

Abstract
Activation of NADPH oxidase 2 (NOX2) triggers reactive oxygen species (ROS) generation, both of which are essential for robust microbial clearance by phagocytes. However, it is unknown whether inhibition of NOX2 activation or ROS generation affects cellular phagocytosis. Here, we found that the classic NOX2 inhibitor diphenyleneiodonium (DPI) induced uptake of E. coli by murine peritoneal macrophages through enhancing phagocytosis, and this effect was temperature-sensitive and attenuated by cytochalasin D as well as chemical inhibition of Syk and PLCγ, two downstream kinases involved in actin polymerization during phagocytosis. DPI also decreased the production of TNF-α and IL-6 resulting from E. coli stimulation. The DPI-induced enhancement of phagocytosis was independent of NOX2 inhibition or ROS generation but depended on increased intracellular calcium and activation of the p38 MAPK signaling pathway. Furthermore, DPI enhanced bacterial elimination and ameliorated inflammation in E. coli-infected mice, leading to improved survival. Our results demonstrate that DPI facilitates ROS-independent bacterial phagocytosis by macrophages through activation of calcium and p38 MAPK signaling pathways.
Keywords: Diphenyleneiodonium, phagocytosis, macrophages, p38 MAPK, E. coli
Introduction
The cellular process of phagocytosis serves to remove pathogens and cell debris and thus plays a critical role in host response against infection [1]. Phagocytosis not only induces non-specific killing but also primes adaptive immunity by facilitating antigen presentation [2]. Macrophages are tissue-resident phagocytes equipped with a variety of phagocytic receptors, such as Fcγ receptors, complement receptors, and scavenger receptors, which sense and interact with microbial components. Ligand binding due to receptor clustering triggers phosphorylation of tyrosine residues in immune receptor tyrosine-based activation motifs and proteins bearing SH2 domains (particularly the spleen tyrosine kinase (Syk)) [3]. This leads to activation of phosphatidylinositol-associated kinases such as PI3K and PLCγ, which induce actin polymerization and cytoskeletal rearrangements essential for membrane invagination and particle engulfment [4].
Activation of phagocytic receptors also leads to production of reactive oxygen species (ROS) such as hydrogen peroxide and superoxide in macrophages and neutrophils [5,6]. This process involves activation of the membrane NADPH oxidase 2 (NOX2) complex, which drives the production of ROS [7]. Myeloperoxidase (MPO), which acts downstream of NOX2 and catalyzes the production of hypochlorous acid from hydrogen peroxide and chloride anions [8]. Importantly, NOX2- or MPO-deficient mice are more susceptible to bacterial and fungal infections, suggesting that ROS generation is essential for the killing of invaded pathogens [9,10].
Although it is well known that ROS are induced by phagocytosis and are critically involved in microbial killing, it is not well understood whether deficiency in ROS generation affects phagocytosis. Recently, MPO deficiency was found to enhance zymosan phagocytosis through upregulation of surface CD11b expression in mouse neutrophils, providing evidence of MPO-independent phagocytosis [10]. However, it is unknown whether NOX2-independent phagocytosis also exists. In the present study, we found that diphenyleneiodonium (DPI), a frequently used NOX2 inhibitor, enhanced uptake of Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) in murine macrophages. This process was independent of ROS but depended on calcium and activation of p38 MAPK. Moreover, DPI enhanced bacterial elimination and ameliorated inflammation in E. coli-infected mice, leading to improved survival.
Materials and methods
Reagents
E. coli and S. aureus were purchased from ATCC (Manassas, VA). Dulbecco’s Modified Eagle’s Medium (DMEM), Fluo-4AM, and FITC-labeled E. coli and S. aureus were purchased from Life Technologies (Eugene, OR). Fetal bovine serum (FBS) was purchased from Sera Best (Bavaria, German). SB203580, U0126, VAS2870 (VA), DPI, Ebselen (EB), EGTA, BAPTA-AM, phorbolmyristateactate (PMA), DCFH-DA, and saponin were purchased from Sigma-Aldrich (St. Louis, MO). R406 and U73122 were purchased from Selleck (Westlake Village, CA). Cytochalasin D was purchased from Aladdin (Shanghai, China). MPO inhibitor-I was purchased from EMD Chemicals (San Diego, CA). p38, p-p38, ERK1/2, p-ERK1/2, and GAPDH primary antibodies and HRP-conjugated secondary IgG antibodies were purchased from Cell Signaling (Danvers, MA). T-PER protein extraction reagent was purchased from Thermo Scientific (Rockford, IL). Protease cocktail and phosphatase inhibitors were purchased from Roche (Basel, Switzerland). TNF-α and IL-6 assay kits were purchased from Affymetrix (Santa Clara, CA). RNA extraction kit, reverse transcription kit, and SYBR® Green real-time PCR master mix were purchased from TOYOBO (Osaka, Japan). Phalloidin was purchased from Abcam (Bristol, UK).
