

Summary
Aging and lipotoxicity are two major risk factors for gout that are linked by the activation of NLRP3 inflammasome. Neutrophil-mediated production of IL-1β drives gouty flares that cause joint destruction, intense pain, and fever. However, metabolites that impact neutrophil inflammasome remain unknown. Here we identified that ketogenic diet (KD) increases beta-hydroxybutyrate (BHB) and alleviates urate crystal induced gout without impairing immune-defense against bacterial infection. BHB inhibited Nlrp3 inflammasome in S100A9 fibril-primed and urate crystal activated macrophages which serve to recruit inflammatory neutrophils in joints. Consistent with reduced gouty flares in rats fed a ketogenic diet, BHB blocked IL-1β in neutrophils in Nlrp3-dependent manner in mice and humans irrespective of age Mechanistically, BHB inhibited the Nlrp3 inflammasome in neutrophils by reducing priming and assembly steps. Collectively, our studies show that BHB, a known alternate metabolic fuel is also an anti-inflammatory molecule that may serve as a treatment for gout.
eTOC Blurb
NLRP3 inflammasome activation in macrophages and neutrophils drives painful inflammation during gout. Goldberg et al., report that ketogenic diet prevents systemic inflammation and joint damage in a rat model of gouty flare. Mechanistically, the ketone body β-hydroxybutyrate, the most abundant ketone in vivo, inhibits NLRP3/caspase-1-dependent IL-1β secretion from neutrophils.
Introduction
Gout is a debilitating chronic inflammatory arthritis that afflicts 4% of adults in the US and is caused by the deposition of monosodium urate (MSU) crystals in the joints (Garrod, 1876; Seegmiller and Howell, 1962). It is widely known that aging and diet-induced lipotoxicity predispose for the development of gout as the prevalence of gout in elderly above the age of 60 years is approximately 10% (Joosten et al., 2010; Li et al., 2013; Roddy and Choi, 2014). MSU crystal-induced gouty flares are characterized by IL-1β-driven acute inflammation, fever and intense pain caused by monocyte-mediated neutrophil accumulation and activation in joints (Duff et al., 1983). Mechanistically, the inflammatory gouty flares caused by MSU crystals are mediated via the activation of NLRP3 inflammasome in myeloid cells that causes the release of bioactive IL-1β and IL-18 (Martinon et al., 2006).
The NLRP3 inflammasome is a carefully regulated inflammatory complex that responds to both endogenous cellular abnormalities and microbial components. Activation of the complex requires 2 signals: a priming signal (signal 1) that licenses NFκB-dependent expression of inflammasome complex proteins, and a secondary signal (signal 2) that promotes assembly of the complex leading to caspase-1 activation. MSU crystals provide signal 2 of NLRP3 activation and NLRP3-deficient mice are protected from MSU-induced inflammation (Martinon et al., 2006). Therefore, gout patients face a vicious feed-forward loop in which chronic deposition and presence of MSU crystals enables constant reactivation of acute inflammatory responses, known as gouty flares which are associated with intense pain and fever due to high systemic IL-1β levels (Dalbeth et al., 2016; Duff et al., 1983).
The long term prophylactic treatment of gout hinges on reducing hyperuricemia. Intriguingly, a major side effect of all current urate-lowering drugs such as xanthine oxidase inhibitors (Allopurinol, Febuxostat), recombinant uricase (Pegloticase) and uricosurics (Probenecid, Benzbromarone) is the induction of gouty flares (Dalbeth et al., 2016). Despite progress in the field, the relief from inflammation during acute gouty flares currently relies on non-NLRP3-specific therapeutic approaches such as the use of ACTH, corticosteroids or NSAIDS (Dalbeth et al., 2016 and references therin). Thus, elucidating the endogenous metabolites that regulate NLRP3 inflammasome activation and resolution of inflammation remains a priority for effective management of gout.
Interestingly, metabolic interventions such as caloric restriction (CR) or moderate carbohydrate restriction reduces gout (Dessein et al., 2000). Of note, a classical feature of adaptive starvation response during negative energy balance is the induction of fatty acid oxidation and production of ketone bodies β-hydroxybutyrate (BHB) and acetoacetate (AcAc) that serve as major substrates for ATP production to support the function of heart and brain (Cahill, 2006). Intriguingly, CR and BHB reduces inflammation and extends lifespan in animal (Edwards et al., 2014; Mitchell et al., 2016), suggesting immune-metabolic interactions driven by BHB may serve as pseudostarvation signals or CR-mimetic that could be harnessed against acute inflammatory diseases such as gouty flares.
