

Abstract
Recently, a new variant of invasive lobular breast cancer (ILBC) with solid‐papillary‐like growth pattern has been described. We present a case of ILBC with solid‐papillary‐like growth pattern in the main tumour mass and classical invasive lobular growth pattern in adjacent satellite foci. The two tumour components were subjected to comprehensive molecular analyses. Both components were ER/PR‐positive, HER2‐negative, and showed a complete loss of E‐cadherin and beta‐catenin protein expression, as determined by immunohistochemistry. Gene expression profiling classified the main tumour and a satellite focus as luminal‐B and luminal‐A subtypes, respectively. Whole‐genome copy number profiles were highly similar in both tumour components. Shared copy number alterations (CNAs) included gains of chromosome 1q21.1–q43 and losses of chromosome 16q11.2–q24.3, the locus of the CDH1/E‐cadherin tumour suppressor gene. CNAs detected only in the main tumour included a gain of chromosome 20q12–q13.33 and a loss of chromosome 1p36.33–p34.3, which has recently been associated with the solid variant of ILBC. Next generation sequencing revealed an identical, truncating CDH1 mutation (p.G169fs*5) in both tumour components confirming a common clonal ancestry. In conclusion, we confirm the recently described variant of ILBC with solid‐papillary‐like growth pattern and provide evidence that it evolves from classical ILBC by subclonal evolution.
Keywords: lobular breast cancer, papillary breast cancer, clonal evolution, E‐cadherin, copy number profiling
Introduction
Breast cancer (BC) is classified according to histomorphological and molecular characteristics. Infiltrating lobular breast cancer (ILBC) is the most common special histological BC entity and accounts for 10–15% of all cases 1, 2, 3. In classical ILBC, tumour cells are often individually dispersed or arranged in single file linear cords. The typical morphological characteristics of classical ILBC are described in detail elsewhere 1, 4, 5, 6. The dispersed growth pattern renders ILBC difficult to detect by mammography 7. ILBC belongs to the luminal molecular subtype 8. Genomic imbalances are similar to those of other luminal BCs, but also show specific features. Recurrent alterations include gains of the long arm of chromosome 1 (1q) and losses of chromosome 6q, 11q, 13q, 16q, and Xq 3, 9, 10, 11, 12. The hallmark of ILBC is mutational inactivation of the CDH1 tumour suppressor gene, which is located on chromosome 16q22.1 and encodes the E‐cadherin cell adhesion molecule 1, 3, 13. Depending on the morphological stringency in the diagnosis of ILBC, lack of E‐cadherin expression is observed in 55–100% of cases 1, 3, 14. Of note, ILBC has a broad morphological spectrum. At least nine different non‐classical histological variants with partly overlapping cytological and/or architectural features have been described. These include pleomorphic, alveolar, trabecular, solid, apocrine, histiocytoid, signet ring cell‐rich, mucinous, and mixed‐type ILBC (recently reviewed in 1). A common denominator of all these morphological variants is that ILBC cells are discohesive and usually do not form higher architectural elements, such as tubular or papillary formations.
The assessment and categorisation of papillary lesions of the breast remains one of the most challenging areas in breast pathology 15. Papillary breast lesions comprise papilloma, papilloma with ductal carcinoma in situ, papillary ductal carcinoma in situ, and various distinctive forms of papillary BCs, which account for approximately 1% of all BC cases, and which are described in detail elsewhere 15, 16, 17. In brief, papillary BC encompasses encapsulated papillary breast cancer (EPBC), solid‐papillary breast cancer (SPBC), and invasive papillary breast cancer (IPBC), which are distinct entities defined by different histomorphological characteristics 17. EPBC forms an expansive nodule composed of delicate, arborising fibrovascular stalks, which are lined by carcinoma cells but lack a myoepihelial cell layer. The nodule is encased in a fibrous capsule. SPBC forms multiple, mostly smaller, nodules with coalescent papillary formations. EPBC or SPBC associated with invasive BC is diagnosed if the tumour margin is infiltrative rather than expansive 17. Papillary BCs belong to the luminal molecular subtype. Genomic imbalances are similar to those of other luminal BCs but also show specific features, including a lower frequency of gains of chromosome 1q and a lower frequency of losses of chromosome 6q 18. Importantly, papillary BCs are E‐cadherin‐positive and mutational alterations are mainly limited to the PIK3CA, AKT, and MET oncogenes 18.
Recently, Rakha et al have reported on three unusual cases of ILBC, which mimicked a solid‐papillary growth pattern and resembled EPBC 19. They have pointed out the difficult histological differential diagnosis and clinical implications of these cases. The present work confirms this new tumour variant and provides molecular evidence that ILBC with solid‐papillary‐like growth pattern evolves from classical ILBC by subclonal evolution.
Materials and methods
Tissue specimens and immunohistochemistry
Formalin‐fixed paraffin‐embedded (FFPE) tissue specimens were prepared and evaluated in the Institute of Pathology of the Hannover Medical School. Immunohistochemistry was performed on whole slide sections (1 µm) of FFPE tissue blocks on a Benchmark Ultra (Ventana, Tucson, USA) automated stainer. The CC1 mild protocol (Ventana) was used for antigen retrieval and the ultraView DAB kit (Ventana) for signal detection. Antibodies are detailed in supplementary material, Table S1. Written informed consent for case presentation was obtained by the corresponding patient. All analyses were conducted in accordance with the guidelines of the local ethics committee (MHH, Hannover, Germany).