Animals
Wild-type BALB/c mice (male, 6-8 weeks) were purchased from HFK Bioscience Co., Ltd. (Beijing, China). All animal experiments were performed in accordance with the National and Institutional Guidelines for Animal Care and Use and approved by the Institutional Animal Ethics Committee of the Third Military Medical University.
Animal experiments
BALB/C mice were intraperitoneally injected with normal saline (NS) or E. coli (1×108 colony forming units [CFU]/kg) in NS or DPI (1 mM/kg). Survival was observed for 72 h. Tissue samples were collected 12 h after injection. Briefly, mice were anesthetized under isoflurane, and the liver, spleen, and lungs were eviscerated and homogenized for cytokine and bacterial load detection.
Isolation of murine peritoneal macrophages
Murine peritoneal macrophages were obtained from peritoneal lavage in BALB/c mice as described previously [11]. Briefly, mice were sacrificed, and 1 ml DMEM was injected into the intraperitoneal cavity. The abdomen was gently massaged for 1 min, and the injected medium was then aspirated. Cell pellets were washed twice with DMEM containing 10% FBS, and peritoneal macrophages were plated in plates or dishes and cultured at 37°C in a humidified incubator supplemented with 5% CO2.
Bacterial culture
E. coli and S. aureus were inoculated in lysogeny broth medium at 37°C for 4 h. Bacteria CFUs were determined by a Bio-RadSmartSpec Plus nucleic acid protein instrument (Hercules, CA). The bacterial solution was diluted to achieve a multiplicity of infection (MOI) of 1 per cell in DMEM before further use.
Phagocytosis assays
For in vitro experiments, murine peritoneal macrophages (5×105/ml) were treated as indicated and infected with bacteria (MOI = 1). Extracellular bacteria were killed by cefmetazole sodium. Cells were then lysed with 0.3% saponin, and the lysate was diluted, added to a nutrient agar plate, and incubated at 37°C for 12 h. CFUs were detected using a SHINESO colony counter (Hangzhou, China). Bacterial uptake is expressed as a phagocytic index, which was calculated as the percentage of intracellular bacteria of the total bacterial input [12]. Phagocytosis of killed bacteria was detected by fluorescence assay. Briefly, murine peritoneal macrophages (1×105/ml) were seeded on glass-bottom dishes and treated as indicated. FITC-labeled E. coli or S. aureus were added and incubated at 37°C for 1 h. Cells were then washed with PBS to remove the non-uptaken bacteria, fixed using 4% paraformaldehyde, and counterstained with DAPI. Images were obtained using a 780 laser scanning confocal microscope (LSCM; Zeiss, Germany). Phagocytosis was expressed as the mean fluorescence intensity (MFI) of FITC-labeled bacteria. At least 100 cells were counted to quantify phagocytosis.
For in vivo experiments, murine peritoneal lavage fluid was acquired 1 h after injection and centrifuged at 1000 rpm for 10 min. Bacterial CFU in the supernatant was determined by the CFU assay. The cell pellet was suspended in DMEM supplemented with cefmetazole sodium at a density of 5×105/ml and incubated for 30 min. Intracellular bacterial counts were quantified as in the in vitro experiments.
ELISA assays
Supernatants from cultured murine peritoneal macrophages or serum/tissue homogenate samples of E. coli-infected mice were collected at the indicated times. TNF-α and IL-6 were detected with ELISA kits according to the manufacturer’s instructions.
Real-time PCR assay
Murine peritoneal macrophages (5×105/ml) were infected with E. coli for the indicated durations. Total RNA was extracted using TRIzol reagent and reverse transcribed into cDNA with a ReverTra Ace-α kit. cDNA templates were mixed with SYBR Green PCR Mastermix and primers for TNF-α, IL-6, and β-actin (Sequences listed in Table S1). Quantitative real-time PCR was performed using an iCycler Thermal Cycler (Bio-Rad).
