

ABSTRACT
Serratia marcescens causes health care-associated opportunistic infections that can be difficult to treat due to a high incidence of antibiotic resistance. One of the many secreted proteins of S. marcescens is the PhlA phospholipase enzyme. Genes involved in the production and secretion of PhlA were identified by screening a transposon insertion library for phospholipase-deficient mutants on phosphatidylcholine-containing medium. Mutations were identified in four genes (cyaA, crp, fliJ, and fliP) that are involved in the flagellum-dependent PhlA secretion pathway. An additional phospholipase-deficient isolate harbored a transposon insertion in the cysE gene encoding a predicted serine O-acetyltransferase required for cysteine biosynthesis. The cysE requirement for extracellular phospholipase activity was confirmed using a fluorogenic phospholipase substrate. Phospholipase activity was restored to the cysE mutant by the addition of exogenous l-cysteine or O-acetylserine to the culture medium and by genetic complementation. Additionally, phlA transcript levels were decreased 6-fold in bacteria lacking cysE and were restored with added cysteine, indicating a role for cysteine-dependent transcriptional regulation of S. marcescens phospholipase activity. S. marcescens cysE mutants also exhibited a defect in swarming motility that was correlated with reduced levels of flhD and fliA flagellar regulator gene transcription. Together, these findings suggest a model in which cysteine is required for the regulation of both extracellular phospholipase activity and surface motility in S. marcescens.
IMPORTANCE Serratia marcescens is known to secrete multiple extracellular enzymes, but PhlA is unusual in that this protein is thought to be exported by the flagellar transport apparatus. In this study, we demonstrate that both extracellular phospholipase activity and flagellar function are dependent on the cysteine biosynthesis pathway. Furthermore, a disruption of cysteine biosynthesis results in decreased phlA and flagellar gene transcription, which can be restored by supplying bacteria with exogenous cysteine. These results identify a previously unrecognized role for CysE and cysteine in the secretion of S. marcescens phospholipase and in bacterial motility.
KEYWORDS: Serratia, cysteine, flagella, phospholipase
INTRODUCTION
Serratia marcescens is a ubiquitous bacterial species that infects organisms ranging from insects to humans (1). In humans, S. marcescens is a health care-associated opportunistic pathogen that causes multiple types of infections, including bacteremia and urinary tract infections (UTI). One of the many secreted factors of S. marcescens is the phospholipase A (PLA) enzyme, PhlA, that is responsible for the lecithinase activity of this organism (2). PhlA is cytotoxic when added to epithelial cells and exhibits hemolytic activity, likely due to the accumulation lysophospholipid cleavage products that can cause membrane destabilization of target cells (3). PhlA is also known to directly interact with the PhlB protein, which is thought to render PhlA inactive when intracellular to prevent bacterial self-membrane damage (4). The phlB gene is located directly downstream of the phlA open reading frame (ORF) and is expected to be cotranscribed.
The transcriptional regulation of phlA in Serratia liquefaciens MG1, which was later reclassified as S. marcescens (5), has been examined by heterologous expression in Escherichia coli. There are two known promoters upstream of phlA, pX and pA, that are involved in controlling phlAB transcription in response to growth phase, temperature, and glucose and oxygen availability (4, 6). Expression from pA accounts for the majority of phlA transcription and is growth-phase dependent, with the highest levels of expression observed during the transition from exponential to stationary phase. The pX promoter controls transcription of the ORF upstream of the phlAB locus but has also been shown to contribute to phlAB expression, particularly under low-oxygen conditions. A predicted Fnr binding site is present within the pX promoter region, suggesting that anaerobic expression of phlAB may also be controlled by this global regulatory protein (6).
A sequence within the pA promoter shares homology with a recognition sequence for binding of the flagellar sigma factor FliA (σ28) (6). Transcription of fliA in related Enterobacteriaceae species is under the control of the master regulator FlhDC, which controls transcription of flagellar genes in a tiered manner (7, 8). Class 2 genes include those that encode flagellar export proteins (e.g., fliJ and fliP) as well as fliA. In turn, FliA is required for the expression of class 3 genes, including the flagellar filament gene fliC. PhlA has been shown to be produced in E. coli when the phlAB genes are provided on a plasmid, but E. coli strains lacking flhD no longer produce PhlA (6). PhlA activity was also not observed when flhD was mutated in S. marcescens; therefore, flhD is required for PhlA activity, likely through transcriptional activation via FliA (9). In S. marcescens and other bacterial species, flhDC is transcriptionally regulated by cyclic AMP (cAMP) receptor protein (CRP) and is subject to catabolite repression in the presence of glucose (8, 10, 11). This paradigm may also hold true for phlA transcription, since heterologous expression in E. coli required cAMP and the cyaA gene encoding adenylate cyclase (6). Taken together, these studies indicate that phlAB is likely a member of the flagellar regulon in S. marcescens and, furthermore, lead to the hypothesis that PhlA is secreted via the flagella assembly apparatus (4, 9).
Yersinia enterocolitica secretes a phospholipase, YplA, that is also flhDC dependent and susceptible to catabolite repression (12, 13). An amino acid alignment of S. marcescens PhlA and Y. enterocolitica YplA shows 74% identity over 244 residues, suggesting that these proteins may have similar functions. However, YplA is reported to have PLA2 enzymatic activity whereas PhlA is reported to have PLA1 activity (14). An additional characterization of YplA revealed that flagellar structural genes were required for extracellular phospholipase activity (12). YplA has also been shown to promote colonization and inflammation in a Y. enterocolitica mouse model of gastrointestinal infection (14).
Although transcriptional regulation of phlA has been studied primarily using a system of heterologous expression in E. coli, the native regulation of phlA in S. marcescens has yet to be fully characterized. Furthermore, the physiological role of S. marcescens PhlA is not known. By screening a transposon mutant library generated in a clinical isolate of S. marcescens, we identified cysE as a novel gene required for extracellular phospholipase activity. Subsequent experiments were designed to characterize the role of CysE on phospholipase and flagellar gene transcription, as well as on the motility of S. marcescens.
RESULTS
Identification of genes required for extracellular phospholipase activity.
To identify S. marcescens genes that are required for PhlA activity and secretion, a transposon insertion library constructed with clinical bacteremia isolate UMH9 and containing ∼32,000 unique mutants (15) was screened for colonies with defects in phospholipase activity on phosphatidylcholine-containing PCY agar. Six mutants having reduced extracellular phospholipase activity were identified from over 15,000 colonies screened. The chromosomal locations of transposon insertions were determined for each mutant (Table 1), and the phospholipase phenotype of each isolate was compared directly with that of wild-type bacteria. The fliP and fliJ genes encode flagellar export and chaperone functions, respectively, and the disruption of either resulted in a complete loss of phospholipase activity (Fig. 1). This observation is consistent with the previously hypothesized role of the flagellar export complex in secretion of PhlA. Three additional phospholipase-negative isolates harbored mutations in either the cyaA or crp gene encoding an adenylate cyclase or cAMP receptor protein, respectively. Both cAMP and CRP are known to modulate the transcription of flagellar genes in Serratia and related bacterial species (8, 10, 11) and are necessary for extracellular phospholipase activity when the S. marcescens phlA gene is expressed in E. coli (6). Lastly, disruption of the BVG96_20000 ORF resulted in a greatly reduced level of extracellular phospholipase activity. BVG96_20000 is predicted to encode a 273-amino-acid protein having serine O-acetyltransferase activity (COG1045). The CysE serine acetyltransferase of E. coli MG1655 and the UMH9 BVG96_20000 predicted protein product share 86% amino acid identity; therefore, BVG96_20000 will be referred to here as cysE. A high level of phospholipase activity, similar to that of the wild-type strain, was restored to the cysE mutant upon genetic complementation, demonstrating that the phospholipase phenotype of this mutant was not the result of polar effects on neighboring genes due to the transposon insertion (Fig. 1). It was determined in previous work using high-throughput sequencing that multiple independent phlA transposon insertion mutants were present in the tested library (15); however, none were identified in this screen, indicating that the number of screened clones was insufficient to achieve saturation. A phlAB deletion mutant was therefore constructed and tested for phospholipase activity in the same assay. As expected, the phlAB mutant exhibited a phospholipase-null phenotype on PCY agar, and activity was restored to high levels when phlAB was provided on a multicopy plasmid (Fig. 1).
