

Abstract
Spinal cord ischemia and reperfusion (SCIR) injury is the major cause of a wide range of complications, including neural degeneration and devastating paraplegia. Decrease of inhibitory neurotransmitters and increase of excitory neurotransmitters are the major cause for the excitotoxicity of neurons. However, no study has reported the temporal loss of motor neuron in the ventral horn of spinal cord area following SCIR-induced spastic paralysis, not even the mechanism under it. In the present study, we found that the rabbits were mainly spastic paralyzed after spinal cord ischemia-reperfusion injury. And the ischemia 60 min group is the optimal treating condition, because of the higher rate of spastic paralysis and lower mortality. Motor neurons in the ventral horn of spinal cord were significant degeneration at 3 h following spastic paralysis and only 12.5% motor neurons were observed at 72 h post-operation, compared with control group. ELISA results indicated that Glycine and GABA were both downregulated following spastic paralysis. But Glycine immediately decreased at 10 min post-operation and lasted for the whole process (at least 72 h). Meanwhile GABA only significantly decreased at 72 h. Furthermore, Glutamic expression was significant upregulation at 3 hours post-operation, and the upregulation back to the base level at 72 h post-operation. Glutamic receptor-(NR1) and Glycine α1 receptor upregulated accordingly, whereas GABBR2 didn’t upregulate significantly until at 72 h post-operation. Abundant extracellular Ca2+ influxed into cytoplasm in neurons following spastic paralysis. The type of paraplegia is mainly spastic paraplegia after SCIR (ischemia 60 min treatment). Following spastic paraplegia, motor neuron in the ventral horn of spinal cord area was significant degeneration at early stage and last for the whole process. It may contribute to the decrease of Glycine at early stage and followed exitotoxicity, which caused intracellular calcium overload to make neurons dead. It would lay the foundation for better understanding the motor neuron degeneration and mechanism following spastic paralysis. And it would supply a novel and effective target for spastic paralysis prevention and therapy.
Keywords: Spinal cord ischemia reperfusion injury, spastic paralysis, motor neurons, Glycine, GABA
Introduction
Spinal cord ischemia and reperfusion (SCIR) injury may develop in a variety of situations, such as vascular pathologies causing acute arterial occlusion, surgical interventions requiring clamping, trauma-causing ischemia, transplantation, and shock [1]. A wide range of complications, including neural degeneration and devastating paraplegia, even death, may develop after SCIR damage. It’s reported about 2%-18% patients displayed neurological deficits due to SCIR damage [2]. Clinically, lower limb movement in patients is observed immediately recover after operation but later deteriorates [3-5]. But the characteristic of paraplegia caused by SCIR is rarely reported.
Motor neurons are nerve cells whose cell body is located in the ventral horn of spinal cord and whose fiber projects outside the spinal cord to directly or indirectly control effector organs, mainly muscles and glands. Motor neurons carry signals from the spinal cord to the effectors to produce effects. Alpha-motor neurons (AMNs or lower motor neurons) reside in the spinal cord. Dysfunction and death of these neurons lead to muscle weakness, atrophy, and spasticity [6,7]. After spinal cord injury (SCI), motor neurons degeneration is observed with time dependent according to neurotransmitters deregulation [8]. BDNF, Glu, GABA, Glycine and other neurotransmitters have been demonstrated to involve in inducing motor neuron degeneration [9-12]. Thus, recovery of motor neurons activity, protecting motor neurons from injury through regulating related neurotransmitters or receptor expression is the widely used and effective strategy [13-15].
However, until now, no study has reported the variation trend of motor neurons in the ventral horn of spinal cord following SCIR-induced spastic paralysis, not even the mechanism under it. In the present study, we aimed to determine the mechanism and effects of motor neurons degeneration.
Materials and methods
Spinal cord ischemia-reperfusion injury model
Adult New Zealand white rabbits (2.2±0.3 kg) were supplied by the Animal Breeding Center of Institute of Surgery Research/Daping Hospital and housed in a light- and temperature-controlled room. All surgical interventions and postoperative animal care were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996, USA) and approved by the Animal Use and Care Committee of School of Medicine, Third Military Medical University. The spinal cord ischemia-reperfusion injury model was established according to the previous study [16]. To established spinal cord ischemia-reperfusion injury model, thirty-two rabbits were divided into four groups: normal group, ischemia 30 min group, ischemia 60 min group and ischemia 90 min group. At 1 day, 3 days and 7 days after reperfusion, the rabbits were photographed and the neurological function was observed. Briefly speaking, thirty-two rabbits were randomly divided into sham group, ischemia/reperfusion injury group (I/R group, ischemia for 30, 60 and 90 min respectively). Each group contained eight rabbits. In I/R group, rabbits were anesthetized with 3% sodium pentobarbital via auricular vein injection, and fixed in the arm recumbent position. The surgical area was disinfected with 75% ethanol and a 6 cm incision was made along the midline of the inferior border of the left ribs and the left kidney and the abdominal aorta is located. The abdominal aorta below the level of the renal artery was occluded for 30, 60 and 90 minutes using a 10 g aortic clamp causing spinal cord ischemic injury respectively. The wounds were covered with saline gauze and the arterial clamp was removed 30, 60 and 90 minutes later. The abdominal cavity was closed after applying and spreading penicillin powder. In the sham group, only the abdominal cavity was opened and the abdominal aorta was not occluded.