We made the surprising discovery that myeloid cells also express ketogenic and ketolytic machinery and that BHB blocks the NLRP3 inflammasome in macrophages (Youm et al., 2015). This suggests that ketones may function as regulatory metabolites, serving as endogenous regulators of inflammation (Goldberg and Dixit, 2015). Given that neutrophils are key instigators of inflammation induced gouty flares, we hypothesized that upregulation of BHB-mediated signals in neutrophils serve as a key immunometabolic checkpoint against gout. Here we report that BHB prevents NLRP3 inflammasome activation in both mouse and human neutrophils irrespective of aging process. We found that BHB inhibits the signals that control the priming as well as assembly of the NLRP3 inflammasome in primary neutrophils and protects against gout.
Results
Elevated BHB protects against acute gout
MSU crystals are the hallmark characteristic of gout (McCarty and Hollander, 1961); the proposed mechanism of a gouty flare is that activation of macrophages by urate crystals recruits neutrophils in an inflammasome-dependent manner, whose accumulation and activation cause substantial pain and swelling (Duff et al., 1983; Martinon et al., 2006). The priming of inflammasome is also a critical step in the process of gouty flare. Interestingly, cytoplasmic S100A8/9 protein heterodimers are highly expressed in neutrophils (Edgeworth et al., 1991) and increase in synovial fluid (Holzinger et al., 2014) and plasma (Ryckman et al., 2003) during a gout flare. Furthermore, S100A8 can activate TLR4 (Vogl et al., 2007) and MSU stimulates the secretion of S100A8/9 from neutrophils (Ryckman et al., 2004). We found that BHB prevented caspase-1 activation and IL-1β secretion in MSU-treated S100A9-primed bone marrow-derived macrophages (BMDM) (Figures 1A, B). While S100A9 fibrils provided a priming signal in BMDM (Figure S1E), they do not function as a DAMP to provide signal 2 for inflammasome activation in LPS-primed BMDM (Figure S1F). Thus, S100A8/9 proteins may represent one of the endogenous sources of priming signal for NLRP3 inflammasome in vivo.
Figure 1. Ketogenic diet protects rats from MSU-induced gouty flare.

(A) AFM height image of S100A9 amyloid fibrils; scale bar=120nm. (B) Western blot of caspase-1 and IL-1β activation in BMDM. (C) Blood BHB levels after 1 week of ketogenic diet feeding, prior to injection with MSU. (D) Serum IL-1β 48hr post-MSU injection. (E) Change in knee thickness 48hr post-MSU injection. (F) Knee thickness was measured daily. (G) Representative sections of femoro-tibial joint. Local tissue reaction (black asterisks) and intrasynovial exudate (white asterisks); scale bar=500μm. (H) Representative images of synovial inflammation. (Hi, ii) Regions of macrophage infiltration (black asterisk) and neutrophil infiltration (black arrows); scale bar=20μm. (Hiii) Higher magnification of MSU-induced inflammatory response; black arrow points to infiltrating neutrophil; scale bar=10μm. (Hiv) Local tissue reaction in control PBS-injected joint; focal fibrin exudate (white asterisk) with surrounding macrophages (black arrows); macrophages within granular amorphous injected material (dashed arrows); scale bar=20μm. (I) Western blot of IL-1β secretion from stimulated BMDM. (J) Body weights and (K) bacterial load after S. aureus infection. Data are pooled from at least 3 independent experiments. Statistical differences calculated by t-test (C–E) or 2-way ANOVA (F). See also Figure S1.
BHB serves as an alternate metabolic fuel during starvation state or in absence of insulin when glucose can’t be utilized. Ketogenic diets (KD) that are rich in fat and low in carbohydrates are routinely employed to induce BHB or nutritional ketosis because sustained elevation of BHB through infusion of sodium salts of BHB is contraindicated due to adverse effects on blood acid-base balance. Thus the KD have been safely employed to treat drug resistant epilepsy (Cahill, 2006). Although IL-1R antagonists have shown benefit in small clinical trials (So et al., 2010; So et al., 2007), the high costs of these biologicals and potential detrimental impact on host-defense have limited their use in treatment of gouty flares. Therefore, we next developed a model of gouty arthritis and investigated the induction of nutritional ketosis by feeding KD as a potential therapy against gout. Humans, but not rodents, are susceptible to gout due to loss of the uricase enzyme, which normally functions to prevent high concentrations of uric acid. Therefore, we developed an in vivo gout model in outbred Sprague Dawley rats by intra-articular injection of MSU crystals in the knee. One week of high-fat, low-carbohydrate KD feeding induced endogenous BHB production (Figure 1C). Rats fed a KD were protected from the MSU-induced elevated serum IL-1β (Figure 1D) and knee swelling observed in the chow-fed rats (Figures 1E, F). Pathology analysis of H&E stained sections of the joints showed that MSU-injected rats displayed combined lesions of intra-articular exudate and synovial soft tissue inflammation (Figure 1G). Intra-articular exudate was characterized by masses of fibrin, amorphous granular foreign material, and clusters of macrophages (Figure 1H). Both the extent of intra-articular exudate and synovial inflammation was less severe in KD-fed animals compared to those on chow diet. Overall, KD reduced the severity of the inflammatory reactions in MSU-injected knees. Foci of frank necrosis are present in animals on the control diet, but not in those on the ketone-rich diet (Figure 1H). Notably, KD results in elevation of blood ketone bodies BHB as well as AcAc. Our data suggest that anti-inflammatory effects of ketones are limited to BHB, as neither AcAc nor microbiota-derived short chain fatty acid, butyrate, prevented IL-1β secretion in inflammasome-activated BMDM (Figure 1I). Importantly, reduced inflammatory responses during KD did not increase disease severity in mice infected with Staphylococcus aureus (Figures 1J, S1G–I) and surprisingly reduced bacterial burdens in the lungs of infected mice (Figure 1K). Taken together, our data shows that elevated BHB levels protects against acute gouty flare without compromising the host-defense functions of the immune system.