TABLE 1.
S. marcescens strains and plasmids
| Name | Relevant genotype | Description | Reference or source |
|---|---|---|---|
| S. marcescens | |||
| UMH9 | Wild type | Bloodstream infection isolate | 15 |
| phlAB | ΔphlAB::nptII | 1,519-bp internal deletion of phlAB locus, insertion of 1,496-bp nptII-containing fragment from pKD4 | This study |
| cysE | cysE::Tn | Insertion at position 138 of 822-bp ORF | This study |
| cyaA | cyaA::Tn | Insertion at position 218 of 2,553-bp ORF | This study |
| cyaA | cyaA::Tn | Insertion at position 104 of 2,553-bp ORF | This study |
| crp | crp::Tn | Insertion at position 123 of 633-bp ORF | This study |
| fliP | fliP::Tn | Insertion at position 471 of 762-bp ORF | This study |
| fliJ | fliJ::Tn | Insertion at position 164 of 447-bp ORF | This study |
| Plasmids | |||
| pKD4 | Source of kanamycin resistance gene | 40 | |
| pSIM19 | Recombineering plasmid | 41 | |
| pBBR1MCS-5 | Genetic complementation plasmid, gentamicin resistant | 42 | |
| pBB1 | phlAB+ | phlAB locus with native promoter sequence ligated to pBBR1MCS-5 | This study |
| pBB2 | cysE+ | cysE ORF with predicted promoter sequence ligated to pBBR1MCS-5 | This study |
FIG 1.
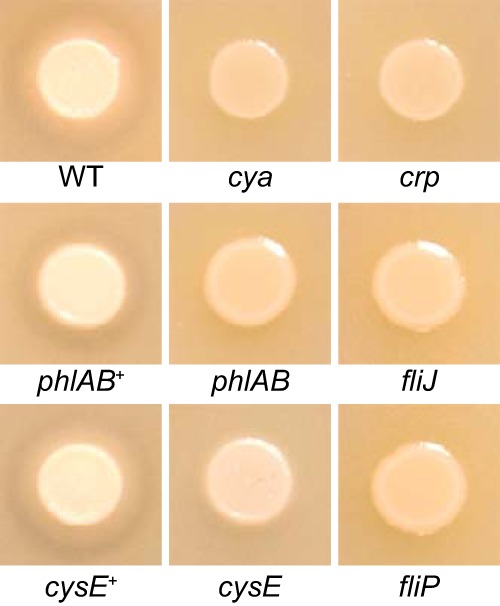
Phospholipase-deficient S. marcescens strains. Bacterial suspensions containing S. marcescens strains were spotted on PCY agar for examination of phospholipase phenotype. Phospholipase activity can be observed as a zone of clearance in the agar surrounding the wild-type (WT) and complemented mutant strains (phlAB+ and cysE+).
A Serratia cysE mutant is auxotrophic for cysteine and O-acetyl-l-serine.
Due to the previously unrecognized requirement for CysE in phospholipase activity, we sought to further characterize the role of this predicted serine acetyltransferase in Serratia PhlA production and secretion. Cysteine biosynthesis from l-serine is accomplished via a two-step reaction involving the production of O-acetyl-l-serine (OAS) as an intermediate. In E. coli, CysE catalyzes the formation of OAS from acetyl coenzyme A (acetyl-CoA) and serine, and cysE mutants are auxotrophic for cysteine (16, 17). The O-acetylserine (thiol)-lyase enzyme catalyzes the second step of the pathway, and together with CysE, forms the multifunctional cysteine synthase complex (17, 18). Based on this information, it was hypothesized that cysE would be required for S. marcescens growth in the absence of exogenous cysteine. The wild-type strain UMH9, the cysE transposon mutant, and the complemented cysE mutant all exhibited robust growth in LB medium, a rich source of exogenous amino acids (Fig. 2A). Similar high levels of growth were observed for the wild-type and complemented cysE mutant strains in defined M9 medium, whereas the cysE mutant failed to grow under these conditions (Fig. 2B). The addition of Casamino Acids (CA) to M9 medium (M9 plus CA) resulted in partial restoration of growth for the cysE mutant (Fig. 2C).
FIG 2.
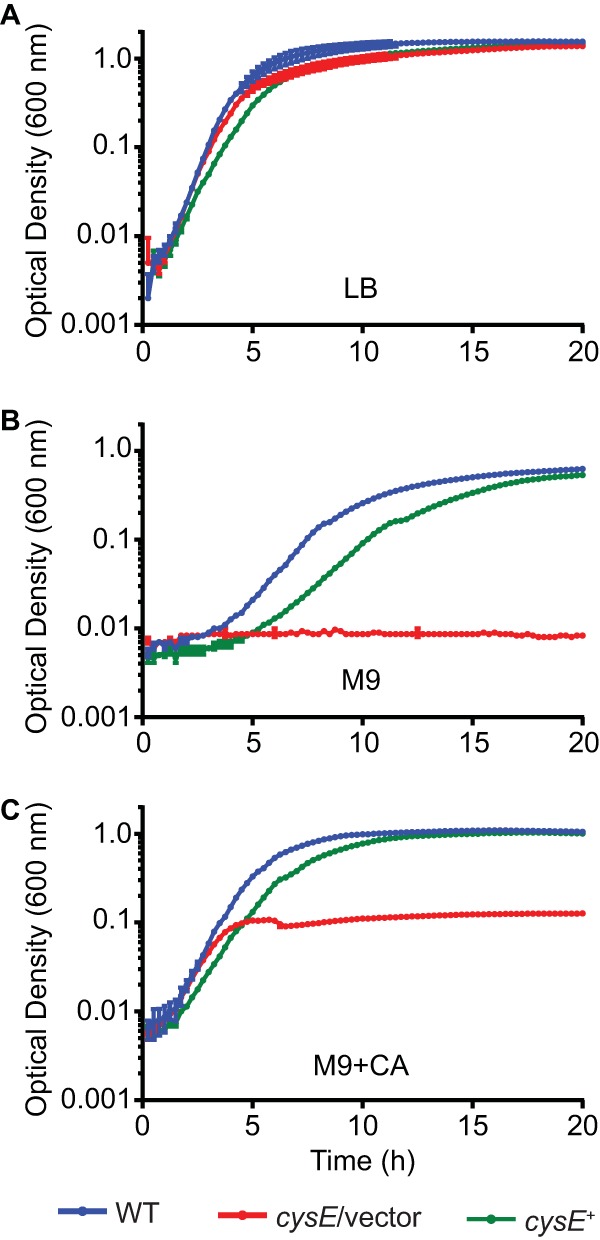
Growth of the S. marcescens cysE mutant requires exogenous amino acids. (A) S. marcescens wild-type (WT), cysE mutant with empty vector (cysE/vector), and complemented cysE mutant (cysE+) strains were inoculated in LB medium and incubated at 37°C. Bacterial growth was measured at an OD600 every 15 min, and the means (±standard deviations) from triplicate cultures are shown. (B and C) Bacteria cultured overnight in LB were harvested by centrifugation, washed in M9 medium, and then subcultured in M9 or M9 plus CA as indicated. Bacterial growth was measured as described for panel A.
To determine whether the S. marcescens cysE-dependent growth defect was due to the lack of cysteine, cultures in defined medium were supplemented with increasing concentrations of the amino acid. Maximal growth was observed when bacteria were provided with 1 mM exogenous cysteine, though enhancement of growth was also observed with as little as 0.001 mM cysteine (Fig. 3A). Since the enzymatic activity of CysE results directly in the production of OAS rather than cysteine, growth levels were also measured in the presence of OAS. The supplementation of culture medium with 1 to 10 mM OAS restored the growth of the cysE mutant; however, the recovery of growth was less efficient than that observed with cysteine (Fig. 3B). Together, these results demonstrate that cysE is required for S. marcescens growth due to its role in cysteine biosynthesis.