Neurologic function assessment
Neurological function was observed at 1 day, 3 days, and 7 days after the procedure. Animals were classified with a 11-point scale according to the method of Reuter’s score [17]: 0: normal function; 11: hind-limb paralysis, the higher score indicates more severe dysfunction. Paraplegia was identified according to the Jacobs score [18]: 0, complete paralysis; 1, minimal functional movement, severe paresis; 2, functional movement, cannot hop; 3, hopping, ataxia and paresis; 4, hopping, mild ataxia and/or paresis; and 5, normal. The detailed rules in Reuter’s score involve in stretch reflex and muscle tonus were used to classify spastic paralysis and flaccid paralysis [17]. Stretch reflex: 0, normal; 1, slightly increase/decrease; 2, excite/disappear. Muscular tension: 0, normal; 1, high/low tension; 2, flaccid/spastic. Two individuals without knowledge of the treatment graded neurological function independently.
Histological analysis
Rabbits were deeply anesthetized by auricular vein injection of sodium pentobarbital and sacrificed by reperfusion with 500 ml of cold 0.01 M phosphate buffered saline (PBS, pH 7.4), and followed by 1,000 ml of 4% paraformaldehyde in cold 0.1 mM PB (pH 7.4). After perfusion, a 30 mm block of the spinal cord containing the injury site in the middle was carefully removed and fixed in the same fixation solution, dehydrated overnight in ethanol, and embedded in a paraffin blocks. Serial transverse sections were made. For further morphological and morphometric analyses, three sets of slides each containing serial sections were stained with hematoxylin and eosin (HE). Morphometric analysis of spinal cord was carried out in the transverse sections of tissue by HE stain. Area measurements were performed using the Image-Pro Plus 5.0 image analysis software (Media Cybernetics Inc., Atlanta, GA, USA). HE staining techniques were used to assess gray and white matters at the lesion site. Sections 5 mm rostral to the injury site were analyzed for residual ventral horn neurons. From each spinal cord, approximately 20 sections were inspected, and all slides were assessed blindly.
Immunofluorescence
The specimen was transferred to a solution containing 25% sucrose in 0.1 M PBS (pH 7.4) at 4°C for frozen section preparation, until it stayed at the bottom of the container. 20 mm spinal cord segment containing the entire injury site 3-6 mm rostral and 3-6 mm caudal to the injury site for transverse sections was embedded in tissue freezing medium (Tissue-Tek, Sakura Finetek Inc, USA). Series 20 μm thick cross sections were obtained using a cryostat (Leica CM1900, Bannockburn, IL) and thaw-mounted on polylysine -coated slides. The frozen sections were stored at 4°C and would be used for immunofluorescence double-labeling. Sections were then placed in primary antibody overnight at 4°C with primary antibodies made in PBS: anti-neurofilament 200 (NF-200) (sigma, N0142, 1:400) anti-Choline Acetyltransferase antibody (ChAT) (abcam, ab18736, 1:100), after incubation with primary antibodies, sections were washed 3 times in PBS and incubated with fluorescent-conjugated secondary antibodies (Alexa 488; FITC; 1:250; Invitrogen Corp., Carlsbad, CA, USA) and DAPI for general nuclear staining. Once staining was complete, sections were mounted on slides, dried at room temperature and covered with glycerin (Beyotime, Beijing, China). Images were captured using a laser scanning confocal microscope (Leica SP-2, Germany). An image post-processing was done with Adobe CS3 (Adobe Systems, Inc., San Jose, CA) with equal changes to any images being compared. Co-labeled cells in per frame were counted.
ELISA
Glycine (No.105632, Lanpai Bio, Shanghai, China), Glutamate (MBS2600824, MYBiosource, San Diego, CA, USA) and GABA (MBS2602301, MYBiosource, San Diego, CA, USA) were assessed by ELISA. The ventral horn of spinal cord was collected from rabbits of different groups. And then the tissues were homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.5), 1 mM EDTA, 5 mM MgCl2, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Pierce, Rockford, IL, USA). The tissue homogenate was centrifuged at 12,000 rpm for 15 min at 4°C, and the protein concentration of the supernatant was measured using a Bradford assay. The protein was added into the plate. The plates were washed 3 in PBS and added with the bound antibody that reacted with HRP-conjugated goat anti-rat IgG (1:10,000; SABC. City, state) for 1 h at 37°C . Optical density (OD) at 570 nm was measured with an ELISA reader.