BHB acts on neutrophils to block IL-1β secretion throughout the lifespan
Gout flares are mediated by both macrophage and neutrophil activation. Because BHB inhibits NLRP3 inflammasome activation in macrophages (Youm et al., 2015) and in an in vitro model of gout (Figure 1B), we next tested whether BHB regulates IL-1β secretion from neutrophils. Indeed, BHB dose-dependently inhibited IL-1β secretion in isolated murine neutrophils (Figure 2A, S2). Aging is a major risk factor for gout and if not properly managed, gouty flares increase in frequency and intensity in the elderly. Importantly, BHB potently inhibited the NLRP3 inflammasome-induced IL-1β secretion in neutrophils of young and elderly humans (Figure 2B) but not secretion of S100A8 protein implicated in the propagation of gouty flares (Figure 2C). BHB inhibited IL-1β secretion in response to classical NLRP3 inflammasome activation (Figure 2D) as well as the age-related lipotoxic DAMP ceramide (Figure 2E) in isolated adult and old murine neutrophils. Ketogenesis is typically induced during hypoglycemia or lack of glucose availability, which may alter neutrophil function. Therefore we next determined whether neutrophils in humans exposed to hypoglycemia in vivo respond to BHB (Figure 2F). Indeed IL-1β secretion is still sensitive to BHB-mediated inhibition regardless of glucose availability in vivo (Figure 2G). These experiments reveal a regulatory role for BHB in neutrophil inflammasome activation regardless of the host’s age.
Figure 2. BHB regulates neutrophil NLRP3 inflammasome activation.

IL-1β secretion by stimulated (A) mouse and (B) human neutrophils. (C) Secretion of S100A8 from stimulated human neutrophils. Different colored symbols connected by dotted line represents a unique individual’s response to all treatments. Columns indicate the mean for each treatment. (D, E) Western blot analysis of IL-1β secretion of adult and old murine neutrophils. (F) Depiction of insulin-induced hypoglycemia in vivo; arrow indicates blood collection time point. (G) Neutrophil IL-1β secretion from hypoglycemic subjects. Lines connect the responses of each individual for each treatment condition. Statistical differences were calculated by paired 1-way ANOVA. See also Figure S2.
BHB reduces urate crystal-induced inflammation during aging
Neutrophils are reported to accumulate several defects during aging, including impaired TLR signaling (Qian et al., 2014) and NETosis (Hazeldine et al., 2014). Neutrophils are also associated with immunopathology during infection in aged hosts (Bou Ghanem et al., 2015; Menter et al., 2014). Bone marrow neutrophils from aged mice exhibited no remarkable differences in abundance or expression of NLRP3 inflammasome proteins compared to young controls (Figures S2). To test neutrophil-intrinsic inflammasome activation defects during aging, isolated neutrophils from adult and old mice were stimulated with LPS+ATP. IL-1β, but not TNFα secretion, was NLRP3- and ASC-dependent and no age-related differences were observed (Figure 3A, B). Due to the potent inhibitory effects of BHB on aged neutrophils in vitro (Figures 2B, D, E), we next sought to determine whether BHB could reduce neutrophillic inflammation in vivo during aging. To induce endogenous ketogenesis, old mice were fed KD for one week. During this time, no alterations to body weight (Figure 3C) were observed and blood BHB levels increased rapidly (Figure 3D). KD did not change peritoneal neutrophil infiltration (Figure S3) but prevented upregulation of Nlrp3 and Il1b gene expression, but not the general inflammatory marker Tnfα in old mice (Figures 3E–G) after MSU-induced peritonitis. This suggests that BHB levels can be elevated during aging to reduce acute urate crystal-induced inflammatory responses.