DNA and RNA extraction
Tumour tissue with either solid‐papillary‐like growth pattern (main tumour) or classical lobular growth pattern (adjacent satellite focus) was marked on a HE‐stained section of a representative FFPE tissue block. Corresponding tissue areas were macrodissected with a precision surgical blade on blank sections (n = 35, 8 µm each) from this FFPE tissue block. Another HE stain prepared after cutting of blank sections confirmed unaltered tumour representation in the depth of the block. As the main tumour and the satellite focus were spatially separated, laser microdissection was not necessary to achieve sufficient purity of tumour tissue with either papillary/solid‐papillary‐like or lobular‐like growth pattern. Subsequently, DNA and RNA were extracted with the Maxwell RSC DNA FFPE kit (Promega, Madison, WI, USA) on a Maxwell RSC instrument according to the manufacturer's recommendations. DNA and RNA were quantified using a Qubit 2.0 fluorometer (Invitrogen, Darmstadt, Germany) and the Qubit dsDNA HS or dsRNA HS assay kits (LifeTechnologies, Carlsbad, CA, USA). Additional samples subjected to DNA extraction included a control sample macrodissected from the centre of the main tumour and a tumour‐free sentinel lymph node of the corresponding patient.
Gene expression profiling
Gene expression profiling was performed with extracted total RNA on a nCounter Flex analysis system (NanoString Technologies, Seattle, WA, USA), using the commercial Prosignia/PAM50 molecular subtyping and prognostication assay according to the manufacturers recommendations 20, 21. The risk of recurrence (ROR) score was calculated as described previously 20.
DNA copy number profiling
Whole‐genome DNA copy number (CN) profiling was performed with molecular inversion probe (MIP) arrays (OncoScan, Affymetrix, Santa Clara, CA, USA) and 80 ng total DNA, as described previously 22. In brief, MIPs were first annealed against genomic DNA. Then, samples were split to separate (A/T) and (G/C) channels. After circularisation, MIPs were linearized by cleavage and amplified by PCR. Another cleavage step with HaeIII divided amplicons into two fragments, which allowed hybridisation of the tags against OncoScan® arrays. Arrays were washed and stained using the GeneChip® Fluidics station 450 and scanned by the GeneChip® scanner 3000 7G (Affymetrix). OSCHP files were produced from the CEL files by OncoScan Console Software (version 1.3.0.39). Further analyses were carried out with Chromosome Analyses Suite (ChAS) software (version 3.1.1.27). Copy number variations were detected using the TuScan algorithm (Affymetrix). Filter settings implemented for annotation of aberrations required gains to span ≥50 markers and ≥50 kb size. Losses were required to span ≥25 markers and regions of loss‐of‐heterozygosity (LOH) were required to span ≥3.000 kb. The MIP array data series is deposited in the Gene Expression Omnibus (GEO) data base (accession number GSE94695). Clonal relatedness of CN profiles corresponding to tumour tissue with solid‐papillary‐like growth pattern (main tumour) or classical lobular growth pattern (adjacent satellite focus) was determined with the likelihood ratio (LR) method 23, 24 using the biostatistic R package 'clonality' provided by Ostrovnaya et al 25. In brief, this method is based on a similarity measure that takes into account both the correlation of the broad pattern of CN gains and losses across chromosome arms and the similarity of specific within‐arm CN changes 23, 24. The likelihood ratio quantifies the odds that the two tumours are clonal and is benchmarked against the distribution of LRs in pairs of independent tumours from a reference cohort 23, 24, 25. As a reference cohort of independent tumours, we utilized a series of n = 29 ER‐positive BCs from n = 29 different patients, which were analysed on the same MIP array platform (GSE83916) 22.
Next generation sequencing
Next generation sequencing (NGS)‐based mutational profiling and bioinformatic data analysis were performed as described previously 26. In brief, two commercially available NGS panels covering mutation hot‐spot regions of n = 52 cancer‐related genes (PIK3CA, ERBB2, TP53, MET, AKT, PTEN, RB1 EGFR, MAP2K1, FGFR1, FGFR2, FGFR3, ABL1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, DDR2, ERBB4, EZH2, FBWX7, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PTPN11, RET, SMAD4, SMARC1B, SMO, SRC, STK11, VHL) with 215 amplicons (Ion AmpliSeq™ cancer hot‐spot panel v2 and the Ion AmpliSeq™ colon/lung panel, LifeTechnologies, Carlsbad, CA, USA) were used. As the commercially available NGS panel did not cover the complete coding sequence of the CDH1 tumour suppressor gene, a customized CDH1 NGS panel was designed with Ion AmpliSeq™ Designer software (pipeline version 5.6). This panel covered the complete protein‐coding sequence of the CDH1 gene (16 exons, 882 codons), the 5´‐UTR sequence of exon 1 and the 3′‐UTR sequence of exon 16 with 26 amplicons. CDH1 amplicon primers are listed in supplementary material, Table S2. All DNA samples were sequenced with both panels on an Ion PGM System (LifeTechnologies, Carlsbad, CA, USA) using 318v2 chips. For the two commercial panels, mean (n = 4) mapped reads per sample was 389 788 (range 296 765–449 285), mean depths per base was 1 832x (range 1 498–2 178) and mean reads on target sequence was 97.69% (range 95.36–98.79%). For the customized CDH1 panel, mean (n = 4) mapped reads per sample was 341 727 (range 197 170–522 830), mean depths per base was 8 481x (range 4 665–13 457) and mean reads on target sequence was 93.07% (range 88.70–95.17%).
Microsatelitte instability
Microsatelitte instability (MSI) was evaluated with 10 microsatellite markers (APC, BAT‐25, BAT‐26, BAT‐40, D10S197, D13S153, D17S250, D18S58, D2S123, and MYCL1) and DNA extracted from tumour and normal lymph node tissue. First, a qPCR analysis of the APP gene (67 bp PCR product) was performed to adjust the DNA input amount for subsequent MSI analysis. APP qPCR primer sequences were 5′‐TCA GGT TGA CGC CGC TGT‐3′ (forward) and 5′‐TTC GTA GCC GTT CTG CTG C‐3′ (reverse). The APP qPCR probe sequence was 5′‐FAM‐ACC CCA GAG GAG CGC CAC CTG‐TAMRA‐3′. Next, MSI analysis was carried out by PCR‐based amplification of microsatellite markers and fragment length analysis of PCR products 27, 28, 29. PCR amplification was performed under the following conditions: 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, 55 °C for 45 s, 72 °C for 30 s and an end‐elongation step at 72 °C for 5 min, with previously published primer pairs 27, 28, 29. PCR product length was assessed on a GenomeLab™ GeXP genetic analysis system (Beckman Coulter, Brea, CA, USA).
