma Cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;40:1141–1152. doi: 10.1159/000453168. [DOI] [PubMed] [Google Scholar]
- 24.Zhang B, et al. Lithium- Sensitive Store-Operated Ca2+ Entry in the Regulation of FGF23 Release. Neurosignals. 2015;23:34–48. doi: 10.1159/000442602. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez G, et al. Regulation of tau phosphorylation and protection against beta-amyloid-induced neurodegeneration by lithium. Possible implications for Alzheimer’s disease. Bipolar Disord. 2002;4:153–165. doi: 10.1034/j.1399-5618.2002.01150.x. [DOI] [PubMed] [Google Scholar]
- 26.Bauer M, Alda M, Priller J, Young LT. International Group For The Study Of Lithium Treated, P. Implications of the neuroprotective effects of lithium for the treatment of bipolar and neurodegenerative disorders. Pharmacopsychiatry. 2003;36(Suppl 3):S250–254. doi: 10.1055/s-2003-45138. [DOI] [PubMed] [Google Scholar]
- 27.Lazzara CA, Kim YH. Potential application of lithium in Parkinson’s and other neurodegenerative diseases. Frontiers in neuroscience. 2015;9:403. doi: 10.3389/fnins.2015.00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang F, et al. Bradykinin-induced oscillations of cell membrane potential in cells expressing the Ha-ras oncogene. J Biol Chem. 1991;266:4938–4942. [PubMed] [Google Scholar]
- 29.Parekh AB. Regulation of CRAC channels by Ca2+ -dependent inactivation. Cell calcium. 2016 doi: 10.1016/j.ceca.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Lang F, et al. Ion channels and cell volume in regulation of cell proliferation and apoptotic cell death. Contrib Nephrol. 2006;152:142–160. doi: 10.1159/000096321. [DOI] [PubMed] [Google Scholar]
- 31.Lang F, Stournaras C. Ion channels in cancer: future perspectives and clinical potential. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2014;369:20130108. doi: 10.1098/rstb.2013.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 33.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 34.Taylor JT, et al. Calcium signaling and T-type calcium channels in cancer cell cycling. World J Gastroenterol. 2008;14:4984–4991. doi: 10.3748/wjg.14.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heise N, et al. Non-selective cation channel-mediated Ca2+ -entry and activation of Ca2+/calmodulin-dependent kinase II contribute to G2/M cell cycle arrest and survival of irradiated leukemia cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2010;26:597–608. doi: 10.1159/000322327. [DOI] [PubMed] [Google Scholar]
- 36.Parkash J, Asotra K. Calcium wave signaling in cancer cells. Life Sci. 2010;87:587–595. doi: 10.1016/j.lfs.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergmeier W, Weidinger C, Zee I, Feske S. Emerging roles of store-operated Ca(2)(+) entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels (Austin) 2013;7:379–391. doi: 10.4161/chan.24302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609–618. doi: 10.1038/nrc3105. [DOI] [PubMed] [Google Scholar]
- 39.Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y. Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2014;369:20130097. doi: 10.1098/rstb.2013.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu B, Al-Ansary D, Kummerow C, Hoth M, Schwarz EC. ORAI-mediated calcium influx in T cell proliferation, apoptosis and tolerance. Cell calcium. 2011;50:261–269. doi: 10.1016/j.ceca.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Somasundaram A, et al. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:9107–9123. doi: 10.1523/JNEUROSCI.0263-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang KM, Chang WL, Wang SM, Su MJ, Wu ML. Arachidonic acid induces both Na+ and Ca2+ entry resulting in apoptosis. J Neurochem. 2008;104:1177–1189. doi: 10.1111/j.1471-4159.2007.05022.x. [DOI] [PubMed] [Google Scholar]
- 43.Lang F, Hoffmann EK. Role of ion transport in control of apoptotic cell death. Compr Physiol. 2012;2:2037–2061. doi: 10.1002/cphy.c110046. [DOI] [PubMed] [Google Scholar]
- 44.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 45.Raimondi C, Falasca M. Targeting PDK1 in cancer. Curr Med Chem. 2011;18:2763–2769. doi: 10.2174/092986711796011238. [DOI] [PubMed] [Google Scholar]
- 46.Lang F, Eylenstein A, Shumilina E. Regulation of Orai1/STIM1 by the kinases SGK1 and AMPK. Cell calcium. 2012;52:347–354. doi: 10.1016/j.ceca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt S, et al. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget. 2014;5:4799–4810. doi: 10.18632/oncotarget.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews. Drug discovery. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ocana A, et al. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: systematic review and meta-analysis. PloS one. 2014;9:e95219. doi: 10.1371/journal.pone.0095219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nature reviews. Clinical oncology. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 51.Zhang L, Zhou F, ten Dijke P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends in biochemical sciences. 2013;38:612–620. doi: 10.1016/j.tibs.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 52.Sedding DG. FoxO transcription factors in oxidative stress response and ageing–a new fork on the way to longevity? Biol Chem. 2008;389:279–283. doi: 10.1515/BC.2008.033. [DOI] [PubMed] [Google Scholar]
- 53.Dudek H, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 54.Arboleda G, Morales LC, Benitez B, Arboleda H. Regulation of ceramide-induced neuronal death: cell metabolism meets neurodegeneration. Brain Res Rev. 2009;59:333–346. doi: 10.1016/j.brainresrev.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 55.Kreis P, Rousseau V, Thevenot E, Combeau G, Barnier JV. The four mammalian splice variants encoded by the p21-activated kinase 3 gene have different biological properties. J Neurochem. 2008;106:1184–1197. doi: 10.1111/j.1471-4159.2008.05474.x. [DOI] [PubMed] [Google Scholar]
- 56.Jia DD, et al. Lithium chloride alleviates neurodegeneration partly by inhibiting activity of GSK3beta in a SCA3 Drosophila model. Cerebellum (London, England) 2013;12:892–901. doi: 10.1007/s12311-013-0498-3. [DOI] [PubMed] [Google Scholar]
- 57.Watase K, et al. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS medicine. 2007;4:e182. doi: 10.1371/journal.pmed.0040182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mao Z, Liu L, Zhang R, Li X. Lithium reduces FoxO3a transcriptional activity by decreasing its intracellular content. Biological psychiatry. 2007;62:1423–1430. doi: 10.1016/j.biopsych.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 59.Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nature methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 60.Hauser S, Hoflinger P, Theurer Y, Rattay TW, Schols L. Generation of induced pluripotent stem cells (iPSCs) from a hereditary spastic paraplegia patient carrying a homozygous Y275X mutation in CYP7B1 (SPG5) Stem cell research. 2016;17:437–440. doi: 10.1016/j.scr.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 61.Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nature protocols. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 62.Yan J, et al. Decrease of Store-Operated Ca2+ Entry and Increase of Na+/Ca2+ Exchange by Pharmacological JAK2 Inhibition. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;38:683–695. doi: 10.1159/000443126. [DOI] [PubMed] [Google Scholar]
- 63.Zeng N, et al. 1alpha, 25(OH) 2D3 Sensitive Cytosolic pH Regulation and Glycolytic Flux in Human Endometrial Ishikawa Cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2017;41:678–688. doi: 10.1159/000458427. [DOI] [PubMed] [Google Scholar]
- 64.Towhid ST, et al. Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2013;32:838–848. doi: 10.1159/000354486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.