FIG 3.
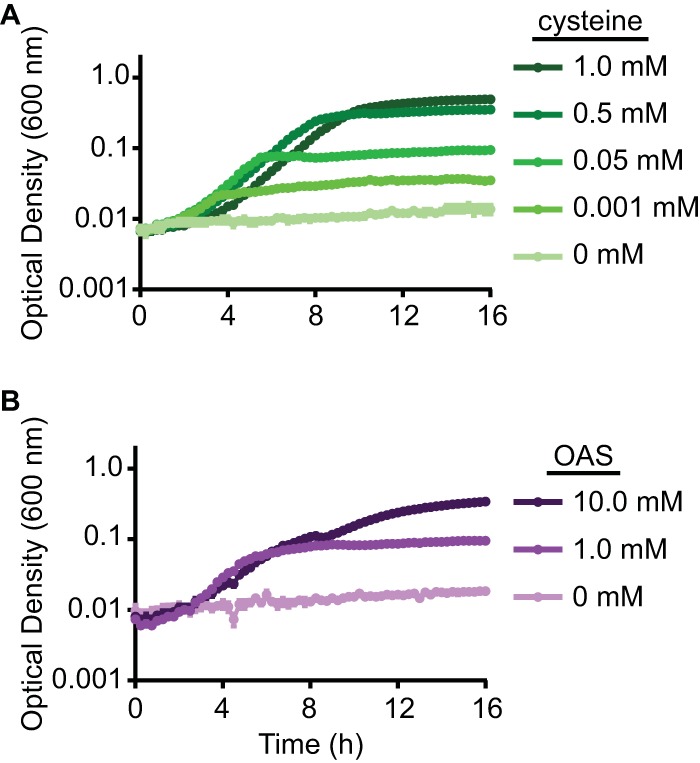
An S. marcescens cysE mutant is auxotrophic for cysteine. The cysE mutant strain was cultured in M9 medium supplemented with either cysteine (A) or OAS (B) at the indicated concentrations. Bacterial growth was measured at an OD600, and the means (±standard deviations) from triplicate cultures are shown.
Phospholipase activity is cysteine dependent.
To quantitate the extracellular phospholipase activity of each mutant identified in the genetic screen, a fluorescent substrate specific for PLA enzymes was used to measure the activity from cell-free supernatants of LB cultures. Each of the five mutant strains identified in the screen (cysE, cyaA, crp, fliP, and fliJ strains) exhibited baseline levels of PLA activity that were similar to that of the phlAB null mutant (Fig. 4A). As was observed on PCY agar, genetic complementation of the phlAB and cysE mutations resulted in significant increases in detected PLA activity compared with that of the respective mutant strain. It is important to note that the phospholipase deficiency of the cysE mutant is not simply due to this strain's reduced growth under cysteine limitation, since cysE mutants exhibit no growth defect when cultured in LB medium (Fig. 2A). These results confirm the findings of the genetic screen and, importantly, establish that both flagellum-associated genes and cysE are required for Serratia phospholipase activity.
FIG 4.
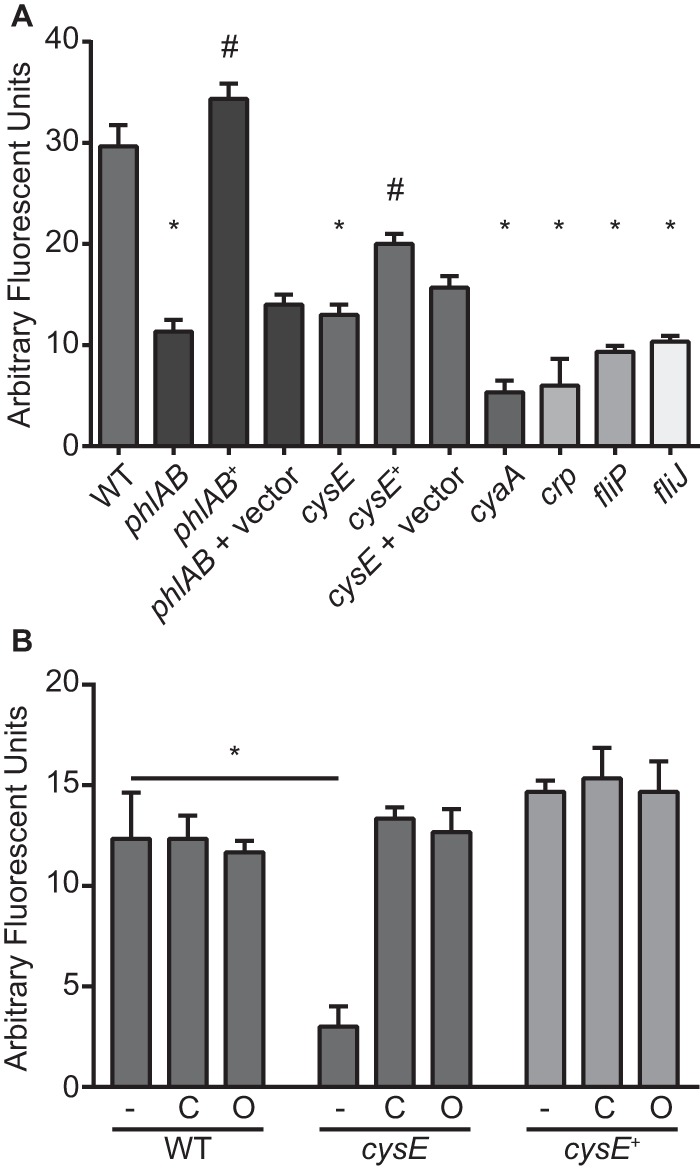
Flagellar and cysteine biosynthesis genes are required for extracellular PLA activity. (A) Cell-free supernatants from S. marcescens strains cultured overnight in LB medium were combined with the fluorescent PLA substrate bis-BODIPYFL C11-PC. Fluorescence was measured after 1 h of incubation at 37°C. Bars represent the means (±standard deviations) from triplicate assays. Asterisks denote a significant reduction in PLA activity compared with that of the wild-type (WT) strain, and the pound symbol indicates a significant increase in PLA activity for complemented mutant strains compared with that of the respective uncomplemented mutant (t test, P ≤ 0.01). (B) PLA activity was determined as described for panel A for bacterial strains cultured in M9 plus CA (−) or the same medium supplemented with 1 mM cysteine (C) or 10 mM OAS (O). The asterisk indicates a significant decrease in PLA activity compared with that of the wild-type strain for the unsupplemented condition (t test, P ≤ 0.01).
Based on the well-studied function of the E. coli CysE protein, the lack of OAS and/or cysteine was hypothesized to be responsible for the loss of phospholipase activity in the S. marcescens cysE mutant. To test this, phospholipase activity from bacterial supernatants cultured in CA-supplemented M9 was measured. Although the tested concentration of CA (0.1%) supplies sufficient amounts of cysteine to partially overcome the auxotrophy of the cysE mutant (Fig. 2C), it was not sufficient to restore phospholipase activity to this strain (Fig. 4B). The addition of 1 mM cysteine or 10 mM OAS to cysE mutant cultures resulted in a complete restoration of the PLA activity to levels observed with wild-type and complemented mutant strains. Since exogenous OAS is likely converted to cysteine intracellularly through the action of an O-acetylserine (thiol)-lyase, and cysteine alone is able to restore PLA activity, it was reasoned that cysteine rather than OAS is the limiting component of extracellular phospholipase activity in these assays. As expected, the supplementation of both the phlAB and fliP mutant cultures with OAS or cysteine did not restore PLA activity to these strains (data not shown). The addition of cysteine or OAS also did not result in a significant change in wild-type or complemented mutant PLA activity, indicating that the levels of cysteine in these cells are sufficient for maximal activity under the tested conditions.
CysE contributes to S. marcescens swarming motility.