Western blotting
Spinal cord tissue blocks were dissected using the same methods as for the histologic study experiment. The tissue blocks were homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl (pH7.5), 1 mM EDTA, 5 mM MgCl2, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail (Pierce, Rockford, IL, USA). The tissue homogenate was centrifuged at 12,000 rpm for 15 min at 4°C, and the protein concentration of the supernatant was measured using a Bradford assay (Beyotime, Beijing, China). Equal amounts of proteins were resolved by SDS-PAGE and transferred to a PVDF membrane (Millipore, Bedford, MA). The membrane was blocked in Tris-buffered saline (TBS) containing 5% milk and probed with anti-Gly α1 (1:500; Santa Cruz, Dallas, USA) and anti-GABAR2B (1:500; Millipore, Bedford, MA, USA) or NMDAR1 (1:1000; BioSS, Beijng, China). After washing, the membranes were incubated for 1 hr. At room temperature with secondary antibodies (1:10000, zombie, Beijing, China). All blots were probed with antibodies against GAPDH (1:3000. Millipore, Bedford, MA, USA) to normalize differences in loading amount. The relative expression is analyzed as previous study indicated [19].
Calcium activity
After the rabbits were anesthetized with 3% sodium pentobarbital, the spinal cord tissues were collected, homogenized in 0°C neural basal medium and cut into the tissue block. Then the tissue block was incubated in 5% CO2 cell incubator for 30 min. After that, the tissue block was incubated with Fura-3AM (Sigma-Aldrich. USA) in Ca2+-free D-Hanks medium for 30 min. After washed with 0.1 M PBS, the medium was replaced with NB + 2% B27 medium. Then the tissue block was observed timely with laser scanning confocal microscope at XYT model to examine the changes in free Ca2+ concentration in the tissue block stimulated by 2,000 nM glutamine.
Statistical analysis
Statistical analyses were performed using SPSS software, version 18.0 (IBM SPSS, Armonk, NY, USA). Statistical analysis of data was performed by one-way analysis of variance and Student’s t-tests for comparing two groups and ANOVA analysis for multiple comparisions. Values are expressed as the mean ± standard error of the mean. P<0.05 was considered to indicate a statistically significant difference.
Results
Spastic paralysis was induced in spinal cord ischemia-reperfusion rabbit
We evaluated locomotor function in spinal cord ischemia-reperfusion rabbit using Jacobs and Reuter’s score. At 1 day post injury (dpi), the mean Reuter’s score was 6.25±0.71, followed by gradual recovery at 3 dpi and 7 dpi in ischemia 30 min group (one rabbit was recovered). In ischemia 60 min and 90 min rabbits, limb rigidity, urinary and fecal incontinence were observed, especially the flexor and extensor muscle were both high tension. Rabbits in these two groups were completely paralysis. The mean Reuter’s score was significant higher in ischemia 60 min and 90 min groups than that in ischemia 30 min group (Figure 1A). According to the Jacobs score among 0-2 was identified as paraplegia, the percentage of paraplegia in ischemia 60 min and 90 min groups were higher than that in ischemia 30 min group (Figure 1B). At 3 dpi and beyond, no significant difference in percentage of paraplegia was observed in ischemia 60 min group and 90 min groups. To further classify the spastic paralysis and flaccid paralysis, stretch reflex and muscular tension was identified according to the Reuter’s score. As the results shown in Figure 1C, the percentage of spastic paralysis is 75% in ischemia 30 min group, while the percentage in ischemia 60 and 90 min group was 82.5% and 100% at 7 dpi. The severity of dysfunction was no significant in ischemia 60 and 90 min group, but 4 rabbits died in ischemia 90 min group and 1 rabbit died in ischemia 60 min group during 7 days. It indicated high mortality in ischemia 90 min group. Therefore, ischemia 60 min group was the optimal treating condition in following experiments.
Figure 1.
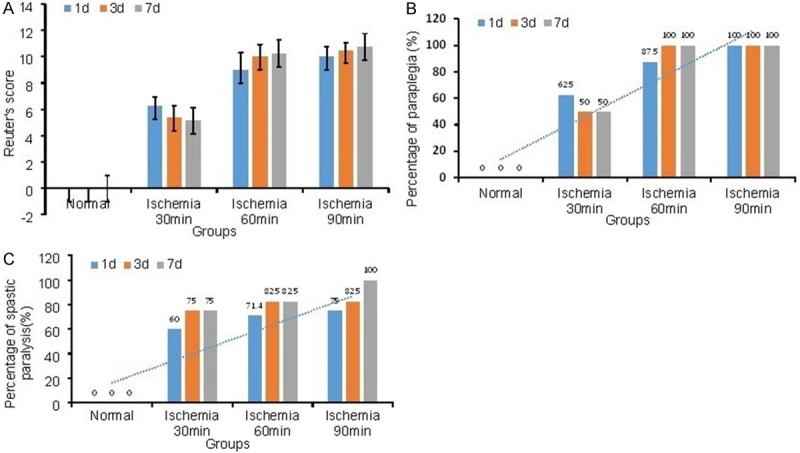
Score of the Rabbits at different time point following SCIR. A. 1 day, 3 days and 7 days post-operation, the neurologic function was recorded according to the Reuter’s score. In Reuter’s score, 0 indicates normal state and 11 indicates most severe dysfunction state. B. Paraplegia rate was statistics. The Jacobs score among 0-1 was identified as paraplegia. C. Spastic paralysis was indentified through stretch reflex: 0, normal; 1, slightly increase/decrease; 2, excite/disappear; and muscular tension: 0, normal; 1, high/low tension; 2, flaccid/spastic. (n=8).