Western blot analysis
Murine peritoneal macrophages (1×106/ml) were treated as indicated. Cells were then lysed with T-PER protein extraction reagent containing protease cocktail and phosphatase inhibitor (Roche, Basel, Switzerland), and proteins were separated by SDS-PAGE and transferred onto PVDF membranes. Blots of protein were incubated with primary antibodies (1:1000 dilution) for p38, p-p38, ERK1/2, p-ERK1/2, and GAPDH at 4°C overnight. Blots were then incubated with HRP-conjugated secondary IgG antibodies (1:2000 dilution) at 37°C for 1 h. Chemiluminescence images were developed with a SuperSignal Sensitivity Substrate kit and detected using a ChemiDoc XRS imaging system (Bio-Rad).
Measurement of intracellular ROS, calcium, and F-actin
Peritoneal macrophages (5×105 cells/ml) was seeded on glass-bottomed culture dishes and preloaded with ROS probe DCFH-DA, calcium probe Fluo-4AM, and fluorescent F-actin probe phalloidin. Cells were then treated as indicated, and intracellular ROS, calcium, and F-actin levels were detected by ACEA NovoCyte flow cytometry (San Diego, CA) or LSCM imaging and quantified by MFI.
Statistical analysis
Quantitative data are expressed as mean ± standard deviation (SD). Student’s t-tests were used for comparisons between two groups. One-way ANOVA with post-hoc Bonferroni corrections were used for comparisons among multiple groups. A P-value less than 0.05 was considered statistically significant.
Results
DPI enhances bacterial uptake in murine macrophages by inducing cellular phagocytosis
To address whether DPI affects cellular uptake of bacteria, we pretreated murine peritoneal macrophages with DPI before incubation with live or fluorescence-labeled killed E. coli and S. aureus. As shown by the CFU assay, extracellular bacteria were eliminated by antibiotics, indicating that remaining bacteria in cell lysates were engulfed by macrophages (Figure S1). DPI significantly increased intracellular E. coli and S. aureus counts in macrophages compared with those in cells treated with bacteria alone (Figure 1A). DPI also enhanced intracellular fluorescence of E. coli and S. aureus, further indicating that DPI upregulated bacterial uptake in murine macrophages (Figure 1B). Moreover, DPI increased E. coli uptake in a dose-dependent (Figure 1C, upper) and time-dependent (Figure 1C, lower) manner. This DPI-induced bacterial uptake was sensitive to low temperature, as incubation at 4°C abolished the increase in E. coli uptake (Figure 1D). Treatment with cytochalasin D, a potent inhibitor of actin polymerization, significantly dampened E. coli uptake (Figure 1E). Accordingly, chemical inhibitors of Syk (R406) and PLCγ (U73122) also reduced the phagocytosis of E. coli (Figure 1F). These results indicate that DPI enhances phagocytosis and thus increases bacterial uptake into macrophages.
Figure 1.

DPI increases bacterial uptake by murine peritoneal macrophages through enhancing phagocytosis. (A) Cells were infected with E. coli (EC) or S. aureus (SA) with or without DPI. Phagocytosis was analyzed by CFU assay. (B) Cells were treated with or without DPI and then treated with FITC-labeled EC or SA. Intracellular fluorescence images were acquired by LSCM. Scale bar, 5 μm. (C) Cells were treated with different concentrations of DPI (0-10 μM) and infected with EC for 0.5-2.5 h. Phagocytosis was analyzed as in (A). (D) Murine peritoneal macrophages were treated with or without DPI and infected with EC at 37°C or 4°C. Phagocytosis was analyzed as in (A). (E) Murine peritoneal macrophages were treated with EC alone, EC with DPI, or EC with DPI and 2.5 μM cytochalasin D. Phagocytosis was analyzed as in (A). (F) Cells were treated with EC, EC with DPI, or EC with DPI and R406 (40 nM) or U73122 (10 μM). Phagocytosis were assessed as in (A and B). The MOI of EC or SA was 1. The dose of DPI was 10 μM and the duration of EC infection was 1 h unless otherwise indicated. **P<0.01. Data are presented as the mean ± standard deviation. n = 4.
DPI inhibits inflammation in E. coli-infected murine macrophages
To determine whether DPI affects proinflammatory reactions in E. coli-infected macrophages, we assessed mRNA levels of TNF-α and IL-6. We found that E. coli infection dramatically upregulated expression of TNF-α and IL-6 mRNA, whereas DPI co-treatment significantly attenuated this increased TNF-α and IL-6 expression (Figure 2A). When we assessed supernatant levels of TNF-α and IL-6 at different time points, we observed an increase in cytokine release between 2 and 8 h after E. coli infection, which was inhibited by DPI co-treatment in a time-dependent manner (Figure 2B).
Figure 2.