It is clear that S. marcescens extracellular phospholipase activity is dependent on both the production of the PhlA protein and its secretion via the flagellar apparatus; therefore, cysteine deficiency could limit phospholipase activity through either or both of these processes. To determine if bacteria lacking cysE were capable of flagellum-mediated motility, S. marcescens strains were cultured on LB medium containing 0.6% agar to facilitate surface motility. Wild-type and phlAB bacteria swarmed readily over the 16-h incubation period, forming a thin film of growth over the surface of the agar (Fig. 5A). This result is in contrast to that from both the fliP and fliJ flagellar mutants that failed to migrate beyond the point of inoculation, as expected. Bacteria lacking cysE exhibited an intermediate phenotype with an approximately 2-fold decrease in swarm diameter (Fig. 5B). The reduced swarming motility observed with the cysE mutant under these conditions was not the result of growth limitation due to a lack of cysteine biosynthesis, since cysE bacteria are capable of robust growth in LB medium (Fig. 2A). Interestingly, the cysE mutant showed flagellum-dependent swimming motility levels that were equivalent to those of wild-type bacteria when tested under similar conditions (LB plus 0.25% agar), demonstrating that the swarming phenotype was not the result of a complete motility defect and that the two forms of flagellum-mediated motility exhibit differential requirements for cysteine (Fig. 5C). Together, these data are consistent with the notion that the loss of secreted PLA activity of cysE mutant bacteria is due in part to the modulation of flagellar apparatus function, though at least some functional flagella must remain in bacteria lacking CysE function.
FIG 5.
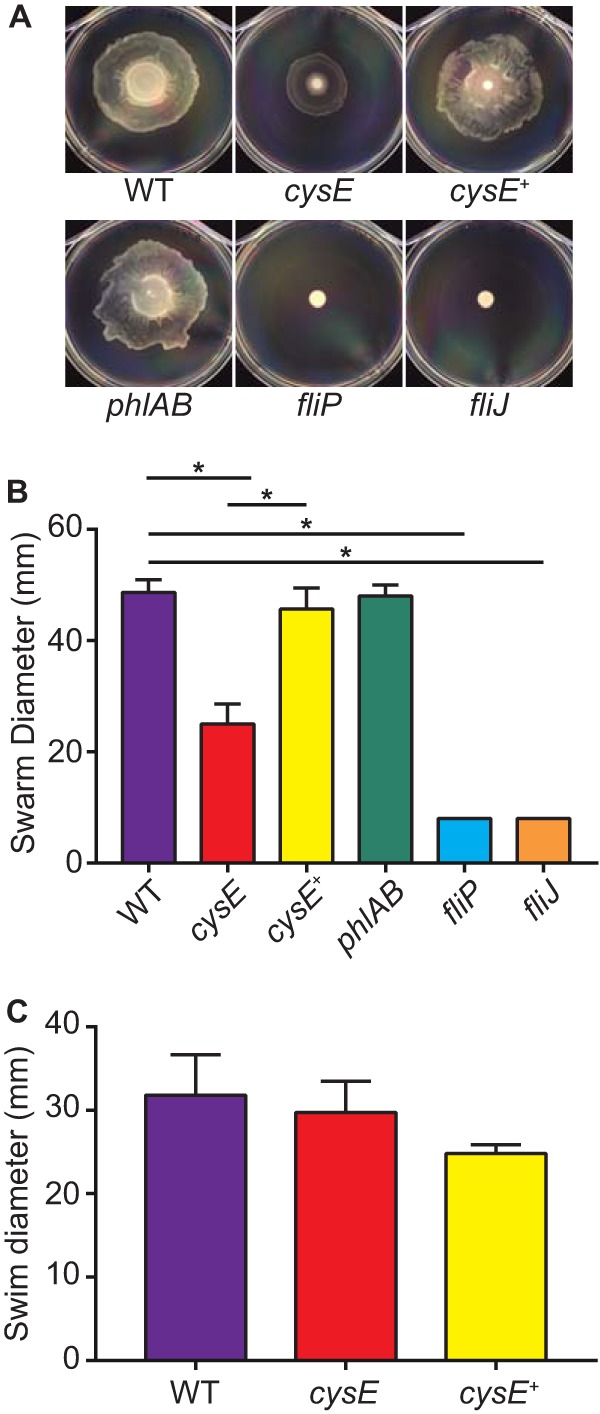
CysE is required for swarming but not swimming motility of S. marcescens in rich medium. (A) Suspensions (10 μl) of bacteria were spotted on LB medium containing 0.6% agar and incubated at 30°C for 16 h to facilitate swarming motility. Images of swarm growth in 100-mm petri dishes were captured from a top-down perspective. (B) Bacterial swarming was quantified by measuring the diameter (mm) of growth for each strain. Bars represent the mean (±standard deviations) swarm diameters from three independent experiments, and asterisks identify swarm diameters that were significantly less than those of the wild-type (WT) or complemented cysE mutant (cysE+) strains as indicated (t test, P < 0.01). (C) Swimming capability of S. marcescens strains was determined as described for panels A and B with the exception that bacteria were cultured on LB medium containing 0.25% agar for 8 h.
cysE mutants have reduced phospholipase and flagellar gene transcription.
The loss of both phospholipase secretion and swarming motility in cysE mutant bacteria was hypothesized to be due to a disruption in transcriptional regulation of either phlA or flagellum-associated genes. To test this hypothesis, the levels of phlA transcripts between wild-type and cysE bacteria were compared by reverse transcription-quantitative PCR (qRT-PCR). In cells harboring the cysE mutation, an ∼6-fold decrease in phlA expression levels was observed for bacteria cultured in M9 plus CA (Fig. 6A). This reduced phlA expression is consistent with the loss of PLA activity observed under the same growth conditions for bacteria lacking cysE (Fig. 4) and indicates that the low level of cysteine supplied by CA is insufficient for optimal phlA expression. Complementation of the cysE mutation in this experiment restored phlA transcription to a level similar to that of the wild-type strain and significantly different than that of the cysE mutant. To determine whether phlA expression was cysteine dependent, transcript abundance was measured from bacteria cultured in M9-plus-CA medium supplemented with either OAS or cysteine, relative to unsupplemented controls. The transcription of phlA was increased in these experiments when cysE mutant bacteria were provided with cysteine or OAS compared with wild-type phlA levels that remained largely unchanged under the same conditions (Fig. 7). These results demonstrate that the phlA transcription deficiency of the cysE mutant strain is due to the lack of cysteine biosynthesis pathway products and not a secondary function of the serine acetyltransferase protein.
FIG 6.
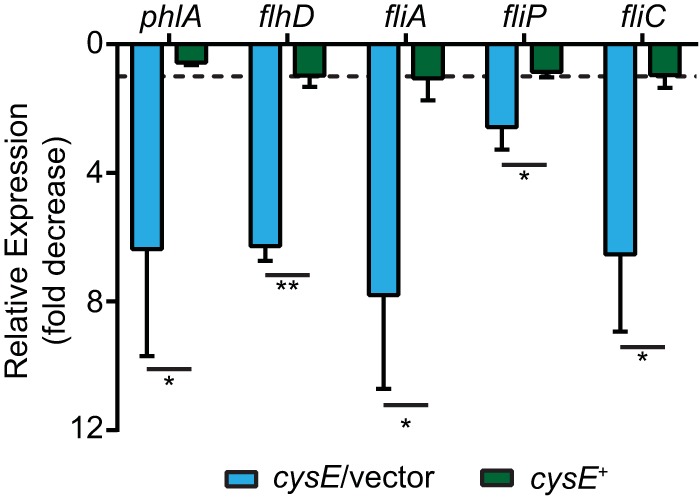
Mutation of cysE results in decreased transcription of phospholipase and flagellar genes. cDNA prepared from S. marcescens strains cultured in M9 plus CA was used to assess relative levels of phospholipase and flagellar gene transcripts (x axis) by qRT-PCR. Transcript levels for the cysE mutant harboring an empty vector (cysE/vector) and the complemented cysE mutant (cysE+) are shown as fold decrease relative to that of the wild-type strain. The dotted line at 1.0 on the y axis indicates a hypothetical value at which there is no change relative to the wild-type strain. Bars represent the means (±standard deviations) from three independent experiments, and asterisks denote significant differences in relative levels between the cysE mutant and complemented cysE mutant strains (t test). *, P < 0.05; **, P < 0.01).