Histology change in the ventral horn of spinal cord after SCIR
To observe the histological change in the ventral horn of spinal cord, the tissue transections at the 3rd day after SCIR were stained with HE. HE stain showed that ventral horn neurons with clear morphous and centered nucleolus were found in the normal group. Edema was also not found around neurons and neurite in the spinal cord (Figure 2A). In the ischemia 30 min group, mild edema was observed around neuron (Figure 2B). In the ischemia 60 and 90 min groups, nucleolus pyknic neuron cells were found, accompanied with thick color in cytoplasm and cellular degeneration (Figure 2C, 2D). Meanwhile, injury on white matters was also observed by HE staining. As showed in Figure 2E, nerve fibers were arranged in white matters, and without inflammatory cell invasion in normal group. In 30 min ischemia groups, disorder nerve fibers were found in white matters, accompanied with the edema around neuraxon following reperfusion (Figure 2F). In the ischemia 60 and 90 min groups, nerve fibers were more disorder and edema in neuraxon was more severe. Some neuraxon disaggregated with vacuole in white matters (Figure 2G, 2H). It suggested that motor neurons soma was more severely damaged than neuraxon.
Figure 2.
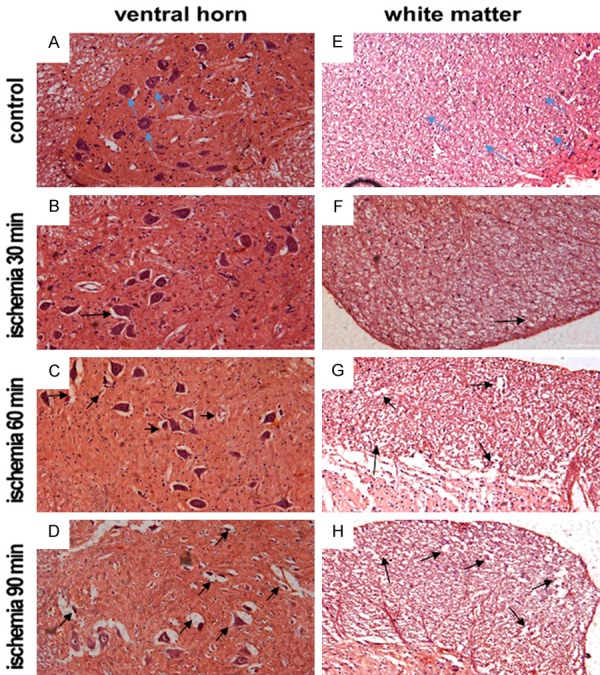
Histological analyses of spinal cord sections following SCIR. A-D. H&E staining of the ventral horn of spinal cord area at 3 days post reperfusion. A. Control group; B. Ischemia for 30 min group; C. Ischemia for 60 min group; D. Ischemia for 90 min group. E, F. H&E staining of the white matter of spinal cord area at 3 days post reperfusion. E. Control group; F. Ischemia for 30 min group; G. Ischemia for 60 min group; H. Ischemia for 90 min group. Blue arrows indicate the normal neurons and neurite; black arrows indicate the edema area and necrotic neurons.
SCIR induced motor neuron death in the ventral horn of spinal cord
As ischemia 60 min group rabbits had higher paraplegia rate and lower mortality, we fundamentally observed rabbit in this group in following researches. To evaluate the morphology of the surviving motor neuron cells in ventral horn, NF-200 and ChAT double staining was performed. The result showed the number of motor neurons in ventral horn reduced along with the elongation of reperfusion. No significant decreasing was observed at 10 min after reperfusion. Whereas, after 3 hours and until 72 hours after reperfusion, the number of motor neurons remarkably decreased (Figure 3A). Only 16.7±1.8 and 3.2±0.6 double labeled cells were found in per field at 6 and 72 hours after reperfusion separately (Figure 3B). Collectively, the above results indicated that the motor neuron cells began to degenerate at 3 hours after reperfusion and last to 72 hours following spastic paralysis.
Figure 3.