Figure 3. Elevated BHB protects against MSU-induced peritonitis in aged mice.

(A) IL-1β and (B) TNFα were measured in culture supernatants after LPS+ATP stimulation of isolated neutrophils from mice of indicated ages and genotypes. Statistical differences were calculated by age-specific t-test (3 months) or 1-way ANOVA (24 months). Data are pooled from 2 independent experiments. (C) Body weights and (D) blood BHB concentrations were measured daily in old mice fed KD, prior to MSU injection. (E–G) Gene expression of Ilb, Nlrp3 and Tnfa within peritoneal cells 4hr post-MSU. Data are represented as fold change relative to sham, and are pooled from 3 independent experiments. Statistical differences were calculated by t-test between MSU-injected groups. See also Figure S3.
β-hydroxybutyrate inhibits NLRP3 inflammasome priming and assembly
Neutrophils contain both inflammasome-dependent and -independent IL-1β cleavage processes (Cassel et al., 2014; Joosten et al., 2009; Karmakar et al., 2015; Mankan et al., 2012). In our experimental conditions, neutrophil IL-1β secretion, in response to several DAMPs including extracellular ATP, the crystalline Silica, or lipotoxic ceramide, was entirely caspase1/11-dependent (Figure 4A). In cell-free assays, BHB had no effect on enzyme activity of neutrophil serine proteases Elastase (Figure 4B) or Cathepsin G (Figure 4C), both of which have been implicated in IL-1β cleavage (Cassel et al., 2014; Guma et al., 2009). We next investigated the mechanism by which BHB inhibits NLRP3 inflammasome activation in neutrophils. The GPR109a can bind BHB on cell surface to inhibit inflammation, however, we found that niacin, a high affinity ligand for Gpr109a, failed to impact neutrophil IL-1β cleavage suggesting that in vitro Gpr109a signaling is dispensable for neutrophil inflammasome activation (Figure 4D). Ketogenesis occurs during energy restriction, a physiological state in which autophagy is induced. However, BHB’s inhibition of IL-1β secretion does not rely on autophagy because treatment of neutrophils with the autophagy inhibitor 3-MA did not prevent BHB-mediated inhibition of IL-1β secretion (Figure 4E). Similarly, treating cells with the TCA cycle entry inhibitor AOA did not prevent BHB’s inhibition of IL-1β secretion, suggesting oxidation of BHB is not required for its effect (Figure 4E). Interestingly, the non-oxidizable chiral enantiomer S-BHB, which cannot enter TCA cycle, also inhibited IL-1β secretion from neutrophils (Figure 4F), suggesting that in the presence of glucose in vitro, the myeloid cells energetically spare BHB to block inflammasome activation.
Figure 4. BHB inhibits inflammasome priming and assembly.

(A) Western blot of culture supernatants from LPS-primed WT and Caspase 1/11−/− neutrophils after ATP, Silica, or ceramide stimulation as indicated. (B, C) Elastase and Cathepsin G activity; n=4. (D–F) Culture supernatants were analyzed for neutrophil IL-1β secretion by Western blot after indicated treatments. (G) Total and acetylated H3 expression in cell lysates. (H) NFκB phosphorylation in neutrophil cell lysates. (I) IL-1β secretion from neutrophils derived from mouse model of FCAS bearing activating mutation of Nlrp3. For all blots, each sample is pooled from at least n=4 mice per experiment. Each blot is representative of at least two independent experiments. See also Figure S4.
Unaltered caspase-11 activation or Gasdermin D expression in the presence of BHB (Figure S4A) suggested that BHB also does not impact neutrophil pyroptosis (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015) and BHB did not alter cell viability (Figure S4B). BHB treatment lead to increased histone H3 acetylation (Figure 4G), probably due to its reported HDAC inhibitor activity (Shimazu et al., 2013). Interestingly, BHB inhibited phosphorylation of NFκB (Figure 4H), a necessary signaling event for inflammasome activation and confirming previous findings (Fu et al., 2014). Finally, BHB was tested in a mouse model of the severe human disease Familial Cold Autoinflammatory Syndrome (FCAS), which contains an L351P nucleotide substitution in Nlrp3 that facilities constitutive inflammasome assembly (Brydges et al., 2009; Yu et al., 2006). IL-1β secretion from neutrophils in this FCAS model was dose-dependently inhibited by treatment with BHB-conjugated nanolipogels (Figure 4I). These data suggest that the inhibitory effect of BHB upon NLRP3 inflammasome activation is two-pronged: (1) prevents TLR4-mediated priming, and (2) blocks the physical assembly of the NLRP3 inflammasome complex.
