

ABSTRACT
R bodies are insoluble large polymers consisting of small proteins encoded by reb genes and are coiled into cylindrical structures in bacterial cells. They were first discovered in Caedibacter species, which are obligate endosymbionts of paramecia. Caedibacter confers a killer trait on the host paramecia. R-body-producing symbionts are released from their host paramecia and kill symbiont-free paramecia after ingestion. The roles of R bodies have not been explained in bacteria other than Caedibacter. Azorhizobium caulinodans ORS571, a microsymbiont of the legume Sesbania rostrata, carries a reb operon containing four reb genes that are regulated by the repressor PraR. Herein, deletion of the praR gene resulted in R-body formation and death of host plant cells. The rebR gene in the reb operon encodes an activator. Three PraR binding sites and a RebR binding site are present in the promoter region of the reb operon. Expression analyses using strains with mutations within the PraR binding site and/or the RebR binding site revealed that PraR and RebR directly control the expression of the reb operon and that PraR dominantly represses reb expression. Furthermore, we found that the reb operon is highly expressed at low temperatures and that 2-oxoglutarate induces the expression of the reb operon by inhibiting PraR binding to the reb promoter. We conclude that R bodies are toxic not only in paramecium symbiosis but also in relationships between other bacteria and eukaryotic cells and that R-body formation is controlled by environmental factors.
KEYWORDS: R body, legume, pathogenesis, reb gene, rhizobia, symbiosis
IMPORTANCE
Caedibacter species, which are obligate endosymbiotic bacteria of paramecia, produce R bodies, and R-body-producing endosymbionts that are released from their hosts are pathogenic to symbiont-free paramecia. Besides Caedibacter species, R bodies have also been observed in a few free-living bacteria, but the significance of R-body production in these bacteria is still unknown. Recent advances in genome sequencing technologies revealed that many Gram-negative bacteria possess reb genes encoding R-body components, and interestingly, many of them are animal and plant pathogens. Azorhizobium caulinodans, a microsymbiont of the tropical legume Sesbania rostrata, also possesses reb genes. In this study, we demonstrate that A. caulinodans has ability to kill the host plant cells by producing R bodies, suggesting that pathogenicity conferred by an R body might be universal in bacteria possessing reb genes. Furthermore, we provide the first insight into the molecular mechanism underlying the expression of R-body production in response to environmental factors, such as temperature and 2-oxoglutarate.
INTRODUCTION
R bodies are bacterial inclusion bodies and are large proteinaceous ribbons that are coiled into cylindrical structures. R bodies were first observed in Caedibacter species, which are obligate endosymbiotic bacteria that inhabit paramecia (1). Paramecia that harbor R-body-producing Caedibacter cells are referred to as killer paramecia and release bacterial cells via the cytopyge. Subsequently, sensitive nonendosymbiont paramecia are killed following ingestion of the released bacteria (2), conferring the “killer trait” of paramecia (3). R bodies play a major role in this trait, because paramecia harboring mutant Caedibacter strains that are defective in R body production do not express the killer trait (4).
The genes involved in R-body production were originally identified in Caedibacter taeniospiralis (5) and include rebA, rebB, rebC, and rebD (5, 6). Moreover, genes that are homologous to rebA, rebB, and rebD of C. taeniospiralis have been found in species of the phylum Proteobacteria and in Kordia algicida OT-1 (7), which belongs to the phylum Bacteroidetes, whereas no rebC-homologous genes have been identified in bacteria other than C. taeniospiralis (8, 9). It is hypothesized that reb-homologous genes were passed on by horizontal gene transfer—i.e., by phages or plasmids (2, 8, 10, 11). Although many bacterial species that carry reb-homologous genes are pathogenic to plants and animals (e.g., Xanthomonas axonopodis pv. citri, Stenotrophomonas maltophilia, Burkholderia pseudomallei, and so on) (8), the rhizobium Azorhizobium caulinodans ORS571, a mutualistic microsymbiont of the tropical legume Sesbania rostrata, possesses reb-homologous genes (8, 12). To date, reb-homologous genes have not been found in rhizobia other than A. caulinodans ORS571.
A. caulinodans ORS571 fixes atmospheric nitrogen in free-living and symbiotic states (13–15) and forms nitrogen-fixing nodules at the sites of adventitious root primordia on roots and stems. Bacteria enter stems via fissures at root primordia and colonize cortical infection pockets (16). From these infection pockets, infection threads guide the bacteria toward the nodule meristematic zone, where they are released into host cells and surrounded by plant-derived peribacteroid membranes (16). Subsequently, infected host cells are filled with differentiated bacteroids and infected areas enlarge with nodule maturation (15, 16).
The A. caulinodans ORS571 strain has a gene cluster containing four reb-homologous genes (8, 12) that are strongly suppressed by PraR, which is a conserved transcription factor among Alphaproteobacteria (8). In a previous study, stem nodules harboring A. caulinodans praR mutants that expressed high levels of reb genes were defective in nitrogen fixation (8). Furthermore, praR knockout in these nodules altered the interactions between these bacteria and their host cells according to two distinctly abnormal patterns. Specifically, host cells maintained normal shapes, and the bacteria disappeared with increasing host vacuolar sizes, or bacteria predominantly occupied host cells that had shrunken gradually (8). These observations suggest that derepression of reb genes at least partially reverts pathogenic traits for the symbiont. The regulatory mechanism underlying the expression of reb genes, however, is yet to be elucidated.
In Caedibacter spp., reb genes are responsible for the production of R bodies, which likely mediate eukaryotic cell death (4, 5). Although reb genes have been identified in many pathogenic bacteria, pathogenic roles of R bodies have not been directly demonstrated. On the other hand, observations of disordered intracellular symbiosis in plant hosts following derepression of reb genes in A. caulinodans suggest that A. caulinodans kills host cells by producing R bodies. Herein, we investigated the regulatory mechanism of reb gene expression in A. caulinodans and defined the roles of R bodies in reb-associated pathogenic traits, with particular emphasis on transcription repressor-activator interactions.
RESULTS
Genetic organization of the reb operon.
The reb operon includes genes from AZC_3781 to AZC_3788 on the genome of A. caulinodans ORS571 (Fig. 1), and transcription units and start sites were determined using reverse transcription-PCR (RT-PCR) and primer extension analyses (see Fig. S1 in the supplemental material). A comprehensive phylogenetic analysis revealed that the proteins encoded by reb genes in A. caulinodans did not belong to clusters, including RebA, RebB, and RebD of C. taeniospiralis (see Fig. S2 in the supplemental material). Accordingly, the reb-homologous genes (AZC_3781, AZC_3782, AZC_3783, and AZC_3786) rebD1, rebA1, rebD2, and rebA2 (8) were renamed rebAZC1, rebAZC2, rebAZC3, and rebAZC4, respectively. An InterProScan analysis revealed that the AZC_3788 gene, which was designated rebR, encoded a putative transcription factor of the cyclic AMP receptor protein-fumarate and nitrate reduction regulator (Crp-Fnr) superfamily (17). The AZC_3784, AZC_3785, and AZC_3787 genes had no similarities to known reb genes, although BLASTp searches identified AZC_3784 homologues in Rhizobium sp. strain AAP43, Oceanicaulis sp. strain HTCC2633, and Oceanicaulis alexandrii DSM 11625, as well as an AZC_3787 homologue in Inquilinus limosus. However, no AZC_3785 homologues were found in the database.
FIG 1 .

Genetic organization of the reb operon on the chromosome of A. caulinodans ORS571. The nucleotide sequence below the map shows the promoter region of the reb operon. Deduced amino acid sequences of the C terminus of the AZC_3780 protein and the N terminus of the RebAZC1 protein are shown under their corresponding nucleotide sequences, respectively. The stop codon of AZC_3780 and the start codon of rebAZC1 are marked by gray and open boxes, respectively. The three PraR binding sites and the RebR binding site are underlined. Transcription start sites of the reb operon are indicated by arrows.
Determination of transcription units and start sites of the reb operon in Azorhizobium caulinodans ORS571. (A) Map of the reb operon of A. caulinodans with target regions and primer positions for RT-PCR and primer extension analyses. (B) RT-PCR analysis to determine transcription units of the reb operon. cDNA was synthesized using total RNA (500 ng) from WT and ΔpraR strains grown in BD medium at 38°C. Subsequent PCRs were performed using the 20-fold-diluted cDNAs and WT genomic DNA (1 × 10−1 ng µl−1) as the templates with the indicated primer pairs. The result showed that AZC_3781 to AZC_3788 were transcribed in a polycistronic manner, indicating that they form an operon. (C) Primer extension analysis to determine transcription start sites of the reb operon. Total RNA isolated from the WT and ΔpraR strains was reverse transcribed using the SuperScript III (Invitrogen) with a fluorescein isothiocyanate (FITC)-labeled primer (P14_FITC). The sequencing reaction for ladders was performed using the Thermo Sequenase primer cycle sequencing kit (GE Healthcare) with the P14_FITC primer and the DNA fragment that was amplified by PCR from WT genomic DNA using primer pair P1-P2. Both primer extension products and sequencing reaction mixtures were electrophoresed in denaturing 6% polyacrylamide gels containing 8 M urea. Gels were scanned for FITC fluorescence using an FLA3000 system (Fujifilm). Transcription start sites are indicated by arrows in the resulting image. This result shows that the reb operon is transcribed from 25, 23, 22, and 19 bp upstream of the AZC_3781 start codon. Download FIG S1, PDF file, 0.2 MB (210.5KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenic relationships among proteins encoded by reb-homologous genes. BLASTp analyses were performed using deduced amino acid sequences encoded by rebA, rebB, and rebD on the pKAP298 plasmid of Caedibacter taeniospiralis and AZC_3781, AZC_3782, AZC_3783, and AZC_3786 of A. caulinodans ORS571 as queries (E < 0.01) in the National Center for Biotechnology Information (NCBI) server (http://www.ncbi.nlm.nih.gov/BLAST/). The resulting sequences were combined, and phylogenic analyses were performed using MEGA5 software (K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei, S. Kumar, Mol Biol Evol 28:2731–2739, 2011, https://doi.org.10.1093/molbev/msr121). Download FIG S2, PDF file, 0.2 MB (195.3KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Contributions of the reb operon to R-body formation.