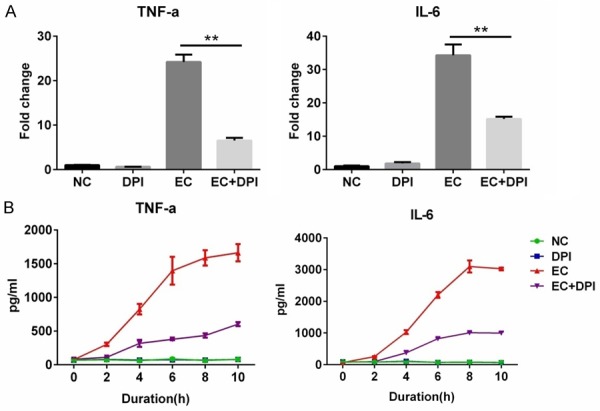
DPI suppresses inflammation in E. coli-infected murine peritoneal macrophages. (A) Cells were untreated (normal control, NC) or treated with DPI, E. coli (EC), or EC with DPI for 4 h. mRNA expression of TNF and IL-6 was detected by RT-PCR. (B) Cells were treated as in (A) for the indicated durations, and supernatant TNF-α and IL-6 levels were detected by ELISA. The dose of DPI was 10 μM, and the MOI for EC was 1. **P<0.01. Data are presented as the mean ± standard deviation. n = 3.
The effect of DPI on phagocytosis is independent of NOX2 and ROS
As DPI is a classic NOX2 inhibitor, we next evaluated whether the DPI-induced enhancement of bacterial phagocytosis depends on NOX2 inhibition. We found that DPI as well as VA and EB, two other NOX2 inhibitors, markedly inhibited the elevated ROS production by macrophages treated with PMA, a representative PKC activator and ROS inducer (Figure S2). E. coli infection also increased intracellular ROS levels as demonstrated by increased DCFH-DA staining (Figure 3A). Treatment with DPI, VA, or EB consistently inhibited ROS production in murine macrophages, demonstrating their comparable effects on ROS activity. However, their effects on phagocytosis differed, as only DPI but not VA or EB significantly enhanced phagocytic index (Figure 3B). Furthermore, DPI but not MPO inhibitor-I significantly enhanced phagocytosis of E. coli by macrophages (Figure 3C). These results suggest that DPI acts independently of NOX2 inhibition.
Figure 3.
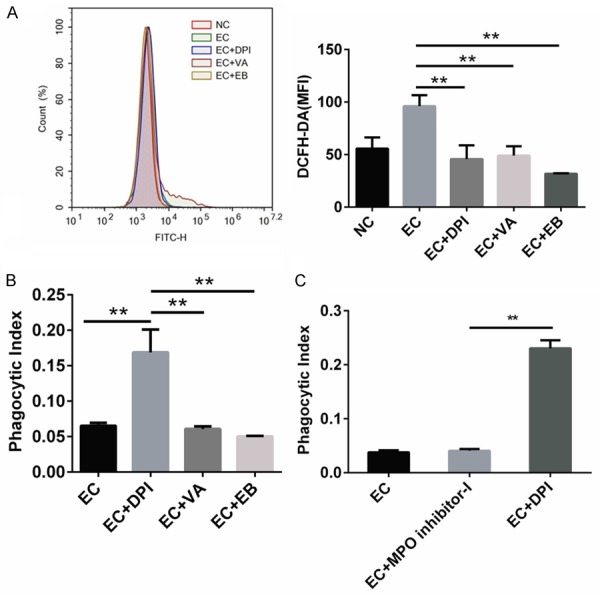
DPI enhances phagocytosis independently of NOX2 in E. coli-infected murine peritoneal macrophages. (A) Cells were pretreated with DPI, VA, or EB for 30 min before E. coli (EC) infection for 1 h. Cells were then stained with 2.5 μM DCFH-DA and detected by flow cytometry. Intracellular ROS levels were expressed as MFI of DCFH-DA. (B) Cells were treated as in (A). Phagocytosis was analyzed by CFU assay. (C) Peritoneal macrophages were pretreated with DPI or MPO inhibitor-I for 30 min and then infected with EC for 1 h. Phagocytosis was detected as in (B). The doses of DPI, VA, EB, and MPO inhibitor-I were 10 μM, and the MOI for EC was 1. **P<0.01. Data are presented as the mean ± standard deviation. n = 3.