The expression of NF-200 and ChAT were detected in the ventral horn of spinal cord area on different time point post-operation. A. anti-NF-200 and anti-ChAT antibody were used for immunofluorescence in the ventral horn of spinal cord area. The green fluorescence signal indicates NF-200 protein and red fluorescence signal indicates ChAT protein. The nuclear was stained by DAPI. The tissues form control group, and 10 min, 3 h, 6 h, 24 h and 72 h post operation group was collected and used for staining. The double positive cells were labeled with white arrow. Scale bar: 150 μm. B. The double positive cells in per field were counted and analyzed. (n=40, *P<0.05, **P<0.01, compared with control group).
Imbalance of EAA/IAA induced excitotoxicity promoted neurons death following spastic paralysis
To reveal the potential mechanism of neurotransmitter under neuron death in spastic paralysis, we conducted tissue ELISA and Western blotting in ventral horn of spinal cord. As showed in Figure 4A-C, Glycine and GABA, which act as an inhibitory amino acid (IAA) in the spinal cord, were both downregulated in the ventral horn of the spinal cord following spastic paralysis. But there was temporal difference between Glycine and GABA downregulating. Glycine was immediately decreased at 10 min post SCIR and kept in a lower expression level lasting 72 hours. Whereas, GABA significant downregulation occurred at the 72 hours post-operation, and there was no significant difference on GABA expression at the early stage. Meanwhile, the excitatory amino acid (EAA), Glutamic expression was dramatically increased at 3 hours following spastic paralysis. But there was reduction at 6 hours and upregulation at 24 hours, then less than normal expression at 72 hours (Figure 4C). These results suggested that Glutamic present a high expression in the oscillator type. Furthermore, Western blotting results indicated that the receptors of Glycine and GABA were both upregulated following spastic paralysis. As showed in Figure 4D-F, Glycine α1 receptor (Gly α1) was significantly increased at 6 hours following spastic paralysis, and lasted to 24 and 72 hours. The expression of GABA B2 receptor (GABBR2) increased at 72 hours post-operation. No significant change was observed at the early stage (Figure 4E). The receptor of Glutamate, NMDA receptor 1(NR1) expression was also induced according to the upregulation of Glutamate (Figure 4F). All these results showed, following spastic paralysis, glutamic and its receptors upregulated, whereas Glycine downregulated at early stage and GABA at later stage.
Figure 4.
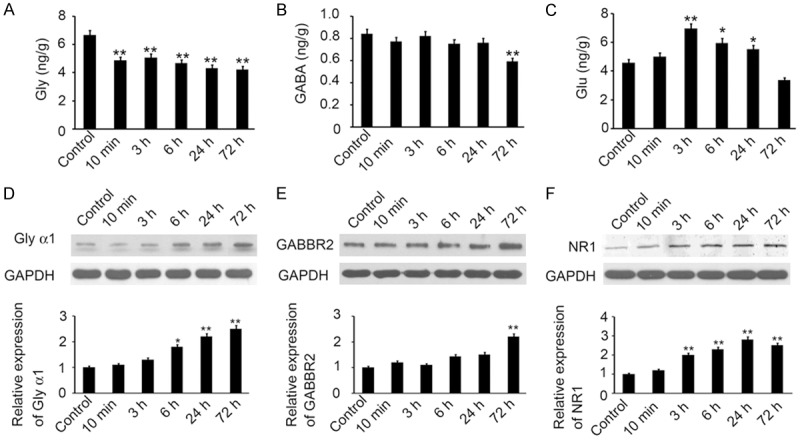
Neurotransmitter and the receptor regulated following spastic paralysis. (A-C) ELISA was performed to determine the expression of Glycine (A), GABA (B) and Glutamic (C) in the ventral horn of spinal cord area of control group, and 10 min, 3 h, 6 h, 24 h and 72 h post reperfusion group. (D-F) The expression of Gly α1, GABBR2 and NR1 in the ventral horn of spinal cord area was also determined by Western blotting. GAPDH was used as a loading control. The relative expression was analyzed. (n=3, *P<0.05, **P<0.01, compared with control group).
Intracellular calcium overload caused the neuron death following spastic paralysis
To further confirm whether the imbalance of EAA/IAA induced neuron death, the neuronal calcium activity was detected. Spinal cord ventral horn tissue with different reperfusion time was collected. The concentration of free calcium ions was indicated by fluorescent density in the cytoplasm. As shown in Figure 5, after adding 2,000 nM glutamine into neuron from ventral horn in cultural conditions, the extracellular Ca2+ influxed into cytoplasm and quickly effluxed in control and reperfusion 10 min group. In contrast, the Ca2+ influxed quickly into the cytoplasm and maintained at relatively high levels in reperfusion 3 hours and 6 hours groups. In reperfusion 24 hours and 72 hours groups, as neurons were severely damaged, the Ca2+ ion channel had no response to glutamine and fluorescent density had no change groups.
Figure 5.