To identify roles of the reb operon in R-body formation, we generated a praR deletion (ΔpraR) mutant, a deletion mutant with deletion of a region from AZC_3781 to AZC_3787 (ΔAZC_3781-7), and a ΔpraR ΔAZC_3781-7 double mutant and observed phenotypes of stem nodules at 14 days postinoculation (dpi) with these mutant and wild-type (WT) bacteria. The nitrogen-fixation-defective (Fix−) phenotype of the stem nodules carrying bacteria with the praR deletion was suppressed by the second deletion in the AZC_3781-7 region (Fig. 2A), as observed previously (8), whereas the AZC_3781-7 deletion did not affect reb operon expression levels (Fig. 2B). Transmission electron microscopy (TEM) observations showed that R bodies were produced in many ΔpraR bacterial cells in shrunken host cells, and many R-body-containing bacterial cells had collapsed appearances (Fig. 2C). R bodies were not observed in stem nodules harboring the double mutant (Fig. 2C), suggesting that genes that are essential for R-body formation are located in the region from AZC_3781 to AZC_3787.
FIG 2 .

Contributions of the reb operon to R-body formation in stem nodules. S. rostrata plants were inoculated with the WT or ΔpraR, ΔAZC_3781-7, or ΔpraR ΔAZC_3781-7 mutant strains and grown at 30°C. Stem nodules were analyzed at 14 days postinoculation (dpi). (A) Hand-cut images (upper) and acetylene reduction activities (ARAs) reflecting nitrogen-fixing activities (lower) of stem nodules. Values are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative reverse transcription-PCR (RT-PCR) analyses of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels, expressed relative to corresponding data from the WT strain, and are presented as means ± standard deviations from three separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (C) Optical microscopic (OM) observations of infected host cells in stem nodules and transmission electron microscopic (TEM) observations of bacterial cells in host cells. Arrowheads indicate R bodies.
To observe the dynamics of host-bacterium interactions in single nodules harboring the ΔpraR mutant, pathogenic roles of R bodies were observed at an early stage of nodule development (7 dpi). Some normal-shape host cells contained bacterial cells that lacked R bodies (Fig. 3A and B), and nuclei in these normal-shape host cells were intact (Fig. 3C). In contrast, R bodies were observed in bacteria within shrunken host cells (Fig. 3A and D), in which nuclei were collapsed (Fig. 3E), indicating that R-body production is associated with host cell death in the nodules.
FIG 3 .

TEM observations of stem nodules harboring the ΔpraR mutant at 7 dpi. (A) Normally shaped host cells (NC) with expanding vacuoles (v) and shrunken host cells (SC). (B and C) Bacterial cells (B) and host nuclei (C) in normally shaped cells. (D and E) Bacterial cells (D) and nuclei (E) in shrunken host cells. Arrows in panel B indicate bacterial cells, and arrowheads in panel D indicate R bodies. v, vacuole; N, nuclei.
Requirements of the non-reb-homologous genes AZC_3784, AZC_3785, and AZC_3787 and the reb-homologous genes rebAZC1, rebAZC2, rebAZC3, and rebAZC4 for R-body production were determined in ΔpraR mutant derivatives with the deletions of these genes. In these experiments, AZC_3784 was essential for R-body formation and AZC_3785 and AZC_3787 were not (see Fig. S3 in the supplemental material). In addition, both rebAZC3 and rebAZC4 and either rebAZC1 or rebAZC2 were essential for R-body production (see Fig. S4 in the supplemental material).
Contributions of AZC_3784, AZC_3785, and AZC_3787 genes to R-body formation. S. rostrata plants were inoculated with the indicated strains and were grown at 30°C and stem nodules were then analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of stem nodules during symbiosis with the indicated strains. Data are presented as means ± standard deviations of five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative RT-PCR analysis of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels and are presented as means ± standard deviations from three separate plants relative to corresponding transcripts from the WT strain. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (C) Optical microscopic (OM) observations of infected host cells in stem nodules and transmission electron microscopic (TEM) observations of residing bacterial cells. Arrowheads indicate R bodies. Deletions of AZC_3785 and AZC_3787 did not suppress the defective nitrogen-fixing phenotype of stem nodules that lacked praR, whereas deletion of AZC_3784 partially restored nitrogen fixation, as indicated by slight red color of the inner region of nodules harboring the ΔpraR ΔAZC_3784 mutant. Expression levels of the reb operon were not affected by the AZC_3784 deletion, suggesting that partial suppression of the defective nitrogen-fixing phenotype of nodules was not caused by low expression of the reb operon. Host cells in nodules harboring the ΔpraR ΔAZC_3784 mutant were oval or elongated, and shrunken host cells were not observed. Bacterial cell densities of the ΔpraR ΔAZC_3784 mutant in host cells were not as high as for the WT strain. R bodies were not observed in ΔpraR ΔAZC_3784 mutant bacteria, suggesting that the AZC_3784 gene is essential for R-body formation. R bodies were observed in ΔpraR ΔAZC_3785 and ΔpraR ΔAZC_3787 mutant bacteria, suggesting that the AZC_3785 and AZC_3787 genes are not essential for R-body formation. Download FIG S3, PDF file, 1.2 MB (1.2MB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Contributions of rebAZC1, rebAZC2, rebAZC3, and rebAZC4 genes to R-body formation. S. rostrata plants were inoculated with the indicated strains and were grown at 30°C. Stem nodules were analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of stem nodules. Values are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative RT-PCR analysis of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels. Values are presented relative to respective WT expression data as means ± standard deviations from three separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (C) OM observations of infected host cells in stem nodules and TEM observations of the bacterial cells in host cells. Arrowheads indicate R bodies. Stem nodules that harbored ΔpraR ΔrebAZC1 and ΔpraR ΔrebAZC2 showed defective nitrogen-fixing phenotypes, and R bodies were observed in the residing bacteria. The ΔpraR ΔrebAZC3, ΔpraR ΔrebAZC4, ΔpraR ΔrebAZC1 ΔrebAZC2, and ΔpraR ΔrebAZC3 ΔrebAZC4 mutants formed normal stem nodules, and R bodies were not observed. Expression levels of the reb operon remained high in normal nodules, eliminating the possibility that the absence of R bodies reflected repression of reb operon expression. These results suggest that rebAZC3 and rebAZC4 and either rebAZC1 or rebAZC2 are essential for R-body production. Download FIG S4, PDF file, 1.1 MB (1.1MB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In investigations of the contributions of RebR to nodule formation, deletion of rebR from the WT strain did not alter the phenotypic expression of stem nodules, but abolished the Fix− phenotype of nodules harboring the ΔpraR mutant so that the ΔpraR ΔrebR mutant produced the Fix+ phenotype (Fig. 4A). R bodies were not observed in the ΔpraR ΔrebR cells in the stem nodules (Fig. 4B). However, in symbiotic stem nodules, reb operon expression in the ΔpraR mutant was more than 10-fold higher than that in the ΔpraR ΔrebR mutant and was about 2 orders of magnitude higher than that in the WT strain, whereas levels of reb operon expression were similar in the WT and ΔrebR mutant strains (Fig. 4C). In contrast, in the free-living state, reb operon expression in the ΔpraR mutant was similar to that in the ΔpraR ΔrebR mutant. These results indicate that although PraR predominantly represses the expression of the reb operon, RebR acts an activator of the operon under conditions of symbiosis.
FIG 4 .
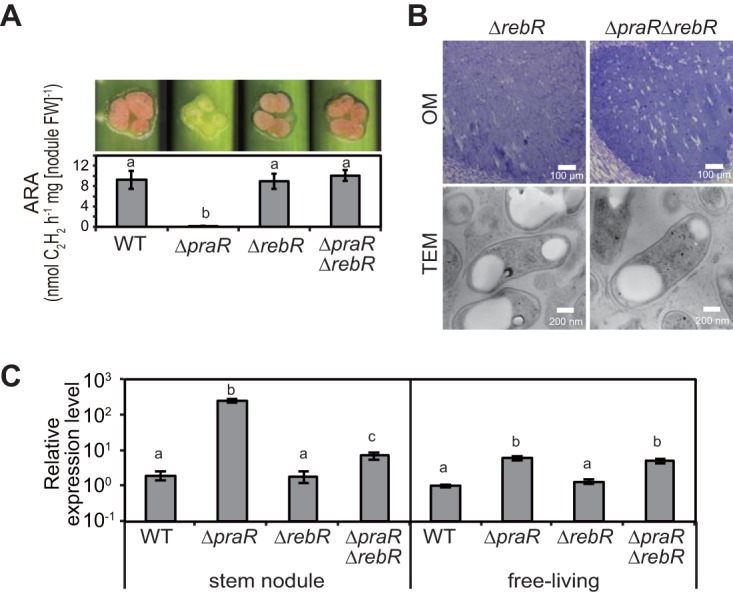
Phenotypes of praR and/or rebR mutants. S. rostrata plants were inoculated with the WT or ΔpraR, ΔrebR, or ΔpraR ΔrebR mutant strains and grown at 30°C, and stem nodules were analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of stem nodules. Values are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) OM observations of infected host cells in stem nodules and TEM observations of harbored bacterial cells. (C) Relative expression levels of the reb operon in stem nodules and free-living cultures. Total RNAs were isolated from bacteria residing in stem nodules and from bacterial cultures after growth to an OD600 of approximately 1.0 at 38°C. Expression levels of the reb operon were estimated using quantitative RT-PCR and were normalized to 16S rRNA. Data are presented as means ± standard deviations of three replicate cultures and plants and are expressed relative to mRNA levels in free-living WT cultures. Statistical analyses were carried out for stem nodules and free-living cultures, respectively. Different letters indicate significant differences (P < 0.05; Tukey-Kramer).