Intracellular calcium and p38 MAPK activation mediate DPI-induced bacterial phagocytosis
As intracellular calcium controls a wide variety of signaling events, we assessed whether DPI affects intracellular calcium levels. We found that E. coli modestly increased the intracellular fluorescence intensity of Fluo-4AM, a specific intracellular calcium probe, in macrophages, which was further enhanced by co-treatment with DPI (Figure 4A). Moreover, co-treatment with EGTA or BAPTA-AM, which chelates extracellular or intracellular calcium, respectively, decreased the uptake of E. coli in DPI-treated macrophages (Figure 4B). These results suggest that calcium mediates the upregulation of phagocytosis. We next assessed activation of p38 MAPK and ERK1/2 in E. coli-infected macrophages. We found that phosphorylation of p38 was upregulated by DPI alone or in combination with E. coli (Figure 4C). E. coli also upregulated phosphorylation of ERK1/2. However, DPI suppressed ERK1/2 phosphorylation when co-treated with E. coli. Concomitantly, inhibition of p38 by SB203580 significantly downregulated E. coli phagocytosis in DPI-treated macrophages, whereas inhibition of ERK1/2 by U0126 did not affect phagocytosis (Figure 4D). We next found that DPI induced actin polymerization as demonstrated by condensed phalloidin staining in macrophages (Figure 4E). Co-treatment with SB203580 markedly inhibited this actin polymerization, whereas co-treatment with U0126 had no effect. These results indicate that DPI-induced enhancement of phagocytosis may by mediated by selective upregulation or activation of calcium and p38 MAPK.
Figure 4.
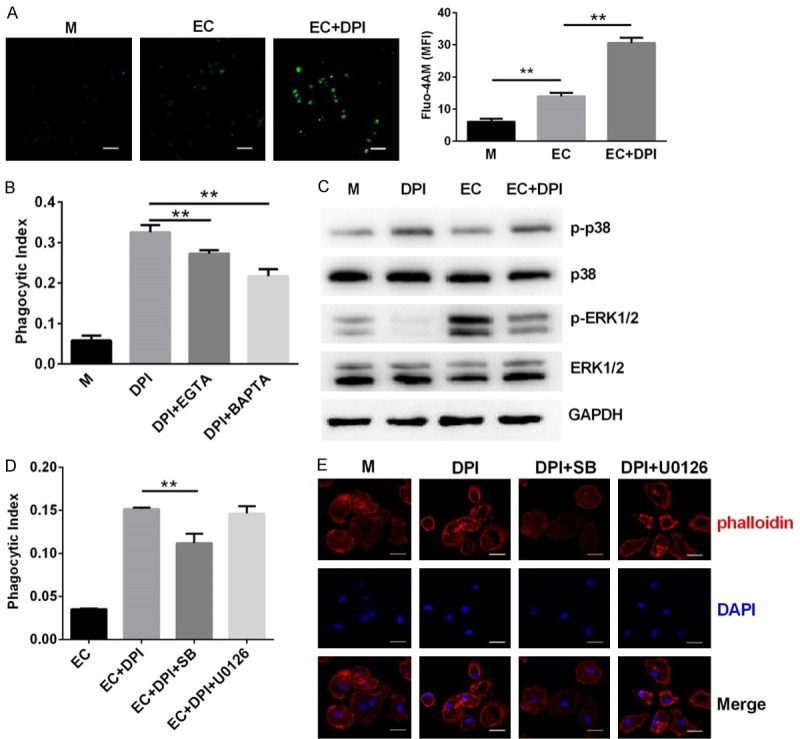
Calcium and p38 MAPK are required for DPI-induced bacterial phagocytosis by murine peritoneal macrophages. (A) Cells were infected with E. coli (EC) alone or EC with DPI for 30 min and then stained by fluo-4 AM for intracellular calcium detection. (B) Cells were treated with DPI alone or DPI with 5 mM EGTA or 10 μM BAPTA-AM before infection with EC for 1 h. Phagocytosis was analyzed by CFU assay. (C) Cells were untreated (medium, M) or treated with DPI, EC, or EC with DPI for 30 min. The phosphorylation of p38 and ERK1/2 was detected by western blot. (D) Cells were treated with DPI alone or with 5 μM SB203580 or 5 μM U0126 before infection with EC for 1 h. Phagocytosis was analyzed as in (B). (E) Cells were treated with DPI alone or DPI with SB203580 or U0126. Cytoskeleton rearrangement was detected by fluorescence staining with phalloidin. **P<0.01. Data are presented as the mean ± standard deviation. n = 4.