Results
Histopathological findings on core needle biopsy
A 74‐year‐old female presented with a well‐circumscribed tumour in the upper/outer quadrant of the left breast. Mammography and Doppler ultra sound revealed a hypoechoic, highly vascularized mass lesion (Figure 1). Core needle biopsy (CNB) sampling resulted in significant bleeding. Histologically, the CNB specimen showed an epithelial neoplasm with cystic areas and necrotic debris in the centre, a fibrous capsule at the periphery and a solid‐papillary‐like histoarchitecture (Figure 2A–C). Delicate, partly coalescent fibrovascular stalks or bands were lined by comparatively small, monomorphic tumour cells with minimal to moderate atypia. Impaired cell cohesion was noted and was interpreted as mechanical or hypoxic injury. The immunophenotype was ER/PR‐positive, HER2‐negative, and the Ki67 index was approximately 35% (Figure 2D). The lesion lacked a CK5/14‐positive myoepithelial cell layer along the fibrous capsule. On CNB, the lesion was classified as BC of indeterminate histological type and EPBC was discussed as the most likely differential diagnosis.
Figure 1.
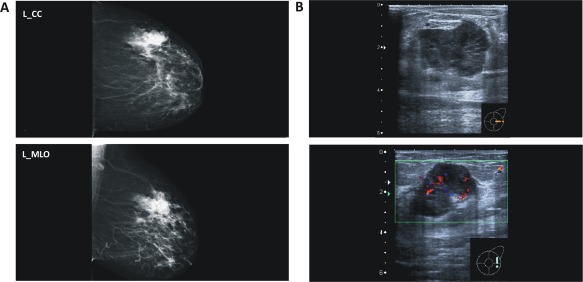
(A) Mammography and (B) Doppler ultrasound showing a highly vascularized mass lesion.
Figure 2.
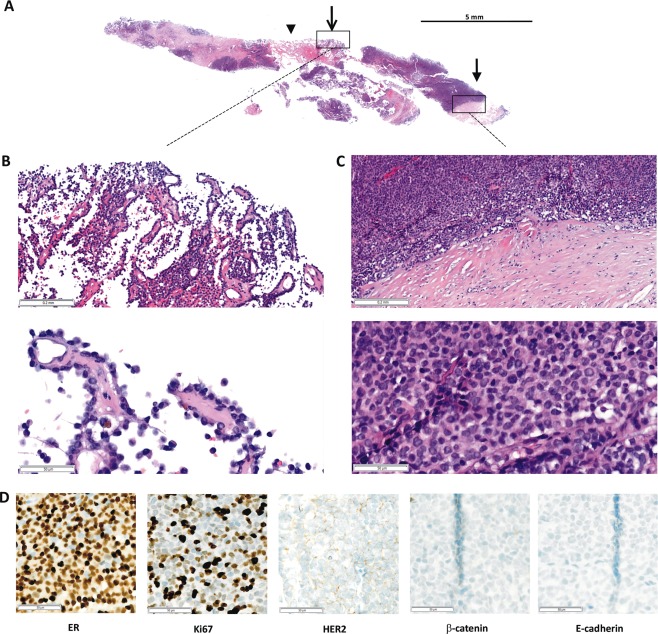
Core needle biopsy (CNB) specimen. (A) Submacroscopic view of the HE stained CNB. Arrow head, cystic areas, and necrotic debris in the centre; open arrow, papillary‐like growth pattern; closed arrow, solid‐papillary‐like growth pattern with coalescent fibrovascular stalks or bands, and the surrounding fibrous capsule. (B and C) Details at x50 and x200 magnification. (D) Immunohistochemical characteristics (x200 magnification). E‐cadherin and β‐catenin expression were determined after the resection specimen had been analysed and found to be E‐cadherin‐negative.
Histopathological findings in the resection specimen
Subsequently, the patient underwent mastectomy. The gross specimen showed a well‐circumscribed main tumour of 4.0 cm diameter and several adjacent invasive satellite foci of 0.5 cm diameter. The maximum diameter of the entire lesion was 5.5 cm. Similar to what was seen on CNB, the main tumour displayed a solid‐papillary‐like growth pattern and was embedded in dense connective tissue. The connective tissue was partly compressed to a fibrous capsule or pseudocapsule (Figure 3). Extensive sampling revealed that the capsule of the main tumour was focally infiltrated by tumour cells and showed a diminutive hemangiosis carcinomatosa. In this area of the main tumour, histomorphology resembled an ILBC of the solid variant (supplementary material, Figure S1).
Figure 3.
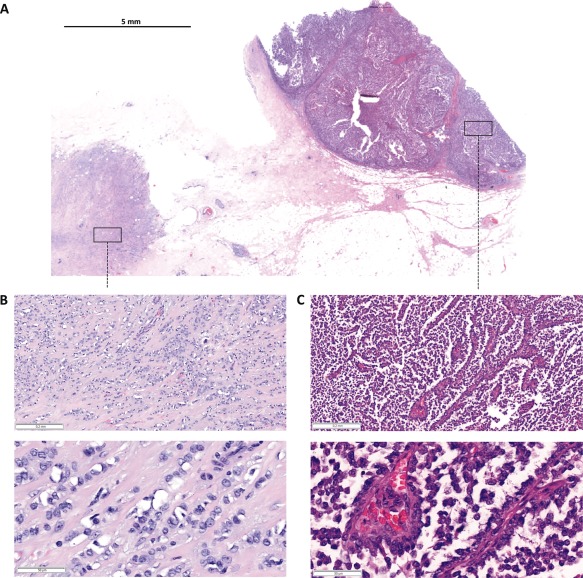
Resection specimen. (A) Submacroscopic view of a representative HE stained FFPE tissue block covering the margin of the main tumour and its capsule (right, solid‐papillary‐like growth pattern) and an adjacent satellite focus (left, classical lobular growth pattern). (B and C) Details at x50 and x200 magnification.