PraR and RebR directly control the reb operon.
Following a systematic evolution of ligands by exponential enrichment (SELEX) analysis, Frederix et al. (18) proposed that PraR of R. leguminosarum binds the consensus palindrome CAAC-N5-GTTG. In the present SELEX analysis using N-terminally His6-tagged PraR (His6-PraR) from A. caulinodans, the consensus PraR sequence of A. caulinodans was also CAAC-N5-GTTG (see Fig. S5A in the supplemental material). However, no sequence on the promoter region of the reb operon (reb promoter) matched this consensus sequence perfectly or with a base substitution, whereas four sequences matched with two or three substitutions, and these were examined as candidates for PraR binding sites. Subsequent electrophoretic mobility shift assays (EMSAs) revealed that His6-PraR binds strongly to a sequence designated PraR-bs-A and weakly to the sequences of PraR-bs-B and -C (Fig. S5B and C). Meanwhile, SELEX analysis using N-terminally His6-tagged RebR (His6-RebR) revealed that RebR potentially binds to a consensus palindrome, GT(A/G)(A/C)C-N4-G(T/G)(T/C)AC (Fig. S5D). A sequence (designated RebR-bs) matching this consensus palindrome was present on the reb promoter, and the EMSA revealed that His6-RebR binds to this sequence (Fig. S5E). The positions of PraR-bs-A, -B, and -C and RebR-bs on the reb promoter are shown in Fig. 1.
Determination of PraR- and RebR-binding sites on the reb promoter. (A) SELEX experiment with His6-PraR of A. caulinodans. A dsDNA library containing a 20-bp random sequence flanked by M13 forward and M13 reverse sequences was prepared by PCR using the primer pair M13F-M13R and the oligonucleotide 5′-TCGAGCTCGGTACCCGACCAGACTG-N20-GTATGTGCGTGGGGATCCTCTAGAG-3′ as a template. The dsDNA library (10 pmol) was incubated with His6-PraR (100 pmol) in 100 µl of SELEX buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM MgCl2, 20 mM imidazole, 0.05% Tween 20) supplied with 1 mM DTT and 10 ng µl−1 poly(dI-dC) for 30 min at 26°C. Ni-charged magnetic beads (His Mag Sepharose Ni; GE Healthcare) were used to capture His6-tagged proteins. Specifically, samples were combined with 1 µl of the magnetic beads (corresponding to 20 µl of 5% medium slurry), which were equilibrated in advance using SELEX buffer and incubated for 30 min at 26°C with gentle mixing by rotation. The beads capturing Hig6-tagged proteins were washed 10 times with 500 µl of the SELEX buffer. After the final washing, the beads were suspended in 50 µl of the SELEX buffer and were incubated at 95°C for 5 min. The eluted dsDNA was diluted 100 times with 10 mM Tris-HCl (pH 8.0) and amplified by PCR using primer pair M13F-M13R for subsequent rounds of selection. After five SELEX rounds, PCR products were cloned into pUC18 (C. Yanisch-Perron, J. Vieira, and J. Messing, Gene 33:103–119, 1985) using the In-Fusion cloning kit (Clontech), and variable regions (A-N20-C) of 48 clones were sequenced. All sequences were aggctaCAACtctagGTTGtgc. This sequence was aligned with the consensus palindrome sequence proposed by Frederix et al. (M. Frederix, A. Edwards, C. McAnulla, J. A. Downie, Mol Microbiol 81:994–1007, 2011, https://doi.org.10.1111/j.1365-2958.2011.07738.x.). No reb promoter sequences matched perfectly or had only one base difference from this consensus sequence, whereas four sequences with two or three different nucleotides (candidates 1, 2, 3, and 4) were present and were considered candidate PraR binding sites. (B) EMSA using FITC-labeled dsDNA probes that contained these candidate sequences. Probes were prepared by PCR using the primer pair M13F-M13R_FITC and oligonucleotides with candidate sequences flanked by M13 forward and reverse sequences. His6-PraR bound strongly to the candidate 2 sequence and weakly to candidate 3 and 4 sequences. His6-PraR did not bind the candidate 1 sequence. Thus, we designated the candidate sequences 2, 3, and 4 as PraR binding sites A, B, and C, respectively. (C) EMSA using FITC-labeled dsDNA probes with point mutations. Probes were prepared by PCR using oligonucleotides of the indicated sequences flanked by M13-forward and M13-reverse sequences and verified that PraR specifically binds these three sites. (D) SELEX experiment with His6-RebR of A. caulinodans. PCR products were cloned into pUC18 after five SELEX rounds, and variable regions of 32 clones were sequenced. Six sequences were thus obtained, and alignments of these suggested that RebR binds the consensus palindrome GT(A/G)(A/C)C-N4-G(T/G)(T/C)AC. A sequence matching this consensus with one different base was present on the reb promoter, and we considered this as a candidate RebR binding site. (E) EMSA using FITC-labeled dsDNA probes that contained this candidate sequence or point-mutated sequences verified that RebR specifically binds this sequence. Download FIG S5, PDF file, 0.3 MB (268.7KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To investigate whether PraR and RebR actually bind to the reb promoter, we carried out EMSAs using a double-stranded DNA (dsDNA) probe that covers the intergenic noncoding region upstream of the reb operon (reb promoter dsDNA probe) as shown in Fig. 5A. The molecular weight (MW) of the reb promoter dsDNA probe increased with the increasing quantity of His6-PraR, whereas addition of His6-RebR to the promoter probe increased not only the MW of the probe but also the amount of stacked probe in the wells of the gel in a concentration-dependent manner (Fig. 5B). Even in the presence of His6-PraR, the addition of His6-RebR to the promoter probe increased the MW of the probe (Fig. 5C), indicating that PraR does not interfere the binding of RebR to the RebR-bs.
FIG 5 .
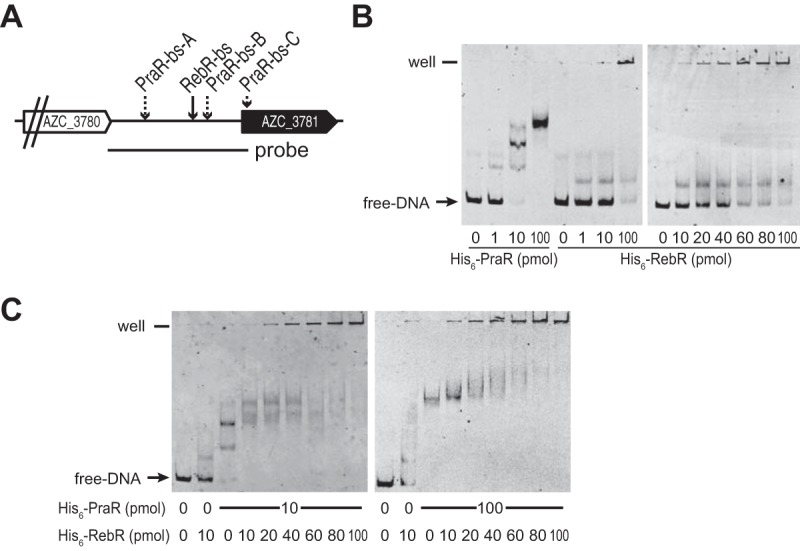
In vitro binding activities of His6-PraR and His6-RebR to the reb promoter. (A) The region of the FITC-labeled dsDNA probe used in EMSA analyses. (B) EMSA analysis of dsDNA probes with increasing levels of purified His6-PraR or His6-RebR. (C) EMSA analysis of dsDNA probes with both His6-PraR and His6-RebR.
To confirm the involvement of PraR and RebR in reb operon expression, the significance of the promoter sequences was assessed in stem nodules that were formed after inoculation with mutants carrying base substitutions of Preb(PraR-bs-A−) and Preb(RebR-bs−) mutants and after inoculation with the Preb(PraR-bs-A− RebR-bs−) double mutant (Fig. 6A). Stem nodules harboring the Preb(PraR-bs-A−) mutant had the Fix− phenotype, whereas those harboring the Preb(PraR-bs-A− RebR-bs−) double mutant had restored nitrogen-fixing activity (Fig. 6B). Accordingly, R bodies were observed in Preb(PraR-bs-A−) mutants but not in Preb(PraR-bs-A− RebR-bs−) double mutants in stem nodules (Fig. 6C). In further experiments, expression levels of the reb operon in Preb(PraR-bs-A−) mutants and Preb(PraR-bs-A− RebR-bs−) double mutants were about 2 orders of magnitude and several times higher than those in the WT strain, respectively, whereas reb operon expression levels in the Preb(RebR-bs−) mutant were similar to those in the WT strain (Fig. 6D). The consistency of phenotypic expression between deletion mutants of praR and rebR (ΔpraR, ΔrebR, and ΔpraR ΔrebR mutants in Fig. 4) and corresponding promoter sequence mutants strongly indicates that PraR and RebR directly control the expression of the reb operon.
FIG 6 .

Phenotypes of PraR-bs-A and/or RebR-bs mutants. (A) Mutants with base substitutions in PraR-bs-A [Preb(PraR-bs-A−) mutant], RebR-bs [Preb(RebR-bs−) mutant], or both genes [Preb(PraR-bs-A− RebR-bs−) double mutant] on the reb promoter. The sequences of the PraR binding site A and RebR binding site regions are shown in the right-hand side. The sequences corresponding to consensus palindromes are indicated in capital letters. Restriction endonuclease recognition sites (XbaI and EcoRI) were generated using the base substitutions. Mutants were inoculated into stems of Sesbania plants grown at 30°C, and stem nodules were analyzed at 14 dpi. (B) Hand-cut images (upper) and ARAs (lower) of the stem nodules formed by the indicated strains. Data are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (C) OM observations of infected host cells in stem nodules and TEM observations of residing symbiotic bacterial cells. Arrowheads indicate R bodies. (D) Relative expression levels of the reb operon in bacteria from stem nodules and free-living cultures. Total bacterial RNA was isolated from bacteria that were grown in stem nodules and from free-living cultures grown in the defined medium at 38°C to an OD600 of approximately 1.0. Expression levels of the reb operon were estimated using quantitative RT-PCR, normalized to 16S rRNA levels, and expressed relative to corresponding mRNA levels in free-living WT cultures and are presented as means ± standard deviations from three replicate cultures and plants. Statistical analyses were carried out for stem nodules and free-living cultures, respectively. Different letters indicate significant differences (P < 0.05, Tukey-Kramer).