DPI enhances bacterial elimination, ameliorates inflammation, and improves survival of E. coli-infected mice
Finally, we examined whether DPI enhances bacterial phagocytosis in vivo. We first found that DPI (without the use of antibiotics) significantly increased the survival of E. coli-injected mice (Figure 5A). We next assessed bacterial load in mouse lung tissue to evaluate bacterial clearance. We found a significant increase in bacterial count 6 h after E. coli injection, whereas DPI co-administration significantly reduced bacterial load in lung homogenates (Figure 5B, left). We also analyzed extracellular and intracellular bacterial counts in peritoneal macrophages of E. coli-infected mice. We found that DPI significantly reduced the number of extracellular bacteria found in the peritoneal fluid (Figure 5B, right). However, DPI significantly increased the number of intracellular bacteria detected in the cell lysate of peritoneal macrophages. These results suggest that DPI increases the uptake of E. coli by macrophages in vivo, thereby ameliorating the bacterial load in E. coli-infected mice. We next measured proinflammatory cytokine levels in mice. Compared with the NS group, E. coli injection elevated TNF-α and IL-6 levels in the serum and tissue homogenates of the lung, spleen, and liver, whereas DPI co-treatment significantly inhibited these elevations (Figure 5C). These results suggest that DPI induces bacteria phagocytosis in vivo, which enhances bacterial elimination, ameliorates inflammation, and improves the survival of E. coli-infected mice.
Figure 5.

DPI enhances bacterial elimination, ameliorates inflammation, and improves survival of E. coli-infected mice. (A) Mice were injected with E. coli (EC) (1×107 CFU/kg) alone or EC with DPI (1 mM/kg). Survival was observed for 72 h. (B, C) Mice were injected with normal saline (NS), EC (1×108 CFU/kg) alone, or EC with DPI (1 mM/kg). Tissue and peritoneal fluid samples were obtained after 6 h. Bacterial load in lung homogenate peritoneal fluid was detected by CFU assay. (B) Peritoneal macrophages were isolated, and extracellular or intracellular bacterial counts were determined by CFU assay. (C) TNF-α and IL-6 levels in serum and homogenates of lung, spleen, and liver were detected by ELISA. *P<0.05, **P<0.01. Data are presented as the mean ± standard deviation. n = 10 for (A), n = 6 for (B and C).
Discussion
Phagocytosis and ROS generation are major cellular mechanisms of microbial clearance [13], although their interplay is not well understood. In the present study, we found that NOX2 inhibition by DPI enhanced phagocytosis of E. coli by murine primary macrophages, and this effect depended on intracellular calcium and activation of p38 MAPK. DPI also facilitated bacterial elimination and ameliorated inflammation, leading to improved survival of E. coli-infected mice.
Receptor-mediated phagocytosis is the primary process of bacterial uptake by phagocytes [14]. In our study, DPI markedly increased cellular uptake of E. coli and S. aureus, an effect that was temperature-sensitive and attenuated by inhibition of actin polymerization. To the best of our knowledge, this is the first study reporting that NOX2 inhibition by DPI increases bacterial uptake by macrophages. In previous studies, the Syk signaling pathway was found to be associated with cytoskeletal rearrangements and Fcγ receptor-mediated phagocytosis in murine macrophage-like RAW 264.7 cells [15]. Moreover, neutrophil-specific deletion of Syk kinase impairs bacterial phagocytosis in response to serum opsonized S. aureus and E. coli [16]. Here, we found that Syk inhibitor reduced the phagocytosis of E. coli and S. aureus by DPI-treated murine macrophages. Other studies show that stimulation of Syk also activates PLCγ, which regulates cell functions such as phagocytosis, adhesion, and migration [17]. We also found that inhibition of PLCγ reduced DPI-induced phagocytosis of E. coli and S. aureus. These findings implicate Syk and PLCγ in mediating bacterial phagocytosis by macrophages, which is facilitated by DPI. We also found that DPI markedly suppressed proinflammatory reactions in E. coli-infected murine macrophages. Such a phenomenon may be attributed to the enhanced uptake of bacteria that quickly enter the degradation process instead of persistently stimulating cytoplasmic immune-responsive receptors such as TLR4 and TLR9. Moreover, these data are consistent with our additional findings that DPI alleviates reductions in cell viability and proliferation of E. coli-infected macrophages (data not shown), further demonstrating that DPI protects against cellular bacterial infection.