FIG 7.
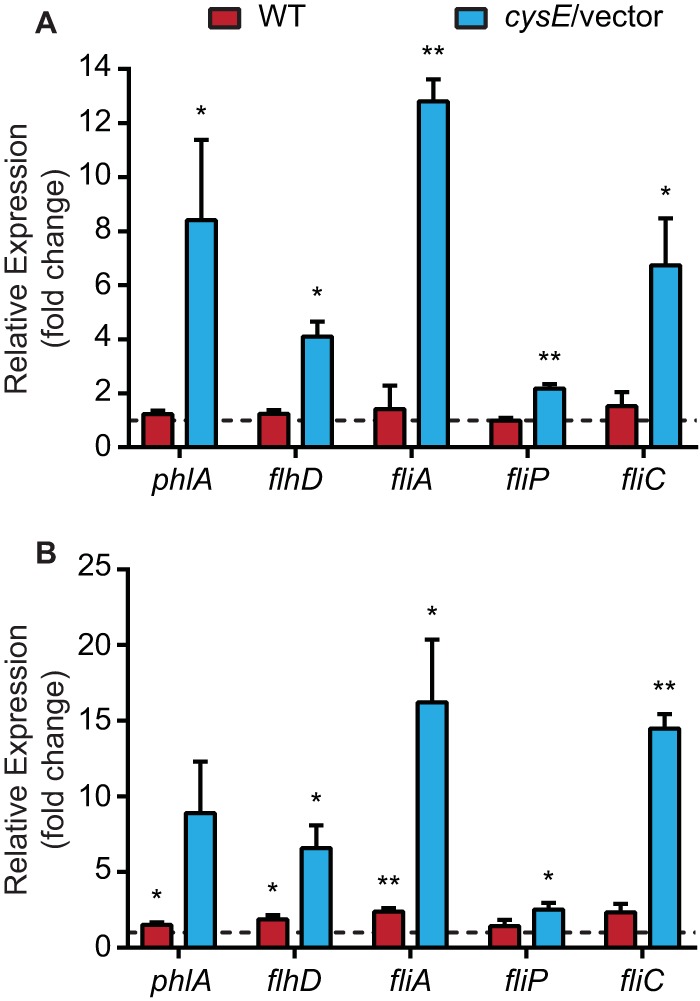
Cysteine and OAS supplementation increase phospholipase and flagellar gene transcription in the cysE mutant. (A) cDNA prepared from S. marcescens strains cultured in M9 plus CA or M9 plus CA supplemented with 1 mM cysteine was used to assess relative levels of phospholipase and flagellar gene transcripts (x axis) by qRT-PCR. Transcript levels for the cysE mutant harboring an empty vector (cysE/vector) and the wild-type strain (WT) are shown as fold change for bacteria cultured in cysteine-supplemented medium relative to that cultured in M9 plus CA. (B) Transcript levels from bacteria cultured in M9 plus CA supplemented with 10 mM OAS relative to those from bacteria cultured in unsupplemented M9 plus CA were determined by qRT-PCR as described for panel A. Results represent the means (±standard deviations) from three independent experiments, and the asterisks denote relative expression levels that are significantly different from the hypothesized value of 1.0 (dotted line, no change) (one-sample t test). *, P < 0.05; **, P < 0.01.
To investigate the possibility that flagellar gene transcription was also dependent on CysE and cysteine levels, fliP and fliC expression levels were measured. By an extension of the paradigm established for other species of the Enterobacteriaceae family, these two genes were chosen as representative examples of class 2 (fliP) and class 3 (fliC) target genes in the hierarchical flagellar regulatory cascade (8). Similar to phlA, both fliP and fliC were demonstrated to have decreased transcript abundance in cysE mutant bacteria compared with that of the wild-type strain (Fig. 6). These flagellar genes also exhibited increased transcription levels relative to unsupplemented control cultures when cysE mutant bacteria were provided with cysteine or OAS (Fig. 7). Wild-type bacteria also exhibited minimal increases in fliP and fliC transcription in the presence of added cysteine or OAS, demonstrating that these products do not stimulate flagellar gene transcription in bacteria competent for serine acetyltransferase.
The transcription of phlA in S. marcescens (9), as well as fliP and fliC in related bacterial species (8, 19), is known to require the FlhDC flagellar master regulator. Therefore, it was hypothesized that the OAS and cysteine deficiency of cysE mutant bacteria could influence the level of flhD transcription, resulting in reduced expression of FlhD-dependent target genes. In bacteria harboring the cysE mutation, the transcription of flhD was reduced ∼6-fold compared with that of wild-type bacteria (Fig. 6). Complementation of the cysE mutation restored flhD expression to approximately wild-type levels and resulted in a significant difference compared with that of the mutant strain. The alternative sigma factor FliA (σ28) is responsible for the transcription of class 3 flagellar genes (8, 19), and likely phlA (6), and the fliA gene itself, is also an FlhDC-dependent class 2 gene. Consistent with the observed transcriptional defect of fhlD in the S. marcescens cysE mutant, fliA expression levels were also reduced at a similar level. Furthermore, both flhD and fliA transcripts increased in abundance when cysE mutant bacteria were cultured in medium supplemented with either cysteine or OAS (Fig. 7). Together, these results strongly suggest that the loss of phospholipase activity and swarming motility in the cysE mutant are due to a role for cysteine at or upstream of the level of flhD transcription.
The role of phospholipase in vivo.
The potential for PhlA to contribute to S. marcescens pathogenesis is an attractive hypothesis given the several well-characterized examples of secreted phospholipases with roles in bacterial virulence (20). Although S. marcescens is known to cause UTI, primarily in health care environments and in patients with urinary catheters (21), the bacterial factors involved in UTI remain uncharacterized for this organism. A murine model of S. marcescens UTI was developed here based on a previously published method (22) to assess the contribution of PhlA to in vivo bacterial fitness. When mice were infected with 5 × 107 CFU, bacteria were routinely detected for at least 4 days postinfection (Fig. 8A), indicating that S. marcescens is capable of establishing a long-term infection in this environment despite the routine voiding of bladder contents. To determine the contribution of PhlA to S. marcescens fitness in this model, a 1:1 mixture of wild-type and phlAB mutant bacteria was used to coinfect mice. The competitive index (CI) was used as a measure of relative fitness and determined from the numbers of phlAB mutant and wild-type bacteria recovered from homogenized bladder tissue. At all tested time points the phlAB mutant exhibited a modest loss of fitness (CI, <1.0) compared with that of the wild-type strain, but only the 24-h time point was statistically significant (Fig. 8B). Although the loss of fitness in the absence of PhlA was minimal, these results warrant further investigation into the potential role of this secreted phospholipase during mammalian infection.
FIG 8.
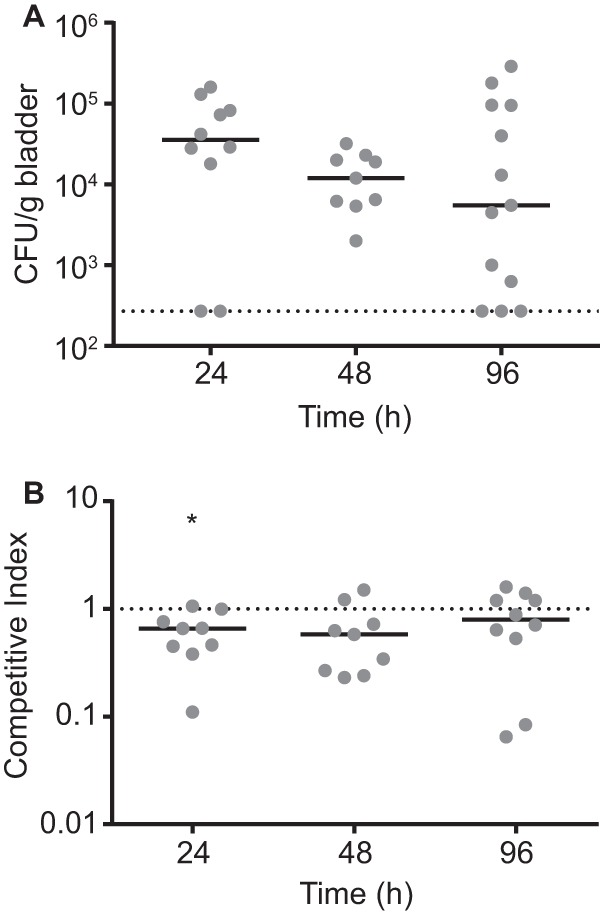
Fitness of the S. marcescens phlAB mutant in a murine model of UTI. (A) C57BL/6 mice were infected with a 1:1 mixture of wild-type and phlAB mutant bacteria via a urethral catheter at a dose of 5 × 107 CFU. At the indicated times, mice were sacrificed and the total bacterial burden in homogenized bladder tissue was determined by serial dilution and plating. Solid lines represent the median bacterial burdens at each time point, and the dotted line indicates the limit of detection. (B) The relative fitness of the phlAB mutant compared with that of the wild type was calculated as a competitive index for the experiment described for panel A. The dotted line represents a CI of 1.0, the level of equivalent fitness between the two strains. Median CI values that were significantly less than a hypothesized value of 1.0 using the Wilcoxon signed rank test are indicated by an asterisk (P < 0.05).