Identification of environmental factors that abolish reb repression by PraR.
In the experiments described above, symbiotic R-body production was observed in ΔpraR and Preb(PraR-bs-A−) mutants but was not present in the WT strain under symbiotic or free-living conditions, suggesting that R-body production is subjected to environmental conditions. Thus, we investigated environmental factors that induce RebR-dependent activation of the reb operon in the ΔpraR mutant and then identified factors that attenuate PraR-dependent repression of the reb operon in WT cells grown under the identified favorable environmental conditions.
Initially, we constructed a reb operon-lacZ transcriptional fusion (reb-lacZ) on chromosomes of the WT and ΔpraR strains, namely reb-lacZ and reb-lacZ ΔpraR strains, respectively. During these manipulations, the reb-lacZ ΔpraR strain expressed β-galactosidase in the free-living state at room temperature (around 26°C) but not at 38°C, suggesting that activation by RebR is temperature dependent. Accordingly, when reb-lacZ and reb-lacZ ΔpraR strains were grown at various temperatures between 26 and 41°C, β-galactosidase activity was highly induced below 35°C in the reb-lacZ ΔpraR strain, but not in the reb-lacZ strain (Fig. 7A). Moreover, expression levels of the reb operon were about 30-fold higher at 26°C than at 38°C in the ΔpraR strain, whereas activation at 26°C was not observed in either the ΔrebR or ΔpraR ΔrebR mutant (Fig. 7B). Under free-living conditions, R bodies were observed in up to 10% of ΔpraR cells grown at 26°C, but not in those grown at 38°C (Fig. 7C). Similarly, ΔpraR cells in stem nodules (symbiotic state) failed to produce R bodies when plants were grown at 38°C, and reb operon expression was lower than that in ΔpraR cells under symbiotic conditions at 30°C (see Fig. S6 in the supplemental material). Taken together, these data indicate that activation of the reb operon by RebR is temperature dependent under both free-living and symbiotic conditions. However, binding of RebR to the reb promoter was not affected by temperature (see Fig. S7 in the supplemental material).
FIG 7 .
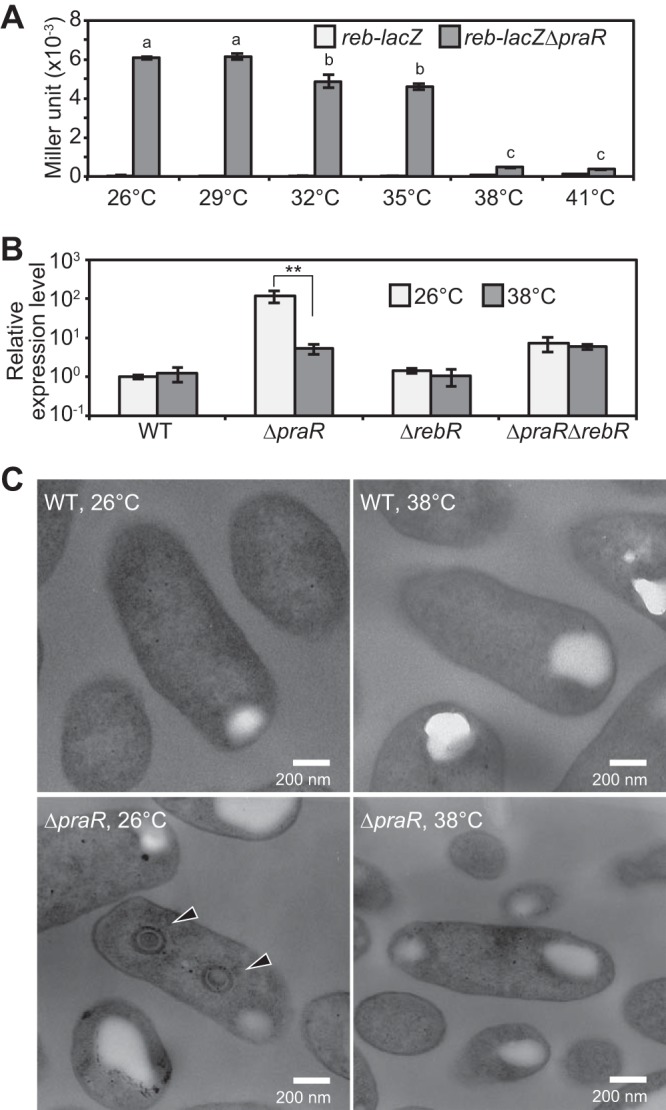
Effects of temperature on the expression of the reb operon and R-body formation in free-living A. caulinodans cells. (A) β-Galactosidase activities of reb-lacZ and reb-lacZ ΔpraR strains. Bacterial cells were grown in the defined medium at the indicated temperatures to an OD600 of approximately 1.0, and β-galactosidase activities were measured. Data are presented as means ± standard deviations from three replicate cultures. Statistical analysis was carried out for the reb-lacZ ΔpraR strain. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative RT-PCR analysis of the reb operon in the WT and ΔpraR, ΔrebR, and ΔpraR ΔrebR mutant strains grown at 26 and 38°C. Data are expressed relative to those from the WT strain grown at 26°C and are presented as means ± standard deviations from three replicate cultures. Statistical analyses were carried out for each strain, respectively. Asterisks indicate significant difference (**, P < 0.01, Student’s t test). (C) TEM observations of free-living WT and ΔpraR strains. Bacterial cells were collected from the same cultures that were used for the quantitative RT-PCR analyses described in panel B. Arrowheads indicate R bodies.
Effects of high temperature on the phenotypes of the stem nodules. S. rostrata plants were inoculated with WT and ΔpraR strains and were grown at 30 or 38°C. Stem nodules were then analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of the stem nodules. Data are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) OM observations of infected host cells in stem nodules and TEM observations of the bacterial cells in host cells. (C) Quantitative RT-PCR analysis of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels and are expressed relative to WT values at 30°C. Values are presented as means ± standard deviations from three separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). Download FIG S6, PDF file, 0.4 MB (408KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effects of temperature and 2OG on in vitro binding activities of His6-RebR to the reb promoter. (A) The region of the dsDNA probes. (B) Quantitative PCR analysis to investigate the effects of temperature on the reb promoter binding activities of His6-RebR. The dsDNA probes (1 pmol) corresponding to the reb promoter of WT and Preb(RebR-bs−) strains were incubated with purified His6-RebR (1, 10, or 100 pmol) in 50 µl of SELEX buffer supplied with 1 mM DTT and 10 ng µl−1 poly(dI-dC) for 1 h at 26 or 38°C. Samples were combined with 0.5 µl of Ni-charged magnetic beads (corresponding to 10 µl of 5% medium slurry of His Mag Sepharose Ni; GE Healthcare) and were incubated at 26 or 38°C for 1 h. Beads that captured His6-RebR were washed 10 times with 500 µl of SELEX buffer at 26 or 38°C. After the final wash, the beads were suspended with 50 µl of the SELEX buffer and incubated at 95°C for 5 min to recover dsDNA from His6-RebR. Recovered dsDNA contents in eluted solutions were measured by quantitative PCR. (C) EMSA analysis to investigate the effects of 2OG on the binding activities of His6-RebR to the reb promoter. An FITC-labeled dsDNA probe corresponding to the reb promoter was incubated with His6-RebR in the presence of 0 to 100 mM 2OG at 26°C. Download FIG S7, PDF file, 0.2 MB (179.9KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In subsequent experiments, the effects of host-derived tricarboxylic acid (TCA) cycle, nitrogen, and oxygen metabolites on reb expression were investigated in reb-lacZ and reb-lacZ ΔpraR strains. In the absence of praR (the reb-lacZ ΔpraR strain), all organic acids except for citrate promoted reb expression under nitrogen-sufficient conditions at either 21 or 3% oxygen (Fig. 8). However, even in the presence of praR (reb-lacZ strain), reb expression was induced in medium containing 2-oxoglutarate (2OG) with sufficient nitrogen sources (Fig. 8). Moreover, induction was increased with 2OG concentrations even in the presence of succinate as a carbon source (Fig. 9A), suggesting that 2OG induces reb irrespective of its metabolism as a carbon source, although these effects of 2OG were observed at 26°C but not at 38°C (Fig. 9B). In agreement, TEM observations showed that R bodies were produced in WT cells grown in the presence of 2OG at 26°C but not at 38°C (Fig. 9C). Taken with the absence of a response to 2OG in ΔrebR mutant cells (Fig. 9D), these observations indicate that rebR is essential for 2OG-mediated induction of the reb operon. Moreover, the ΔpraR mutants did not respond to 2OG, further suggesting that 2OG derepresses the reb operon by attenuating PraR-dependent repression. Western blotting of an rgs-his6-praR transformant (19) showed that expression levels of RGS-His6-PraR protein were not affected by 2OG (Fig. 9E). However, binding of PraR to the reb promoter dsDNA probe decreased with increasing 2OG concentration, whereas RebR binding was impervious to 2OG (Fig. 9F; Fig. S7). These observations strongly suggest that 2OG derepresses the reb operon directly by concentration-dependently inhibiting PraR binding to the reb promoter.
FIG 8 .

