

Abstract
Recurrent triple‐negative breast cancer (TNBC) needs new therapeutic targets. Src homology region 2 domain‐containing phosphatase‐1 (SHP‐1) can act as a tumor suppressor by dephosphorylating oncogenic kinases. One major target of SHP‐1 is STAT3, which is highly activated in TNBC. In this study, we tested a sorafenib analogue SC‐60, which lacks angiokinase inhibition activity, but acts as a SHP‐1 agonist, in TNBC cells. SC‐60 inhibited proliferation and induced apoptosis by dephosphorylating STAT3 in both a dose‐ and time‐dependent manner in TNBC cells (MDA‐MB‐231, MDA‐MB‐468, and HCC1937). By contrast, ectopic expression of STAT3 rescued the anticancer effect induced by SC‐60. SC‐60 also increased the SHP‐1 activity, but this effect was inhibited when the N‐SH2 domain (DN1) was deleted or with SHP‐1 point mutation (D61A), implying that SHP‐1 is the major target of SC‐60 in TNBC. The use of SC‐60 in combination with docetaxel synergized the anticancer effect induced by SC‐60 through the SHP‐1/STAT3 pathway in TNBC cells. Importantly, SC‐60 also displayed a significant antitumor effect in an MDA‐MB‐468 xenograft model by modulating the SHP‐1/STAT3 axis, indicating the anticancer potential of SC‐60 in TNBC treatment. Targeting SHP‐1/p‐STAT3 and the potential combination of SHP‐1 agonist with chemotherapeutic docetaxel is a feasible therapeutic strategy for TNBC.
Keywords: SHP‐1 agonist, STAT3, triple‐negative breast cancer
Abbreviations
- SHP‐1
src homology region 2 domain‐containing phosphatase‐1
- STAT3
signal transducer and activator of transcription 3
- TNBC
triple‐negative breast cancer
1. Introduction
Triple‐negative breast cancer (TNBC), a heterogeneous breast cancer subtype, is known for its poor prognosis and high rate of metastasis. In addition, a considerable proportion of patients with TNBC are also associated with BRCA gene mutation (Lehmann et al., 2011). Despite the discovery of the poly (ADP‐ribose) polymerase (PARP) inhibitors that interfere with DNA damage repair and their implications in BRCA‐deficient TNBCs, currently there are no approved targeted therapies for TNBC (Mayer et al., 2014).
Signal transducer and activator of transcription 3 (STAT3) mediates a plethora of cellular functions in response to cell stimuli by cytokines (such as interleukin 6; Darnell et al., 1994) and growth factors (such as epidermal growth factor; Song and Grandis, 2000). Upon phosphorylation at tyrosine 705 residue, STAT3 transcriptionally regulated genes involved in cell growth, division, cell movement, apoptosis, and so on (Fukada et al., 1996; Kalluri, 2003; Zhang et al., 2005). In cancers, including TNBC, STAT3 is constitutively activated and frequently associated with poor prognosis and tumor resistance to anticancer therapy (Banerjee and Resat, 2015; D'Anello et al., 2010; Marotta et al., 2011; Real et al., 2002; Wei et al., 2014). The tumor resistance to anticancer therapy can be attributed to the participation of STAT3 in antiapoptosis by upregulating antiapoptotic proteins (bcl‐2, Mcl‐1, survivin, etc.; Banerjee and Resat, 2015; Berishaj et al., 2007; Diaz et al., 2006; Gritsko et al., 2006; Hartman et al., 2013) or by activating cell cycle mediators (such as cyclin D1) and many other STAT3‐regulated genes involved in prosurvival signaling and self‐renewal of cancer stem cells (Rajendran et al., 2012; Tan et al., 2014; Yao et al., 2011). Increased STAT3 activity has also been linked to the development of chemoresistance in TNBC (Gariboldi et al., 2007), as well as associated with metastasis promotion in TNBC (Lee et al., 2011). Moreover, newly identified cancer‐promoting functions of STAT3 – its role in mitochondria, epigenetic regulation, cancer stem cells, obesity, and premetastatic niches – further highlight the importance of targeting STAT3 in cancers (Yu et al., 2014). Taken together, these findings suggest that targeting STAT3 has therapeutic potential and might offer clinical benefits for patients with TNBC.