The change of free calcium ions in cytoplasm of neurons collected at different time point following spastic paralysis. A. Control group, n=5; B. 10 min following spastic paralysis group, n=4; C. 3 h following spastic paralysis group, n=7; D. 6 h following spastic paralysis group, n=6; E. 24 h following spastic paralysis group, n=5; F. 72 h following spastic paralysis group, n=4. Each line represents continuous change of free calcium ions concentration in cytoplasm of neurons.
Discussion
In the present study, the rabbit SCIR-induced spastic paralysis model was established and identified by us. We found extensive and dispersivity degeneration in the gray and white matters, and neurons in gray matters was susceptible to ischemia/reperfusion. Further, motor neurons in the ventral horn of spinal cord area were degeneration at 3 hours post-reperfusion, and only 12.5% motor neurons were observed at 72 hours post-reperfusion, compared with the normal group. Mechanism investigations indicated glutamate significantly increased in the oscillator type, while Glycine significantly decreased post-reperfusion immediately at early stage and GABA notablely decreased at later stage. Additionally, higher intracellular calcium activity was found at 3 hours following SCIR, inversely concordance with the decreasing of Glycine expression. Our results would lay the foundation for better understanding of the neuron degeneration and mechanism of spastic paralysis. And it would supply a novel and effective target for prevention and therapy of spastic paralysis.
As we know, excite stretch reflex and spastic muscular tension are the major physiological characteristics in spasticity developing. Thus, stretch reflex and muscular tension are the main characteristics to identify spasticity in clinical [20-22]. In the present study, stretch reflex and muscular tension are introduced to identify whether the paralysis rabbits are spasticity or not. The identifying rules are according to the widely used Reuter’s score [17]. Our results indicated that the percentage of spastic paralysis in ischemic 60 min groups is 82.5% at 7 day post-operation, while the percentage is 75% and 100% in ischemic 30 min group and 90 min group. It indicated that paralysis caused by SCIR was mainly spastic paralysis, and with elongation the time of ischemia and reperfusion, the hind limb dysfunction aggravated. Honestly, higher percentage of spastic paralysis in ischemic 90 min groups was observed, but higher mortality, more severe motor dysfunction and neuropathological damage were also found. Thus, in consideration of the model stability, ischemia 60 min operations were chosen in the subsequent study. Importantly, only the spastic paralysis identified rabbits were used. Collectively, our study established and identified a stable and clinical-like rabbit SCIR-induced spastic paralysis model through ischemia 60 min operations.
In this study, we observed that extensive and dispersivity degeneration in SCIR damage. But motor neurons soma in gray matters was more severely damaged than neuraxon. It indicated that gray matters was more sensitive to ischemia/reperfusion than white matters. Notably, we firstly demonstrated temporal of motor neurons loss in the ventral horn of spinal cord following SCIR-induced spastic paralysis (Figure 3). Previous study by Ling et al has demonstrated the temporal and spatial profiles of cell loss following experimental spinal cord injury [23]. They indicated that decreased motor neurons occurred at the 1 h post-operation and continued over a week and the losses decreased with increasing distance from the epicenter [23]. No recovery of motor neurons has been found during the process [23]. Our study observed the motor neurons by immunofluorescence with NF-200 and ChAT co-labeled, which are the specific biomarker for motor neurons. And our results indicated that the motor neurons began to degenerate significantly at 3 hours after reperfusion, and last to 24 and 72 hours. Meanwhile, there was no significant difference at 10 min after reperfusion. The difference on temporal of motor neurons loss between our study and Ling’s may be according to the different SCI model used. We established the SCI model through occluding the abdominal aorta below the level of the renal artery, causing spinal cord ischemic injury to 60 min, and the arterial clamp was removed, according to the previous study [16]. But the spinal cord injury model used in Ling’s study was established by dropping a weight (10 g) through a guide tube 5 cm directly onto the cord (50 g∙cm). It may cause spinal cord injury more directly, and then induce motor neurons degeneration more quickly. Results of us would provide more evidence for understanding the pathogenesis of SCIR-induced spastic paralysis.
The abundance of GABAergic and Glycinergic synaptic terminals, some of which coexist in the same terminals, and the presence of their respective post synaptic receptors on motor neurons are well documented by morphological evidence [24,25]. Moreover, ex vivo electrophysiological studies in neonatal rat suggest that Glycine and GABA can be co-released in spinal motor neurons with temporal and spatial specificity [26-28]. But in adult rat, Glycine may take a more important role in mediating segmental, proprioceptive, and bulb spinal post synaptic inhibition [29]. In the present study, both of the GABA and Glycine decreasing was found in the ventral horn of spinal cord area following SCIR-induced spastic paralysis (Figure 4). But, importantly, Glycine significantly decreased at early stage and GABA at later stage. It indicated that decrease of Glycine and GABA was not sufficient to antagonism the exitotoxicity inducing by increasing glutamate, which caused motor neurons hyperexcitability, then releasing a large number of acetylcholine to neuromuscular junction to make hypertonia. It suggested that the decrease of inhibitory neurotransmitters make motor neuron disinhibition. According to the difference of temporal expression, Glycine may play a major role at early stage and GABA at later stage. Further evidence by Western blotting indicated that the temporal of Glycine and GABA receptor increase was consistent with the temporal of Glycine and GABA decrease. The increase of receptor may contribute to compensate for the decrease of the ligand. Furthermore, according to the decrease of Glycine and increase of glutamte at the early stage, higher intracellular calcium activity was found after reperfusion (Figure 5). It indicated that the motor neurons in the spinal cord are in a constant state of disinhibition and the neurotransmitter imbalance can induce intracellular calcium overload and neuronal damage.