The invasive satellite foci adjacent to the main tumour showed a cell population with similar cytological features but with a classical lobular growth pattern (Figure 3). E‐cadherin immunohistochemistry was performed to rule out an EPBC‐ILBC collision tumour 30, 31, 32. This revealed a complete loss of E‐cadherin in the main tumour and in the satellite foci (Figure 4A and Table 1). Moreover, both tumour components were β‐catenin‐negative and thus displayed the typical immunophenotype of ILBC (Table 1) 33. This prompted a reanalysis of the tissue initially received by CNB. The solid‐papillary‐like tumour formations in the CNB also lacked E‐cadherin and β‐catenin expression (Figure 2D). Further immunophenotypic characterisation of the resection specimen showed that the Ki67 index was higher in the solid‐papillary‐like main tumour compared with the satellite foci (Table 1). The adjacent mammary gland tissue was atrophic, lacked lobular intraepithelial neoplasia (LIN), and showed no intraductal papilloma or related lesions. Tumour stage was reported as pT3, pN0 (0/2), V1, R0. Histological grading was controversial, but was considered as still compatible with G2. The histological type was reported as ILBC with combined classical and altered solid growth pattern, mimicking an encapsulated papillary carcinoma.
Figure 4.
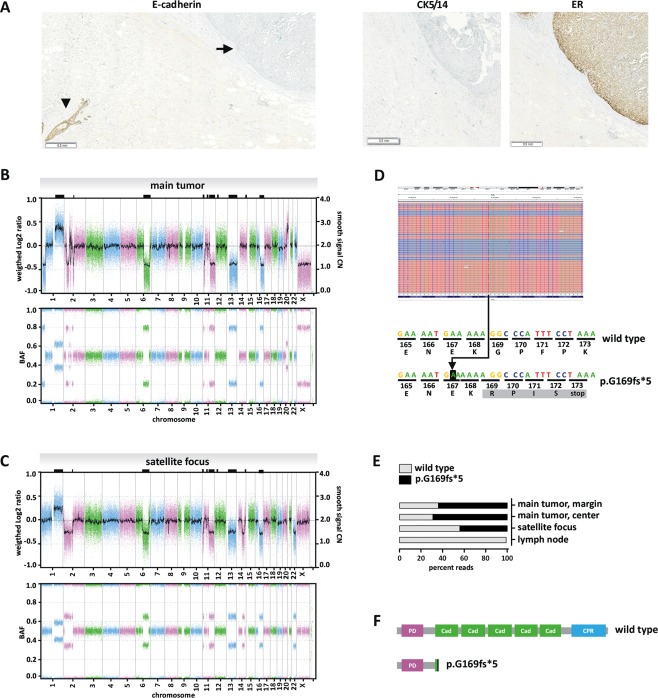
Molecular characteristics. (A) Loss of E‐cadherin protein expression in the solid‐papillary‐like main tumour. The FFPE block shown in Figure 2 was subjected to E‐cadherin immunohistochemical staining. E‐cadherin immunoreactivity was completely lost in the main tumour (closed arrow), while an adjacent normal mammary gland duct was strongly positive for E‐cadherin, which served as internal positive control (arrow head). Please note that the slide is presented at a submacroscopic view to demonstrate that the loss of E‐cadherin expression was complete and uniform. Immunohistochemical staining for ER is provided for comparison (right). Staining for CK5/14 is provided to demonstrate that the main tumour lacked a myoepithelial cell layer along the capsule. (B) Array‐based DNA copy number profiling of the main tumour with solid‐papillary‐like growth pattern. The upper panel presents a whole‐genome overview of copy number alterations (CNAs) with chromosomal locations plotted on the x‐axis. The weighted Log2 ratios and copy numbers (represented as a Gaussian smoothed calibrated copy number estimate) are plotted on the left and right y‐axis, respectively. The lower panel presents the corresponding B‐allele frequencies (BAF). (C) array‐based DNA copy number profiling of the satellite focus with classical lobular growth pattern. Shared CNAs are highlighted by black rectangles on the upper frame of the plot. (D) CDH1 mutational analysis by NGS. Sequencing reads of DNA from the solid‐papillary‐like main tumour were aligned with the IGV Browser, version 2.3.78 (upper panel). Reverse reads are shaded in blue, forward reads in red. Reads with an insertion variant are highlighted by a blue mark (upper panel). Sequence details are shown in the lower panel. The 'A' on black background corresponds to a c.499_500insA insertion mutation. The resulting frameshift generates an altered protein sequence (grey background) and a premature stop at codon 173. (E) Percentage of NGS reads carrying the c.499_500insA mutation in different DNA preparations. (F) Schematic presentation of the human E‐cadherin protein. PD; prodomain, Cad; cadherin domain; CPR, cytoplasmic region. The c.499_500insA mutation results in a heavily truncated E‐cadherin protein (p.G169fs*5).
Table 1.