DISCUSSION
Examples of nonflagellar proteins that are secreted via a flagellar export complex are relatively uncommon in the bacteriological literature, yet they are intriguing given the structural homology between the flagellar basal body and the dedicated machinery of host-associated type III secretion systems (23, 24). This form of flagellum-mediated protein translocation has been previously described with regard to secretion of the Y. enterocolitica YplA phospholipase (12, 25), which shares homology with S. marcescens PhlA (14), and has also been predicted for S. marcescens based on the genetic requirement for the flhD gene when phlA is expressed from an inducible and FlhD-independent promoter (9). Unlike those in Yersinia species, the S. marcescens UMH9 genome does not appear to encode a host-adapted type III secretion system (data not shown). Our data further support flagellum-dependent Serratia phospholipase secretion by demonstrating that transposon insertions in the fliP and fliJ genes, encoding proteins required for flagellar assembly in other species, resulted in the complete loss of extracellular phospholipase activity. This finding is important since it was previously established that the transcription of both phlA and flagellar genes was dependent on FlhD, making it difficult to distinguish between a loss of extracellular phospholipase activity due to decreased transcription of phlA or a loss of the secretory apparatus (4, 6, 9). A second, and perhaps complementary, mechanism by which fliP and fliJ mutants may exhibit reduced phospholipase activity is via FlgM-mediated inhibition of FliA activity. The anti-σ28 factor FlgM is exported once the hook-basal body structure has been assembled, thereby freeing FliA for the transcription of class 3 genes (19, 26, 27). In the absence of a completed flagellar export structure, as is expected with the Serratia fliJ and fliP mutants, FlgM remains capable of sequestering FliA and thus inhibiting σ28-dependent transcription. The present study further links the flagellar export system with phospholipase by demonstrating that cysE and cysteine are required for the transcription of genes controlling both of these biological functions, most likely by acting at or upstream of the level of fhlD transcription. By an extension of the E. coli paradigm, the finding that transcription of both class 2 (FlhD-dependent) and class 3 (FlhD- and FliA-dependent) flagellar genes was altered by disruption of cysE further supports this notion. A similar loss of flagellar function and PhlA secretion has been previously observed in S. marcescens strains that have perturbations in the enterobacterial common antigen synthesis pathway, due to a subsequent repression of the flhDC genes via the Rcs phosphorelay (28, 29).
Our finding that the cysE mutation resulted in a swarming motility defect but not a swimming motility defect suggests a differential requirement for cysteine during these two forms of flagellum-mediated motility. One potential explanation for this observation is that swarming motility, involving hyperflagellated cells (30), may be more sensitive to a modulation of the flagellar transcriptional hierarchy. This hypothesis is consistent with our observation that the cysE mutation results in reduced flhD and fliA transcript levels yet still allows for swimming motility. It is also possible that swarming-specific products, such as biosurfactants (31), require a functional cysE gene for production.
In E. coli and other Enterobacteriaceae, the CysB transcriptional regulator controls the expression of numerous genes involved in the biosynthesis and transport of cysteine and sulfur (17). CysB-mediated transcriptional activation requires the inducer N-acetylserine (NAS), which is derived directly from OAS. Therefore, the addition of OAS to bacteria deficient in CysE could both supply substrate for the production of cysteine and provide a source of NAS inducer. Therefore, it is possible that the lack of phospholipase activity and swarming motility in S. marcescens cysE mutants could be due to a loss of CysB activation. Importantly, we have demonstrated that the phlA and flagellar gene transcriptional defects associated with the Serratia cysE mutation could be reversed with cysteine alone. These results are most consistent with the hypothesis that cysteine is the limiting component of phospholipase activity rather than NAS. However, a role for CysB in the transcriptional activation of flagellar and phospholipase genes has not been formally tested at this point.
The status of PhlA as a Serratia virulence factor has long been speculated. In vitro experiments previously demonstrated that purified PhlA could induce cytotoxicity in human bladder and cervical epithelial cells (3), and the Y. enterocolitica PhlA homolog has been proposed to have a role in pathogenesis during murine gastrointestinal infection (14). The partial loss of fitness for bacteria lacking PhlA observed in this work is intriguing, though not definitive. Since PhlA is secreted from bacterial cells, the production of PhlA by wild-type bacteria may have also benefited phlA mutant bacteria during the coinfection experiment, thereby reducing any potential fitness defect of phlA mutant bacteria. It may be possible to address this issue of cross-complementation in future experiments by performing monoinfections, though the inherent variability in colonization levels for the UTI model makes this experiment challenging. An additional assessment of the contribution of PhlA to virulence was conducted using a murine bacteremia model established for this strain (15), but no significant differences between the wild-type strain and the phlAB mutant were observed (data not shown).
In summary, we have determined that an intact cysteine biosynthesis pathway is required for extracellular phospholipase activity and swarming motility of S. marcescens. Cysteine itself appears to be required for achieving high levels of phospholipase and flagellar gene transcription, resulting in defects in FlhDC-dependent processes when levels of this amino acid are insufficient. These results further support a model of coregulation for flagella and phospholipase in S. marcescens, and future work will be directed at determining the mechanism by which cysteine controls the transcription of flagellar master regulator genes.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
S. marcescens strain UMH9 was isolated from a University of Michigan Hospital System patient with bacteremia. S. marcescens strains and plasmids used in this study are listed in Table 1. E. coli TOP10 (Thermo Fisher) cells were used for routine cloning purposes. S. marcescens and E. coli were cultured in LB medium (32) unless indicated otherwise. The extracellular phospholipase activity of S. marcescens colonies was assayed using PCY agar as described previously (33, 34). Antibiotics were used at the following concentrations: kanamycin, 50 μg/ml; ampicillin, 100 μg/ml; spectinomycin, 100 μg/ml; gentamicin, 6 μg/ml. M9 medium was used to culture S. marcescens strains under defined conditions (35). The M9 base medium contained 0.1 mM CaCl2, 1 mM MgSO4, 5 μg/ml thiamine, and 0.4% glucose and was supplemented with 0.1% Casamino Acids (CA) where indicated. l-Cysteine and OAS were added at the indicated concentrations.
Genetic screen for phospholipase-deficient mutants.
A previously generated transposon mutant library using strain UMH9 (15) was used to identify S. marcescens genes that contribute to extracellular phospholipase activity. Briefly, 0.1 ml of appropriately diluted mutant pools was spread on a PCY agar plate and incubated at 30°C overnight. Colonies lacking a halo of clearance on PCY agar, indicative of a phospholipase defect, were isolated for further analysis. Genomic DNA from these isolates was extracted using published procedures (36), and the location of the transposon insertion in each strain was determined by sequencing the PCR-amplified fragments of transposon-chromosome insertion site junctions (37).
Construction of a phlAB mutant strain.