In summary, in this study, we identify SCIR damage induced spastic paralysis in rabbit. We firstly demonstrated temporal of motor neurons loss in the ventral horn of spinal cord following SCIR. Mechanism results indicated decrease of Glycine at early stage and GABA at later stage was not sufficient to antagonism the exitotoxicity inducing by glutamate, which caused intracellular calcium overload to make neurons dead. Our results would lay the foundation for better understanding of the neuron degeneration and pathogenic mechanism of SCIR damage. And it would supply a novel and effective target for the prevention and therapy to SCIR damage.
Acknowledgements
National Natural Science Foundation of China (No.81272159). We thank Wenhui Hu for helpful scientific discussions. The work was supported by the research grants from the National Natural Science Foundation of China (No.81272159).
Disclosure of conflict of interest
None.
References
- 1.Guven M, Sehitoglu MH, Yuksel Y, Tokmak M, Aras AB, Akman T, Golge UH, Karavelioglu E, Bal E, Cosar M. The neuroprotective effect of coumaric acid on spinal cord ischemia/reperfusion injury in rats. Inflammation. 2015;38:1986–1995. doi: 10.1007/s10753-015-0179-0. [DOI] [PubMed] [Google Scholar]
- 2.Li XQ, Lv HW, Tan WF, Fang B, Wang H, Ma H. Role of the TLR4 pathway in blood-spinal cord barrier dysfunction during the bimodal stage after ischemia/reperfusion injury in rats. J Neuroinflammation. 2014;11:62. doi: 10.1186/1742-2094-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kahn RA, Stone ME, Moskowitz DM. Anesthetic consideration for descending thoracic aortic aneurysm repair. Semin Cardiothorac Vasc Anesth. 2007;11:205–223. doi: 10.1177/1089253207306098. [DOI] [PubMed] [Google Scholar]
- 4.Patel VI, Ergul E, Conrad MF, Cambria M, LaMuraglia GM, Kwolek CJ, Brewster DC, Cambria RP. Continued favorable results with open surgical repair of type IV thoracoabdominal aortic aneurysms. J Vasc Surg. 2011;53:1492–1498. doi: 10.1016/j.jvs.2011.01.070. [DOI] [PubMed] [Google Scholar]
- 5.Smith PD, Puskas F, Meng X, Lee JH, Cleveland JC Jr, Weyant MJ, Fullerton DA, Reece TB. The evolution of chemokine release supports a bimodal mechanism of spinal cord ischemia and reperfusion injury. Circulation. 2012;126(Suppl 1):S110–117. doi: 10.1161/CIRCULATIONAHA.111.080275. [DOI] [PubMed] [Google Scholar]
- 6.Comley L, Allodi I, Nichterwitz S, Nizzardo M, Simone C, Corti S, Hedlund E. Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience. 2015;291:216–229. doi: 10.1016/j.neuroscience.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Shin HY, Kim H, Kwon MJ, Hwang DH, Lee K, Kim BG. Molecular and cellular changes in the lumbar spinal cord following thoracic injury: regulation by treadmill locomotor training. PLoS One. 2014;9:e88215. doi: 10.1371/journal.pone.0088215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winner B, Marchetto MC, Winkler J, Gage FH. Human-induced pluripotent stem cells pave the road for a better understanding of motor neuron disease. Hum Mol Genet. 2014;23:R27–34. doi: 10.1093/hmg/ddu205. [DOI] [PubMed] [Google Scholar]
- 9.Fogarty MJ, Smallcombe KL, Yanagawa Y, Obata K, Bellingham MC, Noakes PG. Genetic deficiency of GABA differentially regulates respiratory and non-respiratory motor neuron development. PLoS One. 2013;8:e56257. doi: 10.1371/journal.pone.0056257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh M, Pearse DD. The role of the serotonergic system in locomotor recovery after spinal cord injury. Front Neural Circuits. 2014;8:151. doi: 10.3389/fncir.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pettersson LM, Geremia NM, Ying Z, Verge VM. Injury-associated PACAP expression in rat sensory and motor neurons is induced by endogenous BDNF. PLoS One. 2014;9:e100730. doi: 10.1371/journal.pone.0100730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamashita T, Kwak S. The molecular link between inefficient GluA2 Q/R site-RNA editing and TDP-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. J Neurosci. 2014;1584:28–38. doi: 10.1016/j.brainres.