Protein expression and mutational characteristics
| Core needle biopsy | Resection specimen | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Solid‐papillary‐like growth pattern | Main tumour Solid‐papillary‐like growth pattern | Satellite focus Classical lobular growth pattern | |||||||
| Immunohistochemistry | Status | Percent | Intensity | Status | Percent | Intensity | Status | Percent | Intensity |
| ER | pos | 100 | + + + | pos | 100 | + + + | pos | 100 | + + + |
| PR | pos | 95 | + + + | pos | 80 | + + | pos | 95 | + + + |
| HER2 | neg | 10 | + (i) | neg | 10 | + (i) | neg | 25 | + (i) |
| Ki67 | pos | 35 | + + + | pos | 35 | + + + | pos | 15 | + + + |
| CK5/14 | neg | 0 | ‐ | neg | 0 | ‐ | neg | 0 | ‐ |
| E‐cadherin | neg | 0 | ‐ | neg | 0 | ‐ | neg | 0 | ‐ |
| Beta‐catenin | neg | 0 | ‐ | neg | 0 | ‐ | neg | 0 | ‐ |
| p53 | neg | 15 | + | neg | 20 | + | neg | 5 | + |
| Expression profiling | |||||||||
| Subtype | na | Luminal‐B | Luminal‐A | ||||||
| ROR [score] | na | 88 | 53 | ||||||
| DRP [%] | na | 38 | 12 | ||||||
| NGS | |||||||||
| CDH1 | na | c.504_505insA | c.504_505insA | ||||||
| E‐cadherin | na | p.G169fs*5 | p.G169fs*5 | ||||||
ROR, risk of recurrence; DRP, distant recurrence probability; i, incomplete staining pattern.
Molecular findings
To further characterize this unusual BC, tumour tissue with either solid‐papillary‐like or classical lobular growth pattern was macrodissected from a representative FFPE block covering the margin of the main tumour and a nearby satellite focus (Figure 3). RNA and DNA were extracted from both tumour components and were subjected to comprehensive molecular analyses. Gene expression profiling classified the molecular subtype of the main tumour and the satellite focus as luminal‐B (ROR score 88) and luminal‐A (ROR score 53) respectively (Table 1).
MIP array analysis showed that whole‐genome DNA copy number profiles were highly similar in both tumour components, suggesting a common clonal origin (Figure 4B,C). Shared copy number alterations (CNAs) included gains of chromosome 1q21.1‐q43 and losses of chromosome 6q14.1–q25.1, 11q13.4–q25, 13q11–q34, and 16q11.2–q24.3 (supplementary material, Table S3). These shared CNAs have all been described as recurrent alterations in classical ILBC 3, 9, 10, 11. Loss of chromosome 16q11.2–q24.3 encompassed the locus of the CDH1/E‐cadherin tumour suppressor gene. CNAs detected only in the main tumour included a gain of chromosome 20q12–q13.33 and a loss of chromosome 1p36.33–p34.3. These CNAs have previously been associated with pleomorphic or solid ILBC 3, 12. The main tumour also showed a loss of chromosome X, which is common in ILBC 11. An additional sample macrodissected from the centre of the main tumour showed identical CNAs and thus verified these findings (supplementary material, Table S3). CNAs detected only in the satellite focus included losses of chromosomes 2p25.1–q12.3, 14q23.2–32.12, and 22q11.1–q13.33. CNAs characteristic for EPBC and SPBC (gains of chromosomes 18q21 and 19p13) were not detected (Figure 4B,C). The main tumour also showed CN losses on chromosome 2p, albeit in smaller regions and with interposed CN gains (Figure 4B). The B‐allele frequency indicated a BBA/BAA allele constellation in the interposed CN gains on chromosome 2, in the main tumour (Figure 4B, lower panel). Accordingly, chromosome 2p alterations have most likely developed independently in the main tumour. Gross loss of chromosome 2p (2p25.1–q12.3) was only detected in the satellite focus.
To analyse the CN profiles more comprehensively, we employed the statistical likelihood ratio (LR) method 23, 24, 25. The LR method determines clonality or independence of two given tumours based on the overall pattern of CN gains and losses 23, 24, 25. A series of n = 29 unrelated ER‐positive BCs served as a reference cohort, providing n = 464 independent/non‐clonal tumour pairs for this analysis 22. LR values, reflecting the odds that two paired tumours are clonal, ranged from 0.000003 to 120 in the reference cohort. The LR obtained by comparison of the solid‐papillary‐like main tumour and the adjacent satellite focus was > 5 × 105, confirming clonality of the two samples (p < 0.001) (supplementary material, Figure S2A).
Next, the mutational status of n = 52 oncogenes and tumour suppressor genes (CDH1, PIK3CA, AKT, MET, TP53, and others) was assessed by NGS using the same DNA preparations subjected to MIP array analysis. A unique CDH1/E‐cadherin mutation (c.499_500insA, p.G169fs*5, 64% mutated reads) was identified in the main tumour (Figure 4D). This insertion mutation results in a frameshift, a premature stop codon and a truncated E‐cadherin protein (Figure 4D,F). The same mutation was also detected in the satellite focus (44% mutated reads), which unambiguously confirmed a common clonal ancestry of the two morphologically distinct tumour components. Normal lymph node tissue from the same patient showed a wild type CDH1 sequence, which verified that the p.G169fs*5 mutation was a somatic alteration (Figure 4E). Except for CDH1, no other mutation was found in any of the n = 52 oncogenes/tumuor suppressor genes tested. Microsatellite instability (MSI) is generally rare in BC 34, 35. However, as the CDH1 mutation detected was an insertion mutation in a (A)5 mononucleotide tract, MSI was considered as a potential mutation mechanism. Therefore, PCR‐based MSI analysis was performed using a consensus panel of 10 microsatellite loci. This revealed an MSI‐low phenotype with instability at 2 of 10 microsatellite loci (D10S197 and MFD15) in both tumour components (supplementary material, Figure S2B).
Discussion
Recently, Rakha et al have described a new ILBC variant which mimics a solid‐papillary growth pattern and resembles EPBC 19. The present work confirms this recently described ILBC variant with solid‐papillary‐like growth pattern and provides further evidence that it evolves from classical ILBC by subclonal evolution.