A number of agents and natural compounds have been reported to inhibit STAT3 using various strategies (Chai et al., 2016; Furtek et al., 2016). Common STAT3‐targeting approaches include inhibiting upstream tyrosine kinases that phosphorylate/activate STAT3 (such as JAKs), and small molecules blocking functional STAT3 dimerization via interfering with the SH2 domains of STAT3 (Furtek et al., 2016). Notably, ruxolitinib, a JAK1/2 tyrosine kinase inhibitor, is currently being tested in phase II trials for solid tumors such as HER‐2‐negative metastatic breast cancers (NCT02120417) and pancreatic cancers (Hurwitz et al., 2014). In addition, compounds suppressing STAT3 phosphorylation independently of JAK inhibition are also attracting attention. Another emerging strategy targeting is the negative regulation of STAT3 signals (Fan et al., 2014; Liu et al., 2013, 2014; Tai et al., 2011, 2014a,b). Src homology region 2 domain‐containing phosphatase 1 (SHP‐1), a nonreceptor protein tyrosine phosphatase, is one of the negative regulators of phosphorylated STAT3 (p‐STAT3; Lopez‐Ruiz et al., 2011). SHP‐1 is composed of two SH‐2 domains (N‐SH2 and C‐SH2) and one catalytic PTP domain (Yang et al., 1998). Studies have shown that the activity of SHP‐1 is also regulated by the conformational rearrangement upon substrate/chemical binding: The N‐SH2 domain is released from the interaction with the PTP domain exposing the catalytic site of the PTP domain, thereby enhancing SHP‐1 activity (Qin et al., 2005; Wang et al., 2011; Yang et al., 2003). Interestingly, we identified that multiangiokinase inhibitor sorafenib also acts as a direct enhancer of SHP‐1 (Tai et al., 2011, 2014b). Accordingly, we developed a series of sorafenib derivatives that are devoid of angiokinase (VEGFR/PDGFR) inhibition and demonstrated that they inhibited p‐STAT3 via increasing SHP‐1 activity (Chen et al., 2011, 2012c; Su et al., 2012). We previously demonstrated the in vitro and in vivo anticancer activity of a SHP‐1 agonist, SC‐43, in TNBC cells (Liu et al., 2013). More recently, we showed a chemical dimeric sorafenib derivative, SC‐60, with survival benefits compared with sorafenib in a hepatocellular carcinoma orthotopic model (Fan et al., 2014). In the current study, we report the effects of SC‐60 in TNBC cells.
2. Materials and methods
2.1. Reagents and antibodies
SC‐60 was synthesized by Hinova Pharmaceuticals Inc. (Chengdu, China), the MW of SC‐60 is 473, and its structure and solubility are described in Fig. S1. For in vitro studies, SC‐60 at various concentrations was dissolved in dimethyl sulfoxide (DMSO) and added to cells in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA). The final DMSO concentration was 0.1% after addition to the medium. For in vivo studies, SC‐60 was dissolved in 50% (v/v) propylene glycol and 50% (v/v) Solutol® HS‐15. The SHP‐1 inhibitor PTP inhibitor III (CAS 29936‐81‐0) was purchased from Cayman Chemical (Ann Arbor, MI, USA). Plasmids of human wild‐type STAT3 were encoded by pCMV6 vector with myc‐tag. The mutant SHP‐1 constructs (DN1 and D61A) have been generated to mimic the open‐form structure of SHP‐1 as previously described (Tai et al., 2014b). Antibodies for immunoblotting such as p‐VEGFR2, VEGFR2, p‐PDGFRβ, PDGFβ, p‐JAK1, JAK1, p‐JAK2, JAK2, p‐SRC, SRC, p‐STAT3, STAT3, and survivin were from Cell Signaling (Danvers, MA, USA). SHP‐1, cyclin D1, and Mcl‐1 antibodies were purchased from Abcam (Cambridge, MA, USA). Other antibodies such as poly (ADP‐ribose) polymerase (PARP) and cleaved caspase 3 were obtained from Cell Signaling Technology.
2.2. Cell culture and western blot analysis
The MCF10A human breast epithelial cell line, MCF7 luminal breast cancer cells, and TNBC (MDA‐MB‐231, MDA‐MB‐468, and HCC‐1937) cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). All breast cancer cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 0.1 mm nonessential amino acids, 2 mm l‐glutamine, 100 U·mL−1 penicillin G, 100 μg·mL−1 streptomycin sulfate, and 25 μg·mL−1 amphotericin B in a 37 °C humidified incubator and an atmosphere of 5% CO2 in air. Lysates of breast cancer cells treated with drugs at the indicated doses and times were prepared for immunoblotting of p‐STAT3, STAT3, and other cells. Western blot analysis was performed as previously reported (Lehmann et al., 2015; Liu et al., 2013).
2.3. DNA fragmentation and apoptosis analysis
Cytoplasmic histone‐associated DNA fragments were determined as a measurement of apoptotic cells by the Cell Death Detection ELISAPLUS Kit (Roche, Indianapolis, IN, USA) according to the manufacturer's instructions. Drug‐induced apoptotic cell death was assessed using sub‐G1 analysis of propidium iodide‐stained cells by flow cytometry and western blot analysis of PARP cleavage.
2.4. Real‐time quantitative PCR
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen), and real‐time quantitative PCR was performed in a LightCycler 480II instrument (Roche Diagnostics) using a LightCycler 480 SYBR Green I Master Kit (Roche Diagnostics), using specific primers for human cyclin D1 (forward primer, 5′‐GGATGCTGGAGGTCTGCGA‐3′; reverse primer, 5′‐AGAGGCCACGAACATGCAAG‐3′; 146 bp), human Mcl‐1 (forward primer, 5′‐GGTGCCTTTGTGGCTAAACA‐3′; reverse primer, 5′‐ACCCATCCCAGCCTCTTTGT‐3′; 133 bp), human survivin (forward primer, 5′‐AGAACTGGCCCTTCTTGGAGG‐3′; reverse primer, 5′‐CTTTTTATGTTCCTCTATGGGGTC‐3′; 170 bp), and the glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) gene was chosen as an internal control (forward primer, 5′‐CGACCACTTTGTCAAGCTCA‐3′; reverse primer, 5′‐AGGGGTCTACAT GGCAACTG‐3′; 228 bp).