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Drachman DB, Adams RN, Balasubramanian U, Lu Y. Strategy for treating motor neuron diseases using a fusion protein of botulinum toxin binding domain and streptavidin for viral vector access: work in progress. Toxins (Basel) 2010;2:2872–2889. doi: 10.3390/toxins2122872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eleftheriadou I, Trabalza A, Ellison S, Gharun K, Mazarakis N. Specific retrograde transduction of spinal motor neurons using lentiviral vectors targeted to presynaptic NMJ receptors. Mol Ther. 2014;22:1285–1298. doi: 10.1038/mt.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Opperman KJ, Grill B. RPM-1 is localized to distinct subcellular compartments and regulates axon length in GABAergic motor neurons. Neural Dev. 2014;9:10. doi: 10.1186/1749-8104-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeGirolami U, Zivin JA. Neuropathology of experimental spinal cord ischemia in the rabbit. J Neuropathol Exp Neurol. 1982;41:129–149. doi: 10.1097/00005072-198203000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Reuter DG, Tacker WA Jr, Badylak SF, Voorhees WD 3rd, Konrad PE. Correlation of motorevoked potential response to ischemic spinal cord damage. J Thorac Cardiovasc Surg. 1992;104:262–272. [PubMed] [Google Scholar]
- 18.Jacobs TP, Shohami E, Baze W, Burgard E, Gunderson C, Hallenbeck JM, Feuerstein G. Deteriorating stroke model: histopathology, edema, and eicosanoid changes following spinal cord ischemia in rabbits. Stroke. 1987;18:741–750. doi: 10.1161/01.str.18.4.741. [DOI] [PubMed] [Google Scholar]
- 19.Dai L, Cui X, Zhang X, Cheng L, Liu Y, Yang Y, Fan P, Wang Q, Lin Y, Zhang J, Li C, Mao Y, Wang Q, Su X, Zhang S, Peng Y, Yang H, Hu X, Yang J, Huang M, Xiang R, Yu D, Zhou Z, Wei Y, Deng H. SARI inhibits angiogenesis and tumour growth of human colon cancer through directly targeting ceruloplasmin. Nat Commun. 2016;7:11996. doi: 10.1038/ncomms11996. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Burke D, Gillies JD, Lance JW. Hamstrings stretch reflex in human spasticity. J Neurol Neurosurg Psychiatry. 1971;34:231–235. doi: 10.1136/jnnp.34.3.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger W, Horstmann G, Dietz V. Tension development and muscle activation in the leg during gait in spastic hemiparesis: independence of muscle hypertonia and exaggerated stretch reflexes. J Neurol Neurosurg Psychiatry. 1984;47:1029–1033. doi: 10.1136/jnnp.47.9.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powers RK, Campbell DL, Rymer WZ. Stretch reflex dynamics in spastic elbow flexor muscles. Ann Neurol. 1989;25:32–42. doi: 10.1002/ana.410250106. [DOI] [PubMed] [Google Scholar]
- 23.Ling X, Bao F, Qian H, Liu D. The temporal and spatial profiles of cell loss following experimental spinal cord injury: effect of antioxidant therapy on cell death and functional recovery. BMC Neurosci. 2013;14:146. doi: 10.1186/1471-2202-14-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Todd AJ, Watt C, Spike RC, Sieghart W. Colocalization of GABA, glycine, and their receptors at synapses in the rat spinal cord. J Neurosci. 1996;16:974–982. doi: 10.1523/JNEUROSCI.16-03-00974.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frazao R, Nogueira MI, Wassle H. Colocalization of synaptic GABA(C)-receptors with GABA (A)-receptors and glycine-receptors in the rodent central nervous system. Cell Tissue Res. 2007;330:1–15. doi: 10.1007/s00441-007-0446-y. [DOI] [PubMed] [Google Scholar]
- 26.Jonas P, Bischofberger J, Sandkuhler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- 27.Russier M, Kopysova IL, Ankri N, Ferrand N, Debanne D. GABA and glycine co-release optimizes functional inhibition in rat brainstem motoneurons in vitro. J Physiol. 2002;541:123–137. doi: 10.1113/jphysiol.2001.016063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bautista W, Aguilar J, Loeza-Alcocer JE, Delgado-Lezama R. Pre- and postsynaptic modulation of monosynaptic reflex by GABAA receptors on turtle spinal cord. J Physiol. 2010;588:2621–2631. doi: 10.1113/jphysiol.2010.188979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang J, Alstermark B. Not GABA but glycine mediates segmental, propriospinal, and bulbospinal postsynaptic inhibition in adult mouse spinal forelimb motor neurons. J Neurosci. 2015;35:1991–1998. doi: 10.1523/JNEUROSCI.1627-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]