Discussion
Gout is a chronic disease characterized by recurrent painful gouty flares. The incidence of gout has steadily increased, and individuals over age 65 account for the majority of gout-related hospitalizations (Lim et al., 2016). Although a primary treatment strategy for gout is to lower uric acid levels, many of these medications induce gouty flares, presumably due to disruption of tophi, which results in poor adherence by patients. We found that elevated blood BHB levels protected outbred rats from joint swelling and systemic inflammation after intra-articular injection of MSU crystals. Reduced joint swelling was due to a qualitative reduction in the inflammatory response, as pathology analysis revealed reduced tissue damage despite similar neutrophil and macrophage infiltration into the joints.
In vitro, BHB also inhibits neutrophil IL-1β secretion from adult and elderly individuals. Notably, elevated BHB levels during KD did not increase disease severity during S. aureus infection in mice and even reduced bacterial burdens in the lungs of infected mice suggesting high translational potential of BHB against gouty flares. It was recently reported that glucose metabolism promotes mortality during bacterial infection and LPS-sepsis (Wang et al., 2016), which may explain the beneficial effects of KD in S. aureus-infected mice as BHB reduces the glucose availability. Notably, other macronutrients in KD can induce hormonal alterations and plasma lipid profile changes in patients. Moreover, at the cellular level, limited glucose availability during KD can induce autophagy which inhibits the inflammasome. Thus, additional studies are required to rule out the exact contribution each of these mechanisms in mediating anti-inflammasome effects of KD in vivo in models of gout. Regardless, our findings that BHB can target neutrophil inflammasome have important clinical implications, as an estimated 8 million Americans have gout (Zhu et al., 2011) and the cumulative nature of the disease causes the risk and frequency of gouty flares to increase during aging.
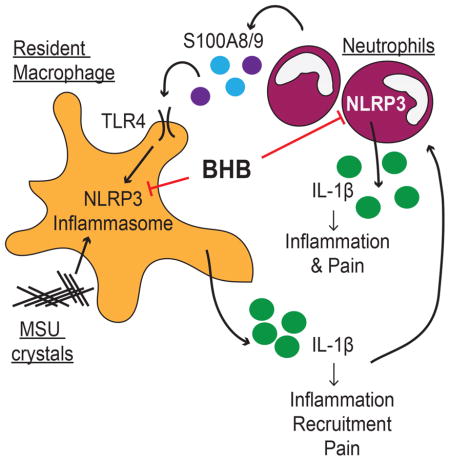
S100A8/9 proteins are also reported to increase during age-related inflammatory diseases including obesity (Nagareddy et al., 2014), cardiovascular disease (Ma et al., 2012), Alzheimer’s disease (Wang et al., 2014), and in the aged prostate (Yanamandra et al., 2009). This implies these proteins may provide an endogenous, local signal 1 for NLRP3 inflammasome activation and may be universal biomarkers of inflammation (Vogl et al., 2007). During a gouty flare, neutrophils are recruited to inflamed joints by resident macrophages and secrete a variety of proinflammatory molecules, including S100A8/9 and IL-1β (Ryckman et al., 2004). BHB inhibited NLRP3 inflammasome activation in S100A9-primed macrophages (Figure 1B). Our data suggest that BHB can break this feed-forward cycle to prevent swelling and inflammation (Figure 5), although defining the exact mechanism will require further experimentation. Neutrophils can secrete IL-1β by caspase 1-independent mechanisms. Although we were not able to detect IL-1β secretion from caspase 1/11 knockout mice, cell-free assays revealed no effect on enzymatic activity of serine proteases neutrophil elastase or cathepsin G. These data highlight the targeted effects of BHB upon the NLRP3 inflammasome, making it an ideal candidate for preventing NLRP3-driven inflammation.
Figure 5. Hypothetical model for dual role of BHB as a regulatory metabolite.

(A) The metabolic role of BHB as an alternative energy source during limited energy availability, coordinated with the anti-inflammatory role of BHB upon NLRP3 inflammasome. (B) BHB inhibits both the priming signal and the secondary assembling signal in neutrophils. (C) Inhibition of IL-1β secretion by BHB does not involve inhibition of serine protease activity, autophagy, oxidation in the TCA cycle, nor the surface receptor GPR109a, although additional experiments will be needed to further clarify the mechanism.