Primers PhlBA-F (5′-ATGCGCCGGCGTTTGCGAGAAATAAAACTGGAACGCAAAGCCCTGGTGTAGGCTGGAGCTGCTTC-3′) and PhlBA-R (5′-TTAAGTTTTACCTCTGCAGTATCCCCGGTGGCCATCCCTACACATATGAATATCCTCCTTAGT-3′) were used to PCR amplify an nptII-containing fragment from pKD4. These primers contain ∼50-bp homology at the 5′ end to the phlAB locus and ∼20-bp homology at the 3′ end to pKD4 (underscored). The PCR product was digested with DpnI and electroporated into UMH9(pSIM19) cells that had been heat treated at 42°C for 20 min prior to transformation. The presence of the ΔphlAB::nptII allele in kanamycin-resistant transformants was confirmed by PCR and sequencing using primers PhlA-355 (5′-TGCGTCGCGAAACCCTGGAAGA-3′) and PhlB plus 72 (5′-CGGCCGGCGACGCGTAATAACC-3′). The pSIM19 plasmid was cured from the recombinant strain using a previously described method prior to phenotypic analysis (38).
Genetic complementation of the phlAB and cysE mutations.
Primers PhlAB_compF (5′-AAGCTTGACGTTATCGCCCGCACCTTTACC-3′) and PhlAB_compR (5′-GAATTCGCACGGCGGACACCTACCTACA-3′) were used to amplify the phlAB ORFs and the 219-bp upstream sequence from UMH9 genomic DNA. Primers CysE_compF (5′-AAGCTTGCGAGCCCGAAAGGACGAAAAACC-3′) and CysE_compR (5′-GAATTCGCTGACCGCCGCCTGAAAAACACA-3′) were used to amplify the cysE ORF along with 134-bp upstream sequence. Both PCR products were amplified with Easy A polymerase (Agilent) for incorporation into plasmid pCR2.1-TOPO (Invitrogen). Recombinant plasmids were digested with HindIII and EcoRI, and the phlAB- and cysE-containing fragments were ligated separately to pBBR1MCS-5. Complementation plasmids were sequenced to verify the proper insertion of phlAB and cysE. Plasmids were transformed into S. marcescens strains via electroporation.
Quantitation of extracellular phospholipase activity.
Extracellular phospholipase activity was measured using the fluorogenic PLA substrate bis-BODIPYFL C11-PC (Invitrogen). Bacteria were cultured overnight at 30°C in LB medium, or M9 plus CA with or without supplementation of 1 mM cysteine or 10 mM OAS. Culture aliquots were centrifuged to remove bacteria, and 50 μl of supernatant was transferred into 96-well microplates. The substrate was prepared and mixed with culture supernatants according to the manufacturer's recommendations. Fluorescence (excitation at 480 nm and emission at 350 nm) was measured after 1 h using a fluorescence plate reader. A PLA2 enzyme from bovine pancreas (Sigma-Aldrich) was used as a positive control in the assay (data not shown).
Motility assays.
Swarming and swimming motility were assayed on LB plates containing 0.6% or 0.25% agar, respectively. To assess swarming motility, 10 μl of bacteria from overnight cultures in LB liquid medium were spotted on plates and allowed to dry, and then incubated for 16 h at 30°C. Swimming motility was assayed by stab-inoculating the center of the swim agar and then incubating for 8 h at 30°C. The motility of each strain was determined by measuring the diameters of the resulting growth zones. Plates were imaged using a Q-Count automated colony counter (Spiral Biotech).
Growth of S. marcescens strains.
For growth analysis in defined medium, bacteria were cultured overnight in LB medium and then collected by centrifugation, washed with M9 base medium, and used to inoculate experimental cultures at an optical density at 600 nm (OD600) of 0.01 in 100-well microplates. M9 medium was supplemented where indicated with 0.1% CA, OAS, or l-cysteine at the given concentrations. For growth analysis in LB medium, bacteria were taken directly from overnight cultures and subcultured to an OD600 of 0.01. Bacterial growth from triplicate cultures was measured every 15 min using a BioScreenC instrument during incubation at 37°C with continuous shaking.
RNA purification and qRT-PCR.
S. marcescens strains were cultured in M9 plus CA overnight and then subcultured in M9 plus CA in the presence or absence of 1 mM cysteine or 10 mM OAS for 4.5 h at 30°C. Cultures were treated with 2 volumes of RNAprotect (Qiagen) at room temperature for 5 min prior to the collection of bacteria by centrifugation. Total RNA was purified using the RNeasy kit (Qiagen) and DNase treated using the Turbo DNA-free kit (Ambion) and repurified.
Each cDNA synthesis reaction mixture consisted of 2 μg of total RNA, random hexamers, 1 U RNaseOUT recombinant RNase inhibitor (Invitrogen), and 200 U Superscript III reverse transcriptase (Invitrogen). Control reaction mixtures for the detection of contaminating genomic DNA were prepared in the same manner except that Superscript III was replaced by diethyl pyrocarbonate (DEPC)-treated water. The remaining RNA template was degraded by the addition of 2 U RNaseH (Invitrogen). The purification of cDNA was accomplished using a QIAquick PCR purification kit (Qiagen).
Purified cDNA was diluted in water 1:5, and 5 μl was aliquoted in triplicates for use as the template for qPCR. The following oligonucleotides were used in the amplification of ∼100-bp internal regions of target genes: gyrBF (5′-CTCTTCTCAGACCAAGGACAAG-3′) and gyrBR (5′-GATTACCTGATGGAGAATCCGG-3′); phlAF (5′-GTTTCCAGGCCGGGATTTA-3′) and phlAR (5′-TGTACTGCACATCGTCATAGC-3′); fliPF (5′-CAAGGTCTATCAGGATGCCTAC-3′) and fliPR (5′-GTTTGGCGCAGCATGAAT-3′); fliCF (5′-GAGAAAGAAGTGGCTCCTACTG-3′) and fliCR (5′-GTCTACGGTCGCCTTGAAATA-3′); flhDF (5′-CGATGTTCCGTCTTGGTATTGA-3′) and flhDR (5′-GTTAAAGCGGAAGTGGCAAAC-3′); and fliAF (5′-GATGCTGGAAGGGCATGAA-3′) and fliAR (5′-ACAGCGTCAGCACCATTT-3′). The gyrB transcript was used as an internal control. Brilliant III Ultra-Fast SYBR green (Agilent) was used for monitoring the product amplification on a Stratagene Mx3000P qPCR system. The relative fold change was calculated using the comparative threshold cycle (CT) method (39). PCR amplification efficiency controls were performed for each primer set and dissociation curves were conducted to verify the amplification of a single product.
Murine UTI model.
Overnight cultures of UMH9 and phlAB in LB were subcultured at an OD600 of 0.1 for ∼2 h at 37°C. Bacteria were collected by centrifugation and resuspended in phosphate-buffered saline (PBS) to a density of 109 CFU/ml. Competition infections were conducted with a 1:1 mixture of the UMH9 parent strain and the phlAB mutant. Female C57BL/6 mice (6 to 8 weeks old) were infected with a 0.05-ml volume containing 5 × 107 total CFU directly into the bladder as described previously (22). Bladder homogenates in PBS were plated on LB agar and in LB containing kanamycin to enumerate CFU. CI values were calculated from the following ratios: (CFUphlAB/CFUwild-type)output/(CFUphlAB/CFUwild-type)input. All animal experiments were conducted using protocols approved by the Institutional Animal Care and Use Committee.
ACKNOWLEDGMENTS
We thank Andrew Machnacki for contributions to the genetic screen and Sara Smith for animal work.
This work was supported by Public Health Service grant AI059722 from the National Institutes of Health (to H.L.T.M).