Effects of carbon and nitrogen sources and oxygen concentrations on the expression of the reb operon under free-living conditions. Mutant reb-lacZ and reb-lacZ ΔpraR strains were grown in medium containing 50 mM pyruvate, citrate, 2-oxoglutarate (2OG), succinate, fumarate, or malate as carbon sources and 10 mM NH4+ or NO3− as nitrogen sources or without a nitrogen source (−N) under aerobic (21% O2) or microaerobic (3% O2) conditions. Initial OD600 values of cultures were adjusted to 0.02 for pyruvate, succinate, fumarate, and malate media with NH4+ or NO3− supplementation, 0.1 for supplementation with 2OG and NH4+ or NO3−, and 0.2 for supplementation with citrate medium and NH4+ or NO3− or nitrogen-deficient media. After incubation at 26°C for 24 h, β-galactosidase activities were measured. Data are presented as means ± standard deviations from three replicate cultures. Different letters indicate significant differences (P < 0.05, Tukey-Kramer).
FIG 9 .

Effects of 2OG on the expression of the reb operon and R-body formation in bacteria under free-living conditions. (A) β-Galactosidase activities of the reb-lacZ strain in the presence of various 2OG concentrations. The reb-lacZ strain was grown in medium with the indicated concentrations of 2OG (0 to 50 mM) at 26°C for 24 h to an OD600 of approximately 1.0. Data are presented as means ± standard deviations from three replicate cultures. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative RT-PCR analysis to investigate the effects of 2OG and temperature on the reb operon expression in the WT strain. Cultures of the WT strain were supplemented with 10 mM 2OG (+2OG) or no 2OG (−2OG) and were grown at 26 or 38°C. The data are expressed relative to those from the cultures under –2OG conditions at 26°C and are presented means ± standard deviations from three replicate cultures. Asterisks indicate significant difference (**, P < 0.01, Student’s t test). (C) TEM observations of the WT cells cultured with 2OG at 26 and 38°C. Bacterial cells were collected from the cultures that were used for the quantitative RT-PCR conducted in panel B. Arrowheads indicate R bodies. (D) Quantitative RT-PCR analysis of the reb operon in the WT, ΔpraR, ΔrebR, and ΔpraR ΔrebR strains. These strains were grown under +2OG or –2OG conditions at 26°C. The data are expressed relative to corresponding values in the WT strain under –2OG conditions at 26°C and are presented as means ± standard deviations from three replicate cultures. Asterisks indicate significant difference (**, P < 0.01, Student’s t test). (E) Western blotting with an anti-His5 antibody to investigate the effects of 2OG on RGS-His6-PraR expression. Whole-cell lysates from the rgs-his6-praR strain grown under +2OG or –2OG conditions and from the WT strain grown under –2OG conditions were electrophoresed. (F) EMSA analysis to investigate the effects of 2OG on binding activities of His6-PraR to the reb promoter. FITC-labeled dsDNA probe corresponding to the reb promoter was incubated with the purified His6-PraR in the presence of 2OG (0.01 to 10 mM) or succinate (10 mM).
DISCUSSION
In this study, we demonstrated that reb-driven pathogenicity is associated with R-body production by A. caulinodans and suggest that R-body production is a widespread trait among bacterial pathogens that carry reb operons. Although the reb operon is predominantly repressed by PraR, we identified biological and environmental factors that derepress reb gene expression and thus R-body production under free-living conditions.
R bodies are rolled up at neutral pH, but reportedly unroll to form needle-shaped structures at low pH (10). Recombinant R bodies from Escherichia coli act as pistons that puncture spheroplasts of E. coli at low pH (20). In paramecia, the role of R bodies in the killer trait follows release of R-body-containing bacteria from killer paramecia and ingestion by sensitive paramecia. Subsequently, internalized bacteria enter acidified food vacuoles, and R bodies are unrolled and penetrate the phagosomal membrane to deliver lethal toxins to the cytoplasm (9, 21). This scenario may also be applicable to interactions of A. caulinodans and S. rostrata, in which the peribacteroid space (microenvironment surrounding bacteroids) is progressively acidified during nodule morphogenesis (22), likely triggering the conformational change of R bodies into the needle-shaped structure that penetrates membranes.
The present series of mutant analyses showed that essential genes for R-body formation in A. caulinodans include both reb-homologous and non-reb-homologous genes. Although the roles of the proteins encoded by the reb-homologous genes remain poorly understood, they are likely to be components of R bodies (5). Among non-reb-homologous genes, AZC_3784 was found in the reb operon and was also essential to R-body formation. In C. taeniospiralis, RebC is encoded by a non-reb-homologous gene that may be involved in the assembly of R bodies (5). Similarly, the AZC_3784 protein is not a component of R bodies but is likely involved in their assembly, although it may lack homology to rebC from C. taeniospiralis. Taken together, these observations warrant further compositional analyses of R bodies and investigations of the molecular mechanisms of R-body formation.
R bodies were frequently produced by ΔpraR mutants in the symbiotic state, but were observed in fewer than 10% of free-living cells, even at the optimum temperature (26°C) for reb operon expression. These results suggest that R-body formation is more strongly regulated (suppressed) in the free-living state—probably at the translation level or at the R-body assembly level—and that A. caulinodan R bodies play more important roles in the symbiotic state.
Although R-body formation was not observed in WT bacterial cells in the symbiotic state, the present environmental factors that derepress the praR regulatory system have significant implications for the understanding of reb operon evolution during microsymbiosis of A. caulinodan with S. rostrata. It is widely accepted that virulence genes are regulated by temperature in many pathogenic bacteria and are upregulated in mammalian bacterial pathogens at around host body temperature (37°C) (23, 24). In contrast, most plant-pathogenic bacteria express virulence genes at ambient temperatures that are generally lower than their optimal growth temperatures (25). For example, Agrobacterium tumefaciens mediates the formation of crown galls at temperatures below 32°C, and the VirA/VirG two-component system regulates the expression of virulence genes according to temperature (26, 27). In agreement, the reb operon was expressed in the ΔpraR mutant of A. caulinodans at temperatures below 35°C and within the optimal range for the growth of the host plant (around 30°C). Moreover, because regulation by PraR was derepressed by 2OG in the present free-living WT cells at 26°C, the reb operon may also be induced during symbiosis in host nodules, wherein the 2OG is accumulated in the host plant cells, although we have not found the conditions under which 2OG actually accumulates in the host plant cells. Plant 2OG levels reflect cellular C/N status and may play a signaling role in the coordination of C and N metabolism (28). Alterations in the activities of nitrogen fixation by bacteria and ammonia assimilation by plant cells may lead to 2OG accumulation in host cells. To elucidate the roles of reb operon in the symbiotic state, we need to conduct more investigations to estimate the conditions wherein 2OG is accumulated in the host plant cells.
Although PraR homologues are widespread among Alphaproteobacteria (8), the roles of PraR have not been well characterized. In particular, the praR homologue phrR was originally identified in the acid-tolerant rhizobium Sinorhizobium medicae WSM419 as a gene that is induced at low pH (29). However, praR expression is not pH sensitive in A. caulinodans and Rhizobium leguminosarum (8, 18). Moreover, R. leguminosarum PraR directly represses the expression of the quorum-sensing genes rhiR and raiR and the biofilm formation genes rapA2, rapB, and rapC (18, 30), whereas the homologous genes in A. caulinodans are not controlled by PraR (8). Hence, although PraR homologues are widely distributed, the roles of PraR have diversified during the evolution of Alphaproteobacteria. The ubiquity of chemical and environmental factors that regulate praR expression in the Alphaproteobacteria, such as the effects of A. caulinodans factors, also requires investigation in the context of the evolution of praR regulatory systems.
RebR belongs to a novel subfamily of the Crp-Fnr superfamily, and all Crp-Fnr members carry putative DNA-binding helix-turn-helix domains on their C terminus and ligand-binding domains on their N terminus (17). Various intracellular and exogenous signals activate Crp-Fnr members via their ligand-binding domains, including 2OG and temperature (17). In A. caulinodans, however, binding activity of RebR to the reb operon was not affected by 2OG and temperature, indicating that in addition to 2OG and temperature, as yet unidentified factors are involved in the activation of reb operon expression via RebR.
Herein, we demonstrated the roles of R bodies in the pathogenicity of bacteria that harbor the reb operon, although the ensuing roles in nodule symbiosis and the related evolutionary implications remain uncharacterized. Because bacterial genomes are plastic, endosymbionts may become pathogenic after acquiring the reb operon if they do not suppress its expression. Although we did not determine whether R bodies threaten biodiversity or ecosystems, this possibility may require solutions in the future. Unlike obligate endosymbionts of paramecia, A. caulinodans can be cultured in vitro and genetic manipulation techniques have been established in this bacterium, warranting further use of A. caulinodans as a model for studies of R-body/reb genes.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are listed in Table S1 in the supplemental material. A. caulinodans strains were grown in tryptone-yeast extract (TY) medium (31) or in basal defined (BD) medium containing 50 mM disodium succinate, 10 mM NH4Cl, 10 mM potassium phosphate (pH 7.0), 100 mg liter−1 MgSO4⋅7H2O, 50 mg liter−1 NaCl, 40 mg liter−1 CaCl2⋅2H2O, 5.4 mg liter−1 FeCl3⋅6H2O, 5 mg liter−1 Na2MoO4⋅2H2O, 2 mg liter−1 biotin, 4 mg liter−1 nicotinic acid, and 4 mg liter−1 pantothenic acid. To vary the carbon and nitrogen contents of BD medium, disodium succinate was replaced with sodium pyruvate, trisodium citrate, disodium 2-oxoglutarate, disodium fumarate, or disodium l-malate, and NH4Cl was replaced with KNO3 or omitted. In some experiments, BD medium was further supplemented with disodium 2OG. To grow A. caulinodans strains under aerobic conditions, test tubes containing medium were sealed with butyl rubber septums, and the contained air was replaced with N2 gas with 3% O2. Before inoculation into BD medium, bacterial cells were cultured overnight in TY medium and were washed twice in 10 mM potassium phosphate buffer (pH 7.0). Unless otherwise noted, initial optical density at 600 nm (OD600) values of cultures were adjusted to 0.1 or 0.02 for growth at 26 or 38°C, respectively, and OD600 values were approximately 1.0 after 24 h of incubation.