The first of the three cases described by Rakha et al and the mammary carcinoma characterized here have several features in common 19. Both cases were initially considered as EPBC on CNB. Both cases were comparatively large and well‐circumscribed. Both cases featured a main tumour with a fibrous capsule or pseudocapsule and a peculiar form of altered solid growth mimicking a solid‐papillary‐like growth pattern. Rakha et al, pointed out that the well‐defined margin, the capsule, intratumoural cystic areas, and supporting fibrovascular cores distinguish this growth pattern from the solid variant of ILBC as described by Fechner et al 6, 19. In the present case, enhanced tumour vascularisation was also noted clinically on pre‐operative Doppler ultrasound and CNB sampling was associated with significant bleeding. Like in the first case reported by Rakha et al, the mammary carcinoma described here featured invasive satellite foci with classical lobular growth pattern as a minor tumour component. Consistent with the previous report 19, both tumour components showed a complete loss of E‐cadherin and β‐catenin expression. Our additional molecular analyses now showed that the solid‐papillary‐like main tumour was a divergent subclone derived from the same ILBC ancestral to the lobular satellite foci (Figure 5). This was proven by shared CNAs consistent with ILBC and identical somatic CDH1/E‐cadherin mutations detected in both tumour components.
Figure 5.
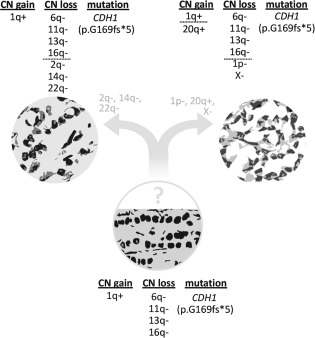
Schematic model for the subclonal evolution of the main tumour with solid‐papillary‐like growth pattern (upper right) from classical ILBC (lower centre). The changes found in the satellite focus are shown upper left. Genomic imbalances have been simplified. For comprehensibility, the complex pattern of copy number alteration of chromosome 2p has not been included in full detail.
The CDH1/E‐cadherin mutation identified in this case (p.G169fs*5) was essentially specific for this individual tumour. In the catalogue of somatic mutations in cancer (COSMIC), the p.G169fs*5 mutation currently accounts for only 1 of 581 listed somatic CDH1 alterations. Because of the rarity of this mutation, it can serve as a clonality marker. Interestingly, the p.G169fs*5 mutation corresponds to an adenine insertion in a (A)5 mononucleotide tract. This (A)5 mononucleotide tract can be interpreted as a short coding microsatellite (cMS) and MSI could be a mutation mechanism 34. Analysis of consensus (non‐coding) microsatellite markers demonstrated an MSI‐low phenotype in both tumour components. The MSI‐low phenotype is rare in BC (<5%) and its significance is controversial 34, 35. Mutation of classical MSI target genes has been reported for some tumours with MSI‐low characteristics 36. However, the global mutational burden is not significantly different in tumours with MSI‐low or MS‐stable phenotypes 34. This argues against a role of MSI for the CDH1/E‐cadherin mutation identified in the present case.
Interestingly, the solid‐papillary‐like main tumour displayed a higher Ki67 cell proliferation index, a higher ROR score and additional CNAs previously associated with pleomorphic and solid ILBC (gain of chromosome 20q12–q13.33 and loss of chromosome 1p36.33–p34.3, the locus of the ARID1A gene) 3. This supports the notion that subclonal evolution had driven the main tumour to become the dominant focus (Figure 5). Remarkably, the main tumour showed a fibrous capsule or pseudocapsule. Although infiltration of the capsule and focal hemangiosis carcinomatosa confirmed invasive capacity, the overall appearance was a well‐circumscribed, large, roundish tumuor nodule. This runs counter to the mechanistic view that ILBC cells are always highly motile because of their lack of E‐cadherin expression 37. In fact, the morphology suggests that the cells in the main tumour were spatially stationary. At least, the tumour cells grew more quickly at their initial location than they could spread into the surrounding tissue. Similar characteristics can be seen in alveolar ILBC, although the individual tumour foci are dimensions smaller 1.
EPBC and SPBC also show a fibrous capsule or encasement in connective tissue. This is the reason for their controversial status as either a variant of carcinoma in situ or a kind of indolent, minimally invasive BC 17. The current WHO classification (4th edition) defines these tumours as in situ lesions, albeit with an absent myoepithelial cell layer 16. Based on the observation of a fibrous capsule around ILBC with solid‐papillary‐like growth pattern, Rakha et al speculated that EPBC and SPBC are also invasive lesions 19. While there is no doubt that the unusual ILBCs described by Rakha et al and us were invasive BCs, it seems uncertain whether a comparison with EPBC and SPBC is valid.
Regarding the histological growth pattern, it may be surprising that E‐cadherin‐defective BC cells can form structures that superficially resemble a papillary‐like growth pattern. However, E‐cadherin expression is not a prerequisite for such a growth pattern. This is exemplified by clear cell sarcomas of the gastrointestinal tract, Frantz tumours of the pancreas, and papillary thyroid carcinomas (diffuse sclerosing variant), which all show papillary‐like growth pattern in the absence of E‐cadherin expression 38, 39, 40, 41. The term pseudopapillary growth pattern is occasionally used to describe these neoplasms. However, the use of this term is not without controversy 42. Rakha et al have considered but rejected complete overgrowth of an intraductal papilloma by LIN as a possible explanation for this unusual growth pattern in the mammary gland 19. With regard to our case, we concur with Rakha's view because of the complete absence of a myoepithelial cell layer, which is usually retained in a papilloma with overgrowth by LIN. In addition, in papillomas with overgrowth by LIN, neighbouring mammary gland lobules also show LIN. In our case, no LIN foci were detected in neighbouring mammary gland lobules. A more probable explanation for the solid‐papillary‐like growth pattern is enhanced neoangiogenesis combined with disassembly of extracellular matrix between sprouting vessels, which leaves thin fibrovascular bands lined by tumour cells running through cystic areas within a formerly solid ILBC. The thin fibrovascular bands cannot be distinguished with certainty from a true papillary growth pattern.