Together with our prior findings that BHB inhibits NLRP3 inflammasome activation in macrophages (Youm et al., 2015) and our current data that BHB also regulates neutrophil inflammasome activation, we propose that strategies to increase BHB levels are likely to be therapeutically beneficial in gout patients. Furthermore, the NLRP3-targeted effects of BHB make it ideal for reducing or preventing many age-related inflammatory disease that have been shown to be driven by chronic NLRP3 inflammasome activation (Youm et al., 2013). Adherence to a high-fat low-carbohydrate ketogenic diet is difficult and can promote dyslipidemia in gout patients. However, ketone esters have been delivered orally to humans, which increased circulating BHB levels and enhanced physical performance in athletes (Clarke et al., 2012; Cox et al., 2016), and should be explored for inducing mild ketosis to prevent inflammation in individuals with gout.
Experimental Procedures
Animals
All animals were housed under specific pathogen-free conditions under 12hr light/dark cycle. All mice used were on the C57BL/6 genetic background. See Supplemental Information for details of mouse and rat strains. Ketogenic diet (Research Diets D12369B) was initiated 1 week prior to experimental manipulation. Blood BHB concentrations were measured with Precision Xtra β-ketone strips. All animal experiments were performed in compliance with the Yale University Institutional Animal Care and Use Committee.
Human Subjects
Healthy adult (18–45 years) and old (65+ years) males and females with no current steroid use were recruited. Individuals were not fasting at time of peripheral blood collection, except for hypoglycemic studies. Insulin was used to induce hypoglycemia, see Supplemental Information for detailed procedure. Informed consent was obtained from all subjects and all studies were approved by the Institutional Review Committee of Yale University.
Cell Isolation and Activation
Neutrophils were isolated from bone marrow (mice) or peripheral blood (humans) by negative magnetic selection (Stem Cell Technology, Figure S2). BMDM were generated and all cells were stimulated as previously described (Youm et al., 2015), see Supplemental Information for detailed treatment conditions.
In vitro activation measurements
All biochemical analysis methods are described in detail in Supplemental Information. Cytokine secretion was measured by Multiplex (Figure 3A, B IL-1β, TNFα from Life Technologies) or ELISA (human IL-1β from eBioscience 88-7261-22; S100A8 from Thermo EHS100A8) according to manufacturers’ protocols. Elastase (ab118971) and Cathepsin G (ab204693) enzymatic activity were assessed using kits (Abcam) according to manufacturer’s instructions.
Peritonitis Model
Peritonitis was induced by injection of MSU crystals i.p. (Invivogen). Mice were injected with 2.5mg MSU in PBS. Four hours later, total peritoneal cells were collected by lavage. Cells were counted by hemacytometer and phenotype and gene expression were analyzed by flow cytometry and RT-PCR, respectively.
Staphylococcus aureus Infection
Mice were infected intranasally with 108 cfu or 106 cfu S. aureus (strain 14458). Bacterial burdens were determined by plating serial dilutions 24hr post-infection. BALF was collected by washing lungs three times with 1mL of sterile PBS.
Gout Model
Gout was induced in rats by intra-articular injection of 1.25mg MSU in the knee. Knee thickness was measured with digital calipers. IL-1β was measured in serum by ELISA (eBioscience BMS630). Knees were fixed and decalcified in Bouins solution (Sigma). Tissue sectioning and H&E staining were performed by the Yale Mouse Research Pathology and Histology Core. For pathology analysis, all sections were taken from the mid-sagittal region of the femoro-tibial joint, encompassing cruciate ligaments and menisci. Images are oriented with patellar ligament on the right. In high power images, all images are taken from the anterior synovial tissue.
Statistical Analysis
Statistical analyses were performed using Graphpad Prism software (Graphpad) as indicated in the text and Figure legends. P<0.05 was considered statistically significant for all tests. All experiments were performed at least twice. All graphs shown are combined from all replicates of each experiment, and each data point represents an individual test subject. All data are expressed as mean ± SEM unless otherwise specified. For all statistical differences *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Western blot experiments were repeated at least three times, each time containing cells pooled from n=3–5 mice.
Supplementary Material
Highlights.
Inflammation and joint pathology during gout flare is prevented by ketogenic diet
BHB inhibits IL-1β secretion from neutrophils
Ketogenic diet and BHB inhibit NLRP3 activation in aged neutrophils
BHB inhibits both priming and assembly steps of NLRP3 activation in neutrophils
Acknowledgments
We thank Kim Nguyen for expert technical assistance. Funding: VDD is supported in part by US National Institutes of Health (NIH) grants AG043608, P01AG051459, AI105097 and The Glenn Foundation for Aging Research. ELG is supported in part by postdoctoral fellowship from American Foundation of Aging Research (AFAR) and American Heart Association (AHA). RDM is supported by NIH T32 grants AI007019-38, AI055403, and the Francis Trudeau Trainee Fellowship. ACS is supported by NIH grants K24 AG042489, U19 AI089992, and P30 AG21342 (Yale Claude D. Pepper Older Americans Independence Center). RIH is supported by NIH/NIDDK through K08 DK082618 and R01 DK101984-02 and the hypoglycemic clamp studies are supported by the Yale Center for Clinical Investigation (YCCI) and Hospital research Unit (HRU) personnel via DK045735, the Yale Diabetes Research Center grant and the Clinical Translational Science Award UL1-RR-024139 from the National Center for Advancing Translational Sciences, a component of the NIH, and NIH Roadmap for Medical Research. AI is supported by the Howard Hughes Medical Institute.