DPI is most frequently investigated in studies of NOX2 modulation [18]. Although ROS production driven by NOX2 is critically upregulated by phagocytosis, the effect of ROS on phagocytosis is not clearly understood. A previous study shows that the vertebrate phagocytic respiratory burst mediated by NOX2 enhances bacterial phagocytosis in epithelial cells [19]. By contrast, activation of NOX due to excessive training, which leads to excessive ROS generation, was recently associated with a decrease in neutrophil phagocytosis function, whereas the combined administration of DPI and glutamine reversed the impaired neutrophil function [20]. In our study, DPI effectively inhibited ROS production by macrophages upon PMA or E. coli stimulation. Although other chemical inhibitors have similar effects on ROS production, they do not increase macrophage phagocytosis as is induced by DPI. This phenomenon indicates that ROS generation due to NOX2 activation is not required for bacterial phagocytosis by macrophages. However, inhibition of ROS itself does not necessarily induce phagocytosis, suggesting that DPI functions through other mechanisms. This possibility is supported by a previous study showing that nonopsonic phagocytosis of Mycobacterium kansasiiby CR3 is not coupled with NADPH oxidase activation [21].
The efficient ingestion of foreign particles requires an elevation of cytosolic calcium to activate phagocytic receptors and control the maturation of phagosomes [22]. In our previous work, we show that E. coli infection increases extracellular calcium influx and thereby induces autophagy [23]. Here, we found that DPI enhanced intracellular calcium levels, and chelation of either extracellular or intracellular calcium impaired the enhanced uptake of E. coli. Therefore, calcium may be required for bacterial phagocytosis. Intracellular calcium is also associated with MAPK activation, which regulates inflammation, cell death, and gene expression [24,25]. In the present study, DPI selectively upregulated p38 activation but inhibited ERK1/2 phosphorylation. Moreover, ERK1/2 was not involved in DPI-mediated phagocytosis, as inhibition of ERK1/2 by U0126 did not alter phagocytosis. Instead, the p38 inhibitor SB203580 significantly reduced phagocytosis, indicating that phagocytosis depends on selective upregulation of p38. These results are supported by a previous observation that p38 MAPK suppression downregulates macrophage phagocytosis to a greater extent than ERK1/2 inhibition [26].
To explore the systemic effects of DPI against infection in vivo, we examined survival, bacterial load, and inflammation in E. coli-infected mice. Our results clearly show that DPI increased the survival of E. coli-infected mice and reduced bacterial load in major organs. DPI-treated mice also exhibited lower levels of proinflammatory cytokines, which could be attributed to the ameliorated bacterial load. Therefore, our results indicate that peritoneal macrophages in DPI-treated mice show enhanced bacterial phagocytosis, suggesting that DPI exerts a systemic protective effect through enhancing bacterial uptake by macrophages.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81202566), Frontier and Applied Basic Research Program of Chongqing (cstc2016jcyjA0105), and Youth Innovation Project of Southwest Hospital (SWH2015QN15).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 2.Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. 2012;12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tohyama Y, Yamamura H. Protein tyrosine kinase, syk: a key player in phagocytic cells. J Biochem. 2009;145:267–273. doi: 10.1093/jb/mvp001. [DOI] [PubMed] [Google Scholar]
- 4.Jongstra-Bilen J, Puig CA, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. 2008;181:288–298. doi: 10.4049/jimmunol.181.1.288. [DOI] [PubMed] [Google Scholar]
- 5.Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 6.Woo CH, Kim TH, Choi JA, Ryu HC, Lee JE, You HJ, Bae YS, Kim JH. Inhibition of receptor internalization attenuates the TNFalpha-induced ROS generation in non-phagocytic cells. Biochem Biophys Res Commun. 2006;351:972–978. doi: 10.1016/j.bbrc.2006.10.154. [DOI] [PubMed] [Google Scholar]
- 7.Petry A, Weitnauer M, Gorlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal. 2010;13:467–487. doi: 10.1089/ars.2009.3026. [DOI] [PubMed] [Google Scholar]