The frequency of ILBC with solid‐papillary‐like growth pattern remains unknown, but it may be very low. The cases described by Rakha et al and us are of importance as, although they do not allow derivation of a general biological principle, they do help to illustrate the heterogeneous presentation of ILBC in the clinic. The implication for BC diagnostics is that invasive ILBC with solid‐papillary‐like growth pattern is a differential diagnosis for EPBC and SPBC (considered in situ lesions), especially on CNB. Shedding and discohesive disarray of tumour cells lining fibrovascular bands may provide a useful diagnostic hint suggesting this ILBC variant. Immunohistochemistry for E‐cadherin is usually not necessary to establish the diagnosis of lobular BC with classical growth pattern. However, immunohistochemistry for E‐cadherin may be prudent in suspicious cases with a solid‐papillary‐like growth pattern. In summary, the present work confirms ILBC with solid‐papillary‐like growth pattern as a distinctive tumour variant and provides further evidence that it evolves from classical ILBC by subclonal evolution.
Author contributions statement
MC, FL and HK assessed histomorphological characteristics. JLvL, MS, SB and DS carried out MIP array analysis. BS and JLvL performed the LR method‐based statistical clonality tests. SB and UL performed NGS‐based mutational analyses. EP was responsible for gene expression profiling. SP and SK provided clinicopathological information. All authors contributed to final data analysis and interpretation. MC, SB, DS and HK wrote the manuscript.
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
Figure S1. Main tumour, infiltration of the capsule and haemangiosis
Figure S2. Likelihood ratio (LR) values (A) and MSI‐low phenotype (B) in both tumour components
Table S1. Antibodies and immunohistochemistry
Table S2. CDH1 PCR primer pairs
Table S3. Copy number alterations (CNAs) in both tumour components
Acknowledgements
The authors thank Henriette Christgen, Luisa Rieger, and Hildegard Frye‐Boukhriss for excellent technical assistance. The authors also thank Daniel Ulbrich‐Gebauer for gene expression profiling and helpful comments. JLvL was supported by the Hannover Biomedical Research School (HBRS) and the MD/PhD program molecular medicine. This work was supported by the Claudia von Schilling breast cancer research junior award granted to MC.
No conflicts of interest were declared.
References
- 1. Christgen M, Steinemann D, Kuhnle E, et al Lobular breast cancer: clinical, molecular and morphological characteristics. Pathol Res Pract 2016; 212: 583–597. [DOI] [PubMed] [Google Scholar]
- 2. Ciriello G, Gatza ML, Beck AH, et al Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015; 163: 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desmedt C, Zoppoli G, Gundem G, et al Genomic characterization of primary invasive lobular breast cancer. J Clin Oncol 2016; 34: 1872–1881. [DOI] [PubMed] [Google Scholar]
- 4. Martinez V, Azzopardi JG. Invasive lobular carcinoma of the breast: incidence and variants. Histopathology 1979; 3: 467–488. [DOI] [PubMed] [Google Scholar]
- 5. Hanby AM, Hughes TA. In situ and invasive lobular neoplasia of the breast. Histopathology 2008; 52: 58–66. [DOI] [PubMed] [Google Scholar]
- 6. Fechner RE. Histologic variants of infiltrating lobular carcinoma of the breast. Hum Pathol 1975; 6: 373–378. [DOI] [PubMed] [Google Scholar]
- 7. Johnson K, Sarma D, Hwang ES. Lobular breast cancer series: imaging. Breast Cancer Res 2015; 17: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weigelt B, Geyer FC, Natrajan R, et al The molecular underpinning of lobular histological growth pattern: a genome‐wide transcriptomic analysis of invasive lobular carcinomas and grade‐ and molecular subtype‐matched invasive ductal carcinomas of no special type. J Pathol 2010; 220: 45–57. [DOI] [PubMed] [Google Scholar]
- 9. Richard F, Pacyna‐Gengelbach M, Schluns K, et al Patterns of chromosomal imbalances in invasive breast cancer. Int J Cancer 2000; 89: 305–310. [PubMed] [Google Scholar]
- 10. Bertucci F, Orsetti B, Negre V, et al Lobular and ductal carcinomas of the breast have distinct genomic and expression profiles. Oncogene 2008; 27: 5359–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reis‐Filho JS, Simpson PT, Turner NC, et al FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res 2006; 12: 6652–6662. [DOI] [PubMed] [Google Scholar]
- 12. Simpson PT, Reis‐Filho JS, Lambros MB, et al Molecular profiling pleomorphic lobular carcinomas of the breast: evidence for a common molecular genetic pathway with classic lobular carcinomas. J Pathol 2008; 215: 231–244. [DOI] [PubMed] [Google Scholar]
- 13. Christgen M, Bruchhardt H, Hadamitzky C, et al Comprehensive genetic and functional characterization of IPH‐926: a novel CDH1‐null tumour cell line from human lobular breast cancer. J Pathol 2009; 217: 620–632. [DOI] [PubMed] [Google Scholar]
- 14. Christgen M, Noskowicz M, Schipper E, et al Oncogenic PIK3CA mutations in lobular breast cancer progression. Genes Chromosomes Cancer 2013; 52: 69–80. [DOI] [PubMed] [Google Scholar]
- 15. Collins LC, Schnitt SJ. Papillary lesions of the breast: selected diagnostic and management issues. Histopathology 2008; 52: 20–29. [DOI] [PubMed] [Google Scholar]
- 16. Lakhani SR, Ellis I, Schnitt S, et al WHO Classification of Tumours of the Breast International Agency for Research on Cancer, WHO Press: Lyon, 2012.