Footnotes
Author Contributions
ELG performed experiments did data analysis, and prepared the manuscript. JLA performed gout experiments and clinical evaluations. RM performed infection experiments. CW and LAM-R provided S100A9 reagents and expertise. ACS provided human samples. RIH provided expertise and samples from hypoglycemic clamp studies. CJZ evaluated gout pathology. AI designed and interpreted the infection experiments. VDD conceived and supervised the project, interpreted the data, and prepared the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data for this study have been included in this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bou Ghanem EN, Clark S, Roggensack SE, McIver SR, Alcaide P, Haydon PG, Leong JM. Extracellular Adenosine Protects against Streptococcus pneumoniae Lung Infection by Regulating Pulmonary Neutrophil Recruitment. PLoS pathogens. 2015;11:e1005126. doi: 10.1371/journal.ppat.1005126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–887. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill GF., Jr Fuel metabolism in starvation. Annual review of nutrition. 2006;26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- Cassel SL, Janczy JR, Bing X, Wilson SP, Olivier AK, Otero JE, Iwakura Y, Shayakhmetov DM, Bassuk AG, Abu-Amer Y, et al. Inflammasome-independent IL-1beta mediates autoinflammatory disease in Pstpip2-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:1072–1077. doi: 10.1073/pnas.1318685111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie TB, et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regulatory toxicology and pharmacology: RTP. 2012;63:401–408. doi: 10.1016/j.yrtph.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell metabolism. 2016;24:256–268. doi: 10.1016/j.cmet.2016.07.010. [DOI] [PubMed] [Google Scholar]
- Dalbeth N, Merriman TR, Stamp LK. Gout Lancet. 2016 doi: 10.1016/S0140-6736(16)00346-9. [DOI] [PubMed] [Google Scholar]
- Dessein PH, Shipton EA, Stanwix AE, Joffe BI, Ramokgadi J. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study. Annals of the rheumatic diseases. 2000;59:539–543. doi: 10.1136/ard.59.7.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff GW, Atkins E, Malawista SE. The fever of gout: urate crystals activate endogenous pyrogen production from human and rabbit mononuclear phagocytes. Transactions of the Association of American Physicians. 1983;96:234–245. [PubMed] [Google Scholar]
- Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. The Journal of biological chemistry. 1991;266:7706–7713. [PubMed] [Google Scholar]
- Edwards C, Canfield J, Copes N, Rehan M, Lipps D, Bradshaw PC. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging. 2014;6:621–644. doi: 10.18632/aging.100683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SP, Li SN, Wang JF, Li Y, Xie SS, Xue WJ, Liu HM, Huang BX, Lv QK, Lei LC, et al. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-kappaB activation. Mediators of inflammation. 2014;2014:983401. doi: 10.1155/2014/983401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod AB. A treatise on gout and rheumatic gout (rheumatoid arthritis) London: Longman Green; 1876. [Google Scholar]
- Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunological reviews. 2015;265:63–74. doi: 10.1111/imr.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis and rheumatism. 2009;60:3642–3650. doi: 10.1002/art.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, Sapey E, Lord JM. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging cell. 2014;13:690–698. doi: 10.1111/acel.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell research. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzinger D, Nippe N, Vogl T, Marketon K, Mysore V, Weinhage T, Dalbeth N, Pool B, Merriman T, Baeten D, et al. Myeloid-related proteins 8 and 14 contribute to monosodium urate monohydrate crystal-induced inflammation in gout. Arthritis & rheumatology. 2014;66:1327–1339. doi: 10.1002/art.38369. [DOI] [PubMed] [Google Scholar]
- Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, Pham CT, van der Meer JW, Dinarello CA, van den Berg WB. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis and rheumatism. 2009;60:3651–3662. doi: 10.1002/art.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten LA, Netea MG, Mylona E, Koenders MI, Malireddi RK, Oosting M, Stienstra R, van de Veerdonk FL, Stalenhoef AF, Giamarellos-Bourboulis EJ, et al. Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis and rheumatism. 2010;62:3237–3248. doi: 10.1002/art.27667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, Camilli A, Kadioglu A, Dubyak GR, Pearlman E. Neutrophil IL-1beta processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. Journal of immunology. 2015;194:1763–1775. doi: 10.4049/jimmunol.1401624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- Li C, Hsieh MC, Chang SJ. Metabolic syndrome, diabetes, and hyperuricemia. Current opinion in rheumatology. 2013;25:210–216. doi: 10.1097/BOR.0b013e32835d951e. [DOI] [PubMed] [Google Scholar]