- 8.Nauseef WM. Myeloperoxidase in human neutrophil host defence. Cell Microbiol. 2014;16:1146–1155. doi: 10.1111/cmi.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto K, Motowaki T, Tamura N, Aratani Y. Myeloperoxidase deficiency enhances zymosan phagocytosis associated with up-regulation of surface expression of CD11b in mouse neutrophils. Free Radic Res. 2016;50:1340–1349. doi: 10.1080/10715762.2016.1244821. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Wang N, Zhu Y, Yang Y, Chen X, Fan S, Chen Q, Zhou H, Zheng J. Inhibition of extracellular calcium influx results in enhanced IL-12 production in LPS-treated murine macrophages by downregulation of the CaMKKbeta-AMPK-SIRT1 signaling pathway. Mediators Inflamm. 2016;2016:6152713. doi: 10.1155/2016/6152713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulye M, Bechill MP, Grose W, Ferreira VP, Lafontaine ER, Wooten RM. Delineating the importance of serum opsonins and the bacterial capsule in affecting the uptake and killing of Burkholderiapseudomallei by murine neutrophils and macrophages. PLoS Negl Trop Dis. 2014;8:e2988. doi: 10.1371/journal.pntd.0002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullarkey CE, Bailey MJ, Golubeva DA, Tan GS, Nachbagauer R, He W, Novakowski KE, Bowdish DM, Miller MS, Palese P. Broadly neutralizing hemagglutinin stalk-specific antibodies induce potent phagocytosis of immune complexes by neutrophils in an Fc-dependent manner. MBio. 2016;7:e01624–16. doi: 10.1128/mBio.01624-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez-Flores G, Hernandez-Silva C, Gutierrez-Escobedo G, De Las PA, Castano I, Arreola J, Perez-Cornejo P. P2X7 from j774 murine macrophages acts as a scavenger receptor for bacteria but not yeast. Biochem Biophys Res Commun. 2016;481:19–24. doi: 10.1016/j.bbrc.2016.11.027. [DOI] [PubMed] [Google Scholar]
- 15.Jongstra-Bilen J, Puig CA, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. 2008;181:288–298. doi: 10.4049/jimmunol.181.1.288. [DOI] [PubMed] [Google Scholar]
- 16.Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood. 2009;114:4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae YS, Lee HY, Jung YS, Lee M, Suh PG. Phospholipase Cγin toll-like receptor-mediated inflammation and innate immunity. Adv Biol Regul. 2016;63:92–97. doi: 10.1016/j.jbior.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Morre DJ. Preferential inhibition of the plasma membrane NADH oxidase (NOX) activity by diphenyleneiodonium chloride with NADPH as donor. Antioxid Redox Signal. 2002;4:207–212. doi: 10.1089/152308602753625960. [DOI] [PubMed] [Google Scholar]
- 19.Yang P, Huang S, Yan X, Huang G, Dong X, Zheng T, Yuan D, Wang R, Li R, Tan Y, Xu A. Origin of the phagocytic respiratory burst and its role in gut epithelial phagocytosis in a basal chordate. Free Radic Biol Med. 2014;70:54–67. doi: 10.1016/j.freeradbiomed.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Dong J, Chen P, Liu Q, Wang R, Xiao W, Zhang Y. Reverse effects of DPI administration combined with glutamine supplementation on function of rat neutrophils induced by overtraining. Int J Sport Nutr Exerc Metab. 2013;23:137–149. doi: 10.1123/ijsnem.23.2.137. [DOI] [PubMed] [Google Scholar]
- 21.Le Cabec V, Cols C, Maridonneau-Parini I. Nonopsonic phagocytosis of zymosan and Mycobacterium kansasii by CR3 (CD11b/CD18) involves distinct molecular determinants and is or is not coupled with NADPH oxidase activation. Infect Immun. 2000;68:4736–4745. doi: 10.1128/iai.68.8.4736-4745.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demaurex N, Nunes P. The role of STIM and ORAI proteins in phagocytic immune cells. Am J Physiol Cell Physiol. 2016;310:C496–C508. doi: 10.1152/ajpcell.00360.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Wang N, Zhu Y, Yang Y, Chen X, Chen Q, Zhou H, Zheng J. Extracellular calcium influx promotes antibacterial autophagy in Escherichia coli infected murine macrophages via CaMKKbeta dependent activation of ERK1/2, AMPK and FoxO1. Biochem Biophys Res Commun. 2016;469:639–645. doi: 10.1016/j.bbrc.2015.12.052. [DOI] [PubMed] [Google Scholar]
- 24.Shie MY, Ding SJ. Integrin binding and MAPK signal pathways in primary cell responses to surface chemistry of calcium silicate cements. Biomaterials. 2013;34:6589–6606. doi: 10.1016/j.biomaterials.2013.05.075. [DOI] [PubMed] [Google Scholar]
- 25.Guo H, Feng P, Chi W, Sun X, Xu X, Li Y, Ren D, Lu C, David RJ, Leister D, Zhang L. Plastid-nucleus communication involves calcium-modulated MAPK signalling. Nat Commun. 2016;7:12173. doi: 10.1038/ncomms12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ninkovic J, Roy S. Morphine decreases bacterial phagocytosis by inhibiting actin polymerization through cAMP-, Rac-1-, and p38 MAPK-dependent mechanisms. Am J Pathol. 2012;180:1068–1079. doi: 10.1016/j.ajpath.2011.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.