Bacterial strains and plasmids used in this study. Download TABLE S1, DOCX file, 0.1 MB (132.8KB, docx) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Construction of deletion and substitution mutants.
The plasmids and primers used for strain construction are listed in Tables S1 and S2 in the supplemental material, respectively.
To construct A. caulinodans deletion mutants of AZC_3784, AZC_3785, AZC_3787, and rebR genes, two DNA fragments containing upstream and downstream regions of each gene were amplified from the WT genomic DNA by PCR using appropriate primer pairs and were then directionally cloned into a suicide vector, pK18mobsacB (32), using the In-Fusion cloning kit (Clontech, Mountain View, CA). The linearization of pK18mobsacB was performed by inverse PCR using the PrimeSTAR Max (TaKaRa-Bio, Shiga, Japan) with primer pair Tp73-Tp74. The resulting plasmids were conjugated into the WT or ΔpraR (8) strains via E. coli S17-1(λpir) (33), and gene deletions were introduced by allelic exchange.
Primers used in this study. Download TABLE S2, DOCX file, 0.1 MB (143.6KB, docx) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To construct deletion mutants of the rebAZC1, rebAZC2, rebAZC3, and rebAZC4 genes, a series of plasmids were constructed as follows. DNA fragments containing the WT AZC_3781-7 region with its upstream and downstream regions were amplified by PCR and cloned into the linearized pK18mobsacB. Genes on plasmids containing the WT region were deleted by inverse PCR using the PrimeSTAR Mutagenesis Basal kit (TaKaRa-Bio). To introduce double deletions, second inverse PCRs were conducted using plasmids harboring single mutations. Constructed plasmids were conjugated into the ΔpraR ΔAZC_3781-7 mutant, and deletion mutants were obtained after allelic exchange.
To construct mutants with base substitutions in PraR-bs-A and/or RebR-bs, a series of plasmids were constructed as follows. A DNA fragment containing the WT reb promoter region was amplified by PCR and cloned into the linearized pK18mobsacB. An XbaI site was generated within the PraR-bs-A on the plasmid containing the reb operon by inverse PCR using the PrimeSTAR Mutagenesis Basal kit. Similarly, an EcoRI site was generated within the RebR-bs on the plasmid containing the reb promoter. The resulting plasmids were conjugated into the WT strain, and mutants were obtained after allelic exchange.
To construct strains that express the reb-lacZ fusion gene, two fragments containing rebR and AZC_3789 and a lacZ fragment were amplified by PCR from the WT genomic DNA and the plasmid pTA-MTL (34), respectively. Fragments were then cloned into the linearized pK18mobsacB in the direction of the rebR, lacZ, and AZC_3789 fragments using the In-Fusion cloning kit. The plasmid containing reb-lacZ was conjugated into the WT and ΔpraR strains, and strains with lacZ at the position immediately downstream of the rebR open reading frame (ORF) were obtained after allelic exchange.
Plant growth and bacterial inoculation for nodule formation.
S. rostrata stems were inoculated with A. caulinodans strains as described previously (19) and were then grown at 30 or 38°C under a 24-h light regimen. Acetylene reduction activities (ARAs) of stem nodules were assayed as described previously (19).
Optical microscopy observation.
Stem nodules were longitudinally cut into three pieces. The middle pieces of each sample were chemically fixed with 4% paraformaldehyde and 2% glutaraldehyde, dehydrated through a graded ethanol series, and then embedded in Technovit 7100 (Heraeus Kulzer). The embedded samples were sliced into 5-µm sections, stained with 0.05% toluidine blue O, and then observed using a bright-field microscope (DMLB; Leica).
TEM observation.
Bacterial cells were collected from culture media by centrifugation. Stem nodules were cut into small pieces. These samples were chemically fixed as described above, postfixed with 2% OsO4, dehydrated through a graded ethanol series, and finally embedded in Spurr low-viscosity embedding medium (Polysciences, Warrington, PA). Embedded samples were then sliced into ultrathin (about 70-nm) sections, stained with uranyl acetate followed by lead citrate, and examined using a JEM-1010 electron microscope (JEOL, Tokyo, Japan) at an accelerating voltage of 100 kV.
β-Galactosidase assay.
β-Galactosidase activity was measured according to a previously reported method (35) with some modifications as follows. Fifty microliters of bacterial cultures was mixed with 450 µl of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4) supplied with 50 mM 2-mercaptoethanol and 0.001% sodium dodecyl sulfate (SDS) and 50 µl of chloroform. The mixture was vortexed for 30 s, and 50 µl of o-nitrophenyl-β-d-galactoside (ONPG [4 mg ml−1 in Z buffer]) was added. After incubation at 25°C, reactions were stopped by adding 250 µl of 1 M Na2CO3. Mixtures were centrifuged, and 200 µl of the supernatants was transferred to 96-well clear plates. Subsequently, 200 µl of the bacterial cultures was transferred to 96-well clear plates. The absorbance of the supernatant at 415 and 540 nm and optical density at 595 nm (OD595) of the cultures were measured using a microplate reader (680 XR; Bio-Rad, Hercules, CA).
Total RNA extraction and quantitative RT-PCR.
Total RNA was isolated from bacterial cultures and stem nodules, and cDNA was synthesized according to previously described methods (19). Quantitative PCR was performed with a LightCycler system (Roche, Basel, Switzerland) using the QuantiTect SYBR green PCR kit (Qiagen, Hilden, Germany) with primer pairs Acp326-Acp714 for the reb operon and Tp35-Tp36 for 16S rNRA. Standard curves were generated using genomic DNA that was isolated from the WT strain using the NucleoSpin tissue kits (Macherey-Nagel, Düren, Germany). To determine expression levels of the reb operon, copy numbers of reb operon transcripts were normalized to those of 16S rRNA.
Purification of His6-tagged PraR and RebR.
The praR ORF was amplified by PCR from the WT genomic DNA using primer pair Acp375-Acp161 and was then cloned into BamHI and XbaI restriction sites of the pCold I vector (TaKaRa-Bio, Shiga, Japan). The resulting plasmid was designated pTAC99. The rebR ORF was amplified by PCR from WT genomic DNA using primer pair Acp669-Acp670 and cloned into pCold I using the In-Fusion cloning kit (Clontech, Mountain View, CA). The linearization of pCold I was performed by inverse PCR using the PrimeSTAR Max (TaKaRa-Bio) with primer pair Tp78-Tp79. The resulting plasmid was designated pTAC133. pTAC99 and pTAC133 were transformed into E. coli BLR(DE3) (Novagen, Darmstadt, Germany). Subsequently, His6-PraR and His6-RebR were extracted and purified using Ni+-charged magnetic beads (His Mag Sepharose Ni; GE Healthcare, Little Chalfont, United Kingdom). Eluted proteins were then concentrated using Vivaspin 500 kits (molecular weight cutoff [MWCO] of 5,000; Sartorius, Göttingen, Germany) and were mixed with 5 volumes of 1.2× storage buffer (24 mM Tris-HCl [pH 8.0], 120 mM NaCl, 1.2 mM dithiothreitol [DTT], 60% glycerol).
EMSA of the reb promoter.
Fluorescein isothiocyanate (FITC)-labeled dsDNA probes corresponding to the reb promoter region were prepared as follows. Initially, the reb promoter region was amplified by PCR using primer pair Acp646-Acp702 and genomic DNA from WT or derivative strains. Acp702 has an M13 reverse sequence at the 5′ end. PCR products from each strain were purified using the MinElute PCR purification kit (Qiagen) and were used as the templates for the second round of PCR using primer pair Acp646-M13R_FITC. Finally, FITC-labeled PCR products were purified. EMSAs were then performed by incubating 1-pmol aliquots of FITC-labeled dsDNA probes with various amounts of His6-PraR and/or His6-RebR in 20 µl of EMSA buffer [20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 60 mM NaCl, 5 mM MgCl2, 1 mM DTT, 10 ng µl−1 poly(dI-dC), 4% glycerol, 0.05% Tween 20] for 30 min at 26°C. Binding reaction mixtures were then electrophoresed in 4% native polyacrylamide gels containing 2.5% glycerol in 0.5× Tris-borate-EDTA (TBE) buffer, and FITC-labeled DNAs were detected using the LAS3000 system (Fuji Film, Tokyo, Japan).
Western blotting analyses of RGS-His6-PraR.
Bacterial cells were collected from cultures by centrifugation, sonicated in phosphate-buffered saline (150 mM NaCl, 10 mM Na2HPO4, 20 mM NaH2PO4 [pH 7.4]), and then fractionated by SDS-PAGE using 14% polyacrylamide gels. Fractionated proteins were electroblotted onto polyvinylidene difluoride (PVDF) membranes, incubated with mouse anti-His5 antibody (Qiagen), and detected using horseradish peroxidase (HRP)-conjugated sheep anti-mouse antibodies (GE Healthcare) and the EzWestLumi Plus reagents (Atto, Tokyo, Japan).
ACKNOWLEDGMENTS
We thank Fumiyasu Endo and Rio Yoguchi for technical assistance.
This work, including the efforts of Toshihiro Aono, was funded by the Japan Society for the Promotion of Science (JSPS) (25450081 and 16K07638).
Footnotes
Citation Matsuoka J-I, Ishizuna F, Kurumisawa K, Morohashi K, Ogawa T, Hidaka M, Saito K, Ezawa T, Aono T. 2017. Stringent expression control of pathogenic R-body production in legume symbiont Azorhizobium caulinodans. mBio 8:e00715-17. https://doi.org/10.1128/mBio.00715-17.