- Lim SY, Lu N, Oza A, Fisher M, Rai SK, Menendez ME, Choi HK. Trends in Gout and Rheumatoid Arthritis Hospitalizations in the United States, 1993–2011. Jama. 2016;315:2345–2347. doi: 10.1001/jama.2016.3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LP, Haugen E, Ikemoto M, Fujita M, Terasaki F, Fu M. S100A8/A9 complex as a new biomarker in prediction of mortality in elderly patients with severe heart failure. International journal of cardiology. 2012;155:26–32. doi: 10.1016/j.ijcard.2011.01.082. [DOI] [PubMed] [Google Scholar]
- Mankan AK, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. European journal of immunology. 2012;42:710–715. doi: 10.1002/eji.201141921. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- McCarty DJ, Hollander JL. Identification of urate crystals in gouty synovial fluid. Annals of internal medicine. 1961;54:452–460. doi: 10.7326/0003-4819-54-3-452. [DOI] [PubMed] [Google Scholar]
- Menter T, Giefing-Kroell C, Grubeck-Loebenstein B, Tzankov A. Characterization of the inflammatory infiltrate in Streptococcus pneumoniae pneumonia in young and elderly patients. Pathobiology: journal of immunopathology, molecular and cellular biology. 2014;81:160–167. doi: 10.1159/000360165. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Madrigal-Matute J, Scheibye-Knudsen M, Fang E, Aon M, Gonzalez-Reyes JA, Cortassa S, Kaushik S, Gonzalez-Freire M, Patel B, et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell metabolism. 2016;23:1093–1112. doi: 10.1016/j.cmet.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell metabolism. 2014;19:821–835. doi: 10.1016/j.cmet.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian F, Guo X, Wang X, Yuan X, Chen S, Malawista SE, Bockenstedt LK, Allore HG, Montgomery RR. Reduced bioenergetics and toll-like receptor 1 function in human polymorphonuclear leukocytes in aging. Aging. 2014;6:131–139. doi: 10.18632/aging.100642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roddy E, Choi HK. Epidemiology of gout. Rheumatic diseases clinics of North America. 2014;40:155–175. doi: 10.1016/j.rdc.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman C, Gilbert C, de Medicis R, Lussier A, Vandal K, Tessier PA. Monosodium urate monohydrate crystals induce the release of the proinflammatory protein S100A8/A9 from neutrophils. Journal of leukocyte biology. 2004;76:433–440. doi: 10.1189/jlb.0603294. [DOI] [PubMed] [Google Scholar]
- Ryckman C, McColl SR, Vandal K, de Medicis R, Lussier A, Poubelle PE, Tessier PA. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis and rheumatism. 2003;48:2310–2320. doi: 10.1002/art.11079. [DOI] [PubMed] [Google Scholar]
- Seegmiller JE, Howell RR. The old and new concepts of acute gouty arthritis. Arthritis and rheumatism. 1962;5:616–623. doi: 10.1002/art.1780050610. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A, De Meulemeester M, Pikhlak A, Yucel AE, Richard D, Murphy V, Arulmani U, Sallstig P, Schlesinger N. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: Results of a multicenter, phase II, dose-ranging study. Arthritis and rheumatism. 2010;62:3064–3076. doi: 10.1002/art.27600. [DOI] [PubMed] [Google Scholar]
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis research & therapy. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, Booth CJ, Medzhitov R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell. 2016;166:1512–1525. e1512. doi: 10.1016/j.cell.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Klechikov AG, Gharibyan AL, Warmlander SK, Jarvet J, Zhao L, Jia X, Narayana VK, Shankar SK, Olofsson A, et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta neuropathologica. 2014;127:507–522. doi: 10.1007/s00401-013-1208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, Alexeyev O, Zamotin V, Srivastava V, Shchukarev A, Brorsson AC, Tartaglia GG, Vogl T, Kayed R, Wingsle G, et al. Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PloS one. 2009;4:e5562. doi: 10.1371/journal.pone.0005562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell metabolism. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, Sagara J, Fernandes-Alnemri T, Alnemri ES. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell death and differentiation. 2006;13:236–249. doi: 10.1038/sj.cdd.4401734. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis and rheumatism. 2011;63:3136–3141. doi: 10.1002/art.30520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.