- 17. Ni YB, Tse GM. Pathological criteria and practical issues in papillary lesions of the breast ‐ a review. Histopathology 2016; 68: 22–32. [DOI] [PubMed] [Google Scholar]
- 18. Duprez R, Wilkerson PM, Lacroix‐Triki M, et al Immunophenotypic and genomic characterization of papillary carcinomas of the breast. J Pathol 2012; 226: 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rakha EA, Abbas A, Sheeran R. Invasive lobular carcinoma mimicking papillary carcinoma: a report of three cases. Pathobiology 2016; 83: 221–227. [DOI] [PubMed] [Google Scholar]
- 20. Dowsett M, Sestak I, Lopez‐Knowles E, et al Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013; 31: 2783–2790. [DOI] [PubMed] [Google Scholar]
- 21. Wuerstlein R, Sotlar K, Gluz O, et al The West German Study Group Breast Cancer Intrinsic Subtype study: a prospective multicenter decision impact study utilizing the Prosigna assay for adjuvant treatment decision‐making in estrogen‐receptor‐positive, HER2‐negative early‐stage breast cancer. Curr Med Res Opin 2016; 32: 1217–1224. [DOI] [PubMed] [Google Scholar]
- 22. Christgen M, van Luttikhuizen JL, Raap M, et al Precise ERBB2 copy number assessment in breast cancer by means of molecular inversion probe array analysis. Oncotarget 2016; 7: 82733–82740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ostrovnaya I, Begg CB. Testing clonal relatedness of tumors using array comparative genomic hybridization: a statistical challenge. Clin Cancer Res 2010; 16: 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ostrovnaya I, Olshen AB, Seshan VE, et al A metastasis or a second independent cancer? Evaluating the clonal origin of tumors using array copy number data. Stat Med 2010; 29: 1608–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ostrovnaya I, Seshan VE, Olshen AB, et al Clonality: an R package for testing clonal relatedness of two tumors from the same patient based on their genomic profiles. Bioinformatics 2011; 27: 1698–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartels S, Schipper E, Hasemeier B, et al Routine clinical mutation profiling using next generation sequencing and a customized gene panel improves diagnostic precision in myeloid neoplasms. Oncotarget 2016; 7: 30084–30093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spirio L, Joslyn G, Nelson L, et al A CA repeat 30–70 KB downstream from the adenomatous polyposis coli (APC) gene. Nucleic Acids Res 1991; 19: 6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dietmaier W, Wallinger S, Bocker T, et al Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997; 57: 4749–4756. [PubMed] [Google Scholar]
- 29. Murphy KM, Zhang S, Geiger T, et al Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn 2006; 8: 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ang D, VanSandt AM, Beadling C, et al Biphasic papillary and lobular breast carcinoma with PIK3CA and IDH1 mutations. Diagn Mol Pathol 2012; 21: 221–224. [DOI] [PubMed] [Google Scholar]
- 31. Nassar H, Qureshi H, Adsay NV, et al Clinicopathologic analysis of solid papillary carcinoma of the breast and associated invasive carcinomas. Am J Surg Pathol 2006; 30: 501–507. [DOI] [PubMed] [Google Scholar]
- 32. Tan BY, Thike AA, Ellis IO, et al Clinicopathologic characteristics of solid papillary carcinoma of the breast. Am J Surg Pathol 2016; 40: 1334–1342. [DOI] [PubMed] [Google Scholar]
- 33. De Leeuw WJ, Berx G, Vos CB, et al Simultaneous loss of E‐cadherin and catenins in invasive lobular breast cancer and lobular carcinoma in situ. J Pathol 1997; 183: 404–411. [DOI] [PubMed] [Google Scholar]
- 34. Hause RJ, Pritchard CC, Shendure J, et al Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 2016; 22: 1342–1350. [DOI] [PubMed] [Google Scholar]
- 35. Halford SE, Sawyer EJ, Lambros MB, et al MSI‐low, a real phenomenon which varies in frequency among cancer types. J Pathol 2003; 201: 389–394. [DOI] [PubMed] [Google Scholar]
- 36. Kim JJ, Baek MJ, Kim L, et al Accumulated frameshift mutations at coding nucleotide repeats during the progression of gastric carcinoma with microsatellite instability. Lab Invest 1999; 79: 1113–1120. [PubMed] [Google Scholar]
- 37. Yang J, Mani SA, Donaher JL, et al Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–939. [DOI] [PubMed] [Google Scholar]
- 38. Antonescu CR, Nafa K, Segal NH, et al EWS‐CREB1: a recurrent variant fusion in clear cell sarcoma–association with gastrointestinal location and absence of melanocytic differentiation. Clin Cancer Res 2006; 12: 5356–5362. [DOI] [PubMed] [Google Scholar]
- 39. Tang WW, Stelter AA, French S, et al Loss of cell‐adhesion molecule complexes in solid pseudopapillary tumor of pancreas. Mod Pathol 2007; 20: 509–513. [DOI] [PubMed] [Google Scholar]
- 40. Chetty R, Serra S. Membrane loss and aberrant nuclear localization of E‐cadherin are consistent features of solid pseudopapillary tumour of the pancreas. An immunohistochemical study using two antibodies recognizing different domains of the E‐cadherin molecule. Histopathology 2008; 52: 325–330. [DOI] [PubMed] [Google Scholar]
- 41. Rocha AS, Soares P, Seruca R, et al Abnormalities of the E‐cadherin/catenin adhesion complex in classical papillary thyroid carcinoma and in its diffuse sclerosing variant. J Pathol 2001; 194: 358–366. [DOI] [PubMed] [Google Scholar]
- 42. Klöppel G, Hruban RH, Klimstra DS, et al Solid‐pseudopapillary neoplasm of the pancreas In WHO Classification of Tumors of the Digestive System (4th edn), Bosman FT, Carneiro F, Hruban RH, Theise ND. (Eds). IARC: Lyon, 2010; 327–330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Figure S1. Main tumour, infiltration of the capsule and haemangiosis
Figure S2. Likelihood ratio (LR) values (A) and MSI‐low phenotype (B) in both tumour components
Table S1. Antibodies and immunohistochemistry
Table S2. CDH1 PCR primer pairs
Table S3. Copy number alterations (CNAs) in both tumour components