REFERENCES
- 1.Anderson TF, Preer JR Jr, Preer LB, Bray M. 1964. Studies on killing particles from Paramecium: the structure of refractile bodies from kappa particles. J Microsc 3:395–402. [Google Scholar]
- 2.Pond FR, Gibson I, Lalucat J, Quackenbush RL. 1989. R-body-producing bacteria. Microbiol Rev 53:25–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonneborn TM. 1938. Mating types in Paramecium aureliae: diverse conditions for mating in different stocks; occurrence, number and interrelations of the types. Proc Am Philos Soc 79:411–434. [Google Scholar]
- 4.Dilts JA, Quackenbush RL. 1986. A mutation in the R body-coding sequence destroys expression of the killer trait in P. tetraurelia. Science 232:641–643. doi: 10.1126/science.3008334. [DOI] [PubMed] [Google Scholar]
- 5.Heruth DP, Pond FR, Dilts JA, Quackenbush RL. 1994. Characterization of genetic determinants for R body synthesis and assembly in Caedibacter taeniospiralis 47 and 116. J Bacteriol 176:3559–3567. doi: 10.1128/jb.176.12.3559-3567.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeblick J, Kusch J. 2005. Sequence, transcription activity, and evolutionary origin of the R-body coding plasmid pKAP298 from the intracellular parasitic bacterium Caedibacter taeniospiralis. J Mol Evol 60:164–173. doi: 10.1007/s00239-004-0002-2. [DOI] [PubMed] [Google Scholar]
- 7.Sohn JH, Lee JH, Yi H, Chun J, Bae KS, Ahn TY, Kim SJ. 2004. Kordia algicida gen. nov., sp. nov., an algicidal bacterium isolated from red tide. Int J Syst Evol Microbiol 54:675–680. doi: 10.1099/ijs.0.02689-0. [DOI] [PubMed] [Google Scholar]
- 8.Akiba N, Aono T, Toyazaki H, Sato S, Oyaizu H. 2010. phrR-like gene praR of Azorhizobium caulinodans ORS571 is essential for symbiosis with Sesbania rostrata and is involved in expression of reb genes. Appl Environ Microbiol 76:3475–3485. doi: 10.1128/AEM.00238-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raymann K, Bobay L-M, Doak TG, Lynch M, Gribaldo S. 2013. A genomic survey of Reb homologs suggests widespread occurrence of R-bodies in proteobacteria. G3 3:505–516. doi: 10.1534/g3.112.005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quackenbush RL, Burbach JA. 1983. Cloning and expression of DNA sequences associated with the killer trait of Paramecium tetraurelia stock 47. Proc Natl Acad Sci U S A 80:250–254. doi: 10.1073/pnas.80.1.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beier CL, Horn M, Michel R, Schweikert M, Görtz HD, Wagner M. 2002. The genus Caedibacter comprises endosymbionts of Paramecium spp. related to the Rickettsiales (Alphaproteobacteria) and to Francisella tularensis (Gammaproteobacteria). Appl Environ Microbiol 68:6043–6050. doi: 10.1128/AEM.68.12.6043-6050.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee KB, De Backer P, Aono T, Liu CT, Suzuki S, Suzuki T, Kaneko T, Yamada M, Tabata S, Kupfer DM, Najar FZ, Wiley GB, Roe B, Binnewies TT, Ussery DW, D’Haeze W, Herder JD, Gevers D, Vereecke D, Holsters M, Oyaizu H. 2008. The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genomics 9:271. doi: 10.1186/1471-2164-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreyfus B, Garcia JL, Gillis M. 1988. Characterization of Azorhizobium caulinodans gen. nov., sp. nov., a stem-nodulating nitrogen-fixing bacterium isolated from Sesbania rostrata. Int J Syst Bacteriol 38:89–98. doi: 10.1099/00207713-38-1-89. [DOI] [Google Scholar]
- 14.Dreyfus BL, Elmerich C, Dommergues YR. 1983. Free-living Rhizobium strain able to grow on N2 as the sole nitrogen source. Appl Environ Microbiol 45:711–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreyfus BL, Dommergues YR. 1981. Nitrogen-fixing nodules induced by Rhizobium on the stem of the tropical legume Sesbania rostrata. FEMS Microbiol Lett 10:313–317. doi: 10.1111/j.1574-6968.1981.tb06262.x. [DOI] [Google Scholar]
- 16.Tsien HC, Dreyfus BL, Schmidt EL. 1983. Initial stages in the morphogenesis of nitrogen-fixing stem nodules of Sesbania rostrata. J Bacteriol 156:888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Körner H, Sofia HJ, Zumft WG. 2003. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev 27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- 18.Frederix M, Edwards A, McAnulla C, Downie JA. 2011. Co-ordination of quorum-sensing regulation in Rhizobium leguminosarum by induction of an anti-repressor. Mol Microbiol 81:994–1007. doi: 10.1111/j.1365-2958.2011.07738.x. [DOI] [PubMed] [Google Scholar]
- 19.Nakajima A, Aono T, Tsukada S, Siarot L, Ogawa T, Oyaizu H. 2012. Lon protease of Azorhizobium caulinodans ORS571 is required for suppression of reb gene expression. Appl Environ Microbiol 78:6251–6261. doi: 10.1128/AEM.01039-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polka JK, Silver PA. 2016. A tunable protein piston that breaks membranes to release encapsulated cargo. Synth Biol 5:303–311. doi: 10.1021/acssynbio.5b00237. [DOI] [PubMed] [Google Scholar]
- 21.Jurand A, Rudman BM, Preer JR Jr. 1971. Prelethal effects of killing action by stock 7 of Paramecium aurelia. J Exp Zool 177:365–387. doi: 10.1002/jez.1401770311. [DOI] [PubMed] [Google Scholar]
- 22.Pierre O, Engler G, Hopkins J, Brau F, Boncompagni E, Hérouart D. 2013. Peribacteroid space acidification: a marker of mature bacteroid functioning in Medicago truncatula nodules. Plant Cell Environ 36:2059–2070. doi: 10.1111/pce.12116. [DOI] [PubMed] [Google Scholar]
- 23.Konkel ME, Tilly K. 2000. Temperature-regulated expression of bacterial virulence genes. Microbes Infect 2:157–166. doi: 10.1016/S1286-4579(00)00272-0. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro RS, Cowen LE. 2012. Thermal control of microbial development and virulence: molecular mechanisms of microbial temperature sensing. mBio 3:e00238-12. doi: 10.1128/mBio.00238-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smirnova A, Li H, Weingart H, Aufhammer S, Burse A, Finis K, Schenk A, Ullrich MS. 2001. Thermoregulated expression of virulence factors in plant-associated bacteria. Arch Microbiol 176:393–399. doi: 10.1007/s002030100344. [DOI] [PubMed] [Google Scholar]
- 26.Riker AJ. 1926. Studies on the influence of some environmental factors on the development of crown gall. J Agric Res 32:83–96. [Google Scholar]
- 27.Jin S, Song YN, Deng WY, Gordon MP, Nester EW. 1993. The regulatory VirA protein of Agrobacterium tumefaciens does not function at elevated temperatures. J Bacteriol 175:6830–6835. doi: 10.1128/jb.175.21.6830-6835.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lancien M, Gadal P, Hodges M. 2000. Enzyme redundancy and the importance of 2-oxoglutarate in higher plant ammonium assimilation. Plant Physiol 123:817–824. doi: 10.1104/pp.123.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeve WG, Tiwari RP, Wong CM, Dilworth MJ, Glenn AR. 1998. The transcriptional regulator gene phrR in Sinorhizobium meliloti WSM419 is regulated by low pH and other stresses. Microbiology 144:3335–3342. doi: 10.1099/00221287-144-12-3335. [DOI] [PubMed] [Google Scholar]
- 30.Frederix M, Edwards A, Swiderska A, Stanger A, Karunakaran R, Williams A, Abbruscato P, Sanchez-Contreras M, Poole PS, Downie JA. 2014. Mutation of praR in Rhizobium leguminosarum enhances root biofilms, improving nodulation competitiveness by increased expression of attachment proteins. Mol Microbiol 93:464–478. doi: 10.1111/mmi.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beringer JE. 1974. R factor transfer in Rhizobium leguminosarum. J Gen Microbiol 84:188–198. doi: 10.1099/00221287-84-1-188. [DOI] [PubMed] [Google Scholar]
- 32.Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pk18 and pk19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 33.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 34.Iki T, Aono T, Oyaizu H. 2007. Evidence for functional differentiation of duplicated nifH genes in Azorhizobium caulinodans. FEMS Microbiol Lett 274:173–179. doi: 10.1111/j.1574-6968.2007.00823.x. [DOI] [PubMed] [Google Scholar]
- 35.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Determination of transcription units and start sites of the reb operon in Azorhizobium caulinodans ORS571. (A) Map of the reb operon of A. caulinodans with target regions and primer positions for RT-PCR and primer extension analyses. (B) RT-PCR analysis to determine transcription units of the reb operon. cDNA was synthesized using total RNA (500 ng) from WT and ΔpraR strains grown in BD medium at 38°C. Subsequent PCRs were performed using the 20-fold-diluted cDNAs and WT genomic DNA (1 × 10−1 ng µl−1) as the templates with the indicated primer pairs. The result showed that AZC_3781 to AZC_3788 were transcribed in a polycistronic manner, indicating that they form an operon. (C) Primer extension analysis to determine transcription start sites of the reb operon. Total RNA isolated from the WT and ΔpraR strains was reverse transcribed using the SuperScript III (Invitrogen) with a fluorescein isothiocyanate (FITC)-labeled primer (P14_FITC). The sequencing reaction for ladders was performed using the Thermo Sequenase primer cycle sequencing kit (GE Healthcare) with the P14_FITC primer and the DNA fragment that was amplified by PCR from WT genomic DNA using primer pair P1-P2. Both primer extension products and sequencing reaction mixtures were electrophoresed in denaturing 6% polyacrylamide gels containing 8 M urea. Gels were scanned for FITC fluorescence using an FLA3000 system (Fujifilm). Transcription start sites are indicated by arrows in the resulting image. This result shows that the reb operon is transcribed from 25, 23, 22, and 19 bp upstream of the AZC_3781 start codon. Download FIG S1, PDF file, 0.2 MB (210.5KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenic relationships among proteins encoded by reb-homologous genes. BLASTp analyses were performed using deduced amino acid sequences encoded by rebA, rebB, and rebD on the pKAP298 plasmid of Caedibacter taeniospiralis and AZC_3781, AZC_3782, AZC_3783, and AZC_3786 of A. caulinodans ORS571 as queries (E < 0.01) in the National Center for Biotechnology Information (NCBI) server (http://www.ncbi.nlm.nih.gov/BLAST/). The resulting sequences were combined, and phylogenic analyses were performed using MEGA5 software (K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei, S. Kumar, Mol Biol Evol 28:2731–2739, 2011, https://doi.org.10.1093/molbev/msr121). Download FIG S2, PDF file, 0.2 MB (195.3KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Contributions of AZC_3784, AZC_3785, and AZC_3787 genes to R-body formation. S. rostrata plants were inoculated with the indicated strains and were grown at 30°C and stem nodules were then analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of stem nodules during symbiosis with the indicated strains. Data are presented as means ± standard deviations of five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative RT-PCR analysis of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels and are presented as means ± standard deviations from three separate plants relative to corresponding transcripts from the WT strain. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (C) Optical microscopic (OM) observations of infected host cells in stem nodules and transmission electron microscopic (TEM) observations of residing bacterial cells. Arrowheads indicate R bodies. Deletions of AZC_3785 and AZC_3787 did not suppress the defective nitrogen-fixing phenotype of stem nodules that lacked praR, whereas deletion of AZC_3784 partially restored nitrogen fixation, as indicated by slight red color of the inner region of nodules harboring the ΔpraR ΔAZC_3784 mutant. Expression levels of the reb operon were not affected by the AZC_3784 deletion, suggesting that partial suppression of the defective nitrogen-fixing phenotype of nodules was not caused by low expression of the reb operon. Host cells in nodules harboring the ΔpraR ΔAZC_3784 mutant were oval or elongated, and shrunken host cells were not observed. Bacterial cell densities of the ΔpraR ΔAZC_3784 mutant in host cells were not as high as for the WT strain. R bodies were not observed in ΔpraR ΔAZC_3784 mutant bacteria, suggesting that the AZC_3784 gene is essential for R-body formation. R bodies were observed in ΔpraR ΔAZC_3785 and ΔpraR ΔAZC_3787 mutant bacteria, suggesting that the AZC_3785 and AZC_3787 genes are not essential for R-body formation. Download FIG S3, PDF file, 1.2 MB (1.2MB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Contributions of rebAZC1, rebAZC2, rebAZC3, and rebAZC4 genes to R-body formation. S. rostrata plants were inoculated with the indicated strains and were grown at 30°C. Stem nodules were analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of stem nodules. Values are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) Quantitative RT-PCR analysis of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels. Values are presented relative to respective WT expression data as means ± standard deviations from three separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (C) OM observations of infected host cells in stem nodules and TEM observations of the bacterial cells in host cells. Arrowheads indicate R bodies. Stem nodules that harbored ΔpraR ΔrebAZC1 and ΔpraR ΔrebAZC2 showed defective nitrogen-fixing phenotypes, and R bodies were observed in the residing bacteria. The ΔpraR ΔrebAZC3, ΔpraR ΔrebAZC4, ΔpraR ΔrebAZC1 ΔrebAZC2, and ΔpraR ΔrebAZC3 ΔrebAZC4 mutants formed normal stem nodules, and R bodies were not observed. Expression levels of the reb operon remained high in normal nodules, eliminating the possibility that the absence of R bodies reflected repression of reb operon expression. These results suggest that rebAZC3 and rebAZC4 and either rebAZC1 or rebAZC2 are essential for R-body production. Download FIG S4, PDF file, 1.1 MB (1.1MB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Determination of PraR- and RebR-binding sites on the reb promoter. (A) SELEX experiment with His6-PraR of A. caulinodans. A dsDNA library containing a 20-bp random sequence flanked by M13 forward and M13 reverse sequences was prepared by PCR using the primer pair M13F-M13R and the oligonucleotide 5′-TCGAGCTCGGTACCCGACCAGACTG-N20-GTATGTGCGTGGGGATCCTCTAGAG-3′ as a template. The dsDNA library (10 pmol) was incubated with His6-PraR (100 pmol) in 100 µl of SELEX buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM MgCl2, 20 mM imidazole, 0.05% Tween 20) supplied with 1 mM DTT and 10 ng µl−1 poly(dI-dC) for 30 min at 26°C. Ni-charged magnetic beads (His Mag Sepharose Ni; GE Healthcare) were used to capture His6-tagged proteins. Specifically, samples were combined with 1 µl of the magnetic beads (corresponding to 20 µl of 5% medium slurry), which were equilibrated in advance using SELEX buffer and incubated for 30 min at 26°C with gentle mixing by rotation. The beads capturing Hig6-tagged proteins were washed 10 times with 500 µl of the SELEX buffer. After the final washing, the beads were suspended in 50 µl of the SELEX buffer and were incubated at 95°C for 5 min. The eluted dsDNA was diluted 100 times with 10 mM Tris-HCl (pH 8.0) and amplified by PCR using primer pair M13F-M13R for subsequent rounds of selection. After five SELEX rounds, PCR products were cloned into pUC18 (C. Yanisch-Perron, J. Vieira, and J. Messing, Gene 33:103–119, 1985) using the In-Fusion cloning kit (Clontech), and variable regions (A-N20-C) of 48 clones were sequenced. All sequences were aggctaCAACtctagGTTGtgc. This sequence was aligned with the consensus palindrome sequence proposed by Frederix et al. (M. Frederix, A. Edwards, C. McAnulla, J. A. Downie, Mol Microbiol 81:994–1007, 2011, https://doi.org.10.1111/j.1365-2958.2011.07738.x.). No reb promoter sequences matched perfectly or had only one base difference from this consensus sequence, whereas four sequences with two or three different nucleotides (candidates 1, 2, 3, and 4) were present and were considered candidate PraR binding sites. (B) EMSA using FITC-labeled dsDNA probes that contained these candidate sequences. Probes were prepared by PCR using the primer pair M13F-M13R_FITC and oligonucleotides with candidate sequences flanked by M13 forward and reverse sequences. His6-PraR bound strongly to the candidate 2 sequence and weakly to candidate 3 and 4 sequences. His6-PraR did not bind the candidate 1 sequence. Thus, we designated the candidate sequences 2, 3, and 4 as PraR binding sites A, B, and C, respectively. (C) EMSA using FITC-labeled dsDNA probes with point mutations. Probes were prepared by PCR using oligonucleotides of the indicated sequences flanked by M13-forward and M13-reverse sequences and verified that PraR specifically binds these three sites. (D) SELEX experiment with His6-RebR of A. caulinodans. PCR products were cloned into pUC18 after five SELEX rounds, and variable regions of 32 clones were sequenced. Six sequences were thus obtained, and alignments of these suggested that RebR binds the consensus palindrome GT(A/G)(A/C)C-N4-G(T/G)(T/C)AC. A sequence matching this consensus with one different base was present on the reb promoter, and we considered this as a candidate RebR binding site. (E) EMSA using FITC-labeled dsDNA probes that contained this candidate sequence or point-mutated sequences verified that RebR specifically binds this sequence. Download FIG S5, PDF file, 0.3 MB (268.7KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effects of high temperature on the phenotypes of the stem nodules. S. rostrata plants were inoculated with WT and ΔpraR strains and were grown at 30 or 38°C. Stem nodules were then analyzed at 14 dpi. (A) Hand-cut images (upper) and ARAs (lower) of the stem nodules. Data are presented as means ± standard deviations from five separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). (B) OM observations of infected host cells in stem nodules and TEM observations of the bacterial cells in host cells. (C) Quantitative RT-PCR analysis of the reb operon in stem nodules. Expression levels of the reb operon were normalized to 16S rRNA levels and are expressed relative to WT values at 30°C. Values are presented as means ± standard deviations from three separate plants. Different letters indicate significant differences (P < 0.05, Tukey-Kramer). Download FIG S6, PDF file, 0.4 MB (408KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Effects of temperature and 2OG on in vitro binding activities of His6-RebR to the reb promoter. (A) The region of the dsDNA probes. (B) Quantitative PCR analysis to investigate the effects of temperature on the reb promoter binding activities of His6-RebR. The dsDNA probes (1 pmol) corresponding to the reb promoter of WT and Preb(RebR-bs−) strains were incubated with purified His6-RebR (1, 10, or 100 pmol) in 50 µl of SELEX buffer supplied with 1 mM DTT and 10 ng µl−1 poly(dI-dC) for 1 h at 26 or 38°C. Samples were combined with 0.5 µl of Ni-charged magnetic beads (corresponding to 10 µl of 5% medium slurry of His Mag Sepharose Ni; GE Healthcare) and were incubated at 26 or 38°C for 1 h. Beads that captured His6-RebR were washed 10 times with 500 µl of SELEX buffer at 26 or 38°C. After the final wash, the beads were suspended with 50 µl of the SELEX buffer and incubated at 95°C for 5 min to recover dsDNA from His6-RebR. Recovered dsDNA contents in eluted solutions were measured by quantitative PCR. (C) EMSA analysis to investigate the effects of 2OG on the binding activities of His6-RebR to the reb promoter. An FITC-labeled dsDNA probe corresponding to the reb promoter was incubated with His6-RebR in the presence of 0 to 100 mM 2OG at 26°C. Download FIG S7, PDF file, 0.2 MB (179.9KB, pdf) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bacterial strains and plasmids used in this study. Download TABLE S1, DOCX file, 0.1 MB (132.8KB, docx) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers used in this study. Download TABLE S2, DOCX file, 0.1 MB (143.6KB, docx) .
Copyright © 2017 Matsuoka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.