REFERENCES
- 1.Mahlen SD. 2011. Serratia infections: from military experiments to current practice. Clin Microbiol Rev 24:755–791. doi: 10.1128/CMR.00017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Givskov M, Olsen L, Molin S. 1988. Cloning and expression in Escherichia coli of the gene for extracellular phospholipase-A1 from Serratia liquefaciens. J Bacteriol 170:5855–5862. doi: 10.1128/jb.170.12.5855-5862.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimuta K, Ohnishi M, Iyoda S, Gotoh N, Koizumi N, Watanabe H. 2009. The hemolytic and cytolytic activities of Serratia marcescens phospholipase A (PhlA) depend on lysophospholipid production by PhlA. BMC Microbiol 9:261. doi: 10.1186/1471-2180-9-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Givskov M, Molin S. 1993. Secretion of Serratia liquefaciens phospholipase from Escherichia coli. Mol Microbiol 8:229–242. doi: 10.1111/j.1365-2958.1993.tb01567.x. [DOI] [PubMed] [Google Scholar]
- 5.Labbate M, Zhu H, Thung L, Bandara R, Larsen MR, Willcox MD, Givskov M, Rice SA, Kjelleberg S. 2007. Quorum-sensing regulation of adhesion in Serratia marcescens MG1 is surface dependent. J Bacteriol 189:2702–2711. doi: 10.1128/JB.01582-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Givskov M, Molin S. 1992. Expression of extracellular phospholipase from Serratia liquefaciens is growth-phase-dependent, catabolite-repressed and regulated by anaerobiosis. Mol Microbiol 6:1363–1374. doi: 10.1111/j.1365-2958.1992.tb00857.x. [DOI] [PubMed] [Google Scholar]
- 7.Komeda Y. 1982. Fusions of flagellar operons to lactose genes on a Mu lac bacteriophage. J Bacteriol 150:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg HC. 2003. The rotary motor of bacterial flagella. Annu Rev Biochem 72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 9.Givskov M, Eberl L, Christiansen G, Benedik MJ, Molin S. 1995. Induction of phospholipase and flagllear synthesis in Serratia liquefaciens is controlled by expression of the flagellar master operon flhD. Mol Microbiol 15:445–454. doi: 10.1111/j.1365-2958.1995.tb02258.x. [DOI] [PubMed] [Google Scholar]
- 10.Komeda Y, Suzuki H, Ishidsu JI, Iino T. 1976. The role of cAMP in flagellation of Salmonella typhimurium. Mol Gen Genet 142:289–298. [DOI] [PubMed] [Google Scholar]
- 11.Stella NA, Kalivoda EJ, O'Dee DM, Nau GJ, Shanks RM. 2008. Catabolite repression control of flagellum production by Serratia marcescens. Res Microbiol 159:562–568. doi: 10.1016/j.resmic.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young GM, Schmiel DH, Miller VL. 1999. A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc Natl Acad Sci U S A 96:6456–6461. doi: 10.1073/pnas.96.11.6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen S, Young GM. 2002. Essential role for cyclic AMP and its receptor protein in Yersinia enterocolitica virulence. Infect Immun 70:3665–3672. doi: 10.1128/IAI.70.7.3665-3672.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmiel DH, Wagar E, Karamanou L, Weeks D, Miller VL. 1998. Phospholipase A of Yersinia enterocolitica contributes to pathogenesis in a mouse model. Infect Immun 66:3941–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson MT, Mitchell LA, Zhao L, Mobley HLT. 2017. Capsule production and glucose metabolism dictate fitness during Serratia marcescens bacteremia. mBio 8:e00740-17. doi: 10.1128/mBio.00740-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denk D, Bock A. 1987. l-cysteine biosynthesis in Escherichia coli: nucleotide sequence and expression of the serine acetyltransferase (cysE) gene from the wild-type and a cysteine-excreting mutant. J Gen Microbiol 133:515–525. [DOI] [PubMed] [Google Scholar]
- 17.Kredich NM. 2008. Biosynthesis of cysteine. EcoSal Plus 2008. doi: 10.1128/ecosalplus.3.6.1.11. [DOI] [PubMed] [Google Scholar]
- 18.Kredich NM, Tomkins GM. 1966. The enzymic synthesis of l-cysteine in Escherichia coli and Salmonella typhimurium. J Biol Chem 241:4955–4965. [PubMed] [Google Scholar]
- 19.Chevance FF, Hughes KT. 2008. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol 6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flores-Diaz M, Monturiol-Gross L, Naylor C, Alape-Giron A, Flieger A. 2016. Bacterial sphingomyelinases and phospholipases as virulence factors. Microbiol Mol Biol Rev 80:597–628. doi: 10.1128/MMBR.00082-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su LH, Ou JT, Leu HS, Chiang PC, Chiu YP, Chia JH, Kuo AJ, Chiu CH, Chu C, Wu TL, Sun CF, Riley TV, Chang BJ, Infection Control Group . 2003. Extended epidemic of nosocomial urinary tract infections caused by Serratia marcescens. J Clin Microbiol 41:4726–4732. doi: 10.1128/JCM.41.10.4726-4732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane MC, Lockatell V, Monterosso G, Lamphier D, Weinert J, Hebel JR, Johnson DE, Mobley HL. 2005. Role of motility in the colonization of uropathogenic Escherichia coli in the urinary tract. Infect Immun 73:7644–7656. doi: 10.1128/IAI.73.11.7644-7656.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubori T, Matsushima Y, Nakamura D, Uralil J, Lara-Tejero M, Sukhan A, Galan JE, Aizawa SI. 1998. Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science 280:602–605. doi: 10.1126/science.280.5363.602. [DOI] [PubMed] [Google Scholar]
- 24.Abby SS, Rocha EP. 2012. The non-flagellar type III secretion system evolved from the bacterial flagellum and diversified into host-cell adapted systems. PLoS Genet 8:e1002983. doi: 10.1371/journal.pgen.1002983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young BM, Young GM. 2002. YplA is exported by the Ysc, Ysa, and flagellar type III secretion systems of Yersinia enterocolitica. J Bacteriol 184:1324–1334. doi: 10.1128/JB.184.5.1324-1334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlinsey JE, Tanaka S, Bettenworth V, Yamaguchi S, Boos W, Aizawa SI, Hughes KT. 2000. Completion of the hook-basal body complex of the Salmonella typhimurium flagellum is coupled to FlgM secretion and fliC transcription. Mol Microbiol 37:1220–1231. doi: 10.1046/j.1365-2958.2000.02081.x. [DOI] [PubMed] [Google Scholar]
- 27.Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. 1993. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science 262:1277–1280. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- 28.Castelli ME, Fedrigo GV, Clementin AL, Ielmini MV, Feldman MF, Garcia Vescovi E. 2008. Enterobacterial common antigen integrity is a checkpoint for flagellar biogenesis in Serratia marcescens. J Bacteriol 190:213–220. doi: 10.1128/JB.01348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castelli ME, Vescovi EG. 2011. The Rcs signal transduction pathway is triggered by enterobacterial common antigen structure alterations in Serratia marcescens. J Bacteriol 193:63–74. doi: 10.1128/JB.00839-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eberl L, Molin S, Givskov M. 1999. Surface motility of Serratia liquefaciens MG1. J Bacteriol 181:1703–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuyama T, Bhasin A, Harshey RM. 1995. Mutational analysis of flagellum-independent surface spreading of Serratia marcescens 274 on a low-agar medium. J Bacteriol 177:987–991. doi: 10.1128/jb.177.4.987-991.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chrisope GL, Fox CW, Marshall RT. 1976. Lecithin agar for detection of microbial phospholipases. Appl Environ Microbiol 31:784–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song JK, Kim MK, Rhee JS. 1999. Cloning and expression of the gene encoding phospholipase A1 from Serratia sp. MK1 in Escherichia coli. J Biotechnol 72:103–114. [DOI] [PubMed] [Google Scholar]
- 35.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 36.Wilson K. 2001. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol 56:2.4.1–2.4.5. [DOI] [PubMed] [Google Scholar]
- 37.Goodman AL, Wu M, Gordon JI. 2011. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc 6:1969–1980. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomason LC, Sawitzke JA, Li X, Costantino N, Court DL. 2014. Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 106:1.16.11–1.16.39. [DOI] [PubMed] [Google Scholar]
- 39.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 40.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Datta S, Costantino N, Court DL. 2006. A set of recombineering plasmids for Gram-negative bacteria. Gene 379:109–115. doi: 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 42.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]