2.5. Gene knockdown using small interfering RNA
Smart‐pool small interfering RNAs (siRNAs) including the control (D‐001810‐10) and SHP‐1 (PTPN6, L‐009778‐00‐0005) were purchased from Dharmacon (Chicago, IL, USA). The knockdown procedure was as described previously. Cells were transfected with siRNA (final concentration of 100 nm) in six‐well plates using the liposome transfection reagent Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 72 h, the medium was replaced and the breast cancer cells were incubated with nintedanib, harvested, and separated for western blot analysis and apoptosis analysis by flow cytometry.
2.6. In vitro STAT3 Activity Assay
A Cignal Stat3 Reporter Kit (SABiosciences, Valencia, CA, USA) was used to measure the in vitro Stat3 activity. Cells were seeded in a 96‐well plate and transfected with reference pCMV‐Renilla luciferase plasmid with a plasmid driven by the promoter region containing STAT3‐specific binding sites and the constitutively expressing Renilla construct encodes the Renilla luciferase reporter gene and acts as an internal control for normalizing transfection efficiencies. After incubation for 48 h, the cells were treated with SC‐60 for 6 h and lysed with passive buffer. The lysates were transferred to a glass tube, and promoter activity was determined by Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer's instructions. Luciferase activities were measured on a GloMax 20/20 Luminometer (Promega). All the luciferase activities of samples were normalized with cells treated with DMSO.
2.7. SHP‐1 phosphatase activity
A RediPlate 96 EnzChek Tyrosine Phosphatase Assay Kit (R‐22067) was used for SHP‐1 activity assay (Molecular Probes, Carlsbad, CA, USA). The method was as described previously (Liu et al., 2013). Briefly, the protein extracts of breast cancer cells were incubated with anti‐SHP‐1 antibody in immunoprecipitation buffer overnight. Then, the Protein G‐Sepharose 4 Fast Flow (GE Healthcare, Piscataway, NJ, USA) was added to each sample followed by incubation for three hours at 4 °C with rotation and then assayed for phosphatase activity.
2.8. Xenograft tumor growth
The animal experiments were approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital. All experimental procedures using mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Taipei Veterans General Hospital. Female NCr athymic nude mice (5–7 weeks of age) were obtained from the National Laboratory Animal Center (Taipei, Taiwan, Republic of China). The mice were housed in groups and maintained in a specific pathogen‐free environment. Each mouse was inoculated subcutaneously in the dorsal flank under isoflurane anesthesia with 2 × 106 breast cancer cells suspended in 0.1 mL of serum‐free medium containing 50% Matrigel (BD Biosciences, San Jose, CA, USA). Tumors were measured using calipers, and their volumes were calculated using a standard formula: width2 × length × 0.52. When tumors reached around 100 mm3, mice were administered SC‐60 (20 mg·kg−1 oral) three times a week. Controls received vehicle (50% (v/v) propylene glycol and 50% (v/v) Solutol® HS‐15). Upon termination of treatment, mice were sacrificed and xenografted tumors were harvested and assayed for subsequent experiments.
2.9. Statistical analysis
Data are expressed as mean ± SD or SE. Statistical comparisons were based on nonparametric tests, and statistical significance was defined as a P value of less than 0.05. For survival analysis, progression‐free survival curves of patients were generated using the Kaplan–Meier method and compared by performing a log‐rank test. All statistical analyses were carried out using spss for Windows software, version 12.0 (SPSS, Chicago, IL, USA).
3. Results
3.1. SC‐60 shows a growth inhibitory effect in human breast cancer cells
To evaluate the efficacy of SC‐60 on human TNBC cells, we treated three human TNBC lines with different doses of SC‐60 for 72 h, and the cell viability was analyzed by MTT assay. As shown in Fig. 1A, SC‐60 dose dependently inhibited cell growth of MDA‐MB‐468, HCC1937, and MDA‐MB‐231 cells. In addition, flow cytometry assay revealed that SC‐60 treatment resulted in increases in the percentage of apoptotic cells in the aforementioned cell lines in a dose (Fig. 1B)‐ and time (Fig. 1C)‐dependent manner. In accordance with the above results, SC‐60 treatment also led to increased DNA fragmentation in human breast cancer cells in a dose‐dependent manner (Fig. 1D). To further test the cytotoxic effect of SC‐60 on normal MCF‐10A human breast epithelial cells and MCF‐7 luminal breast cancer cells, we also performed the MTT assay and flow cytometry analysis. As shown in Fig. S2A, SC‐60 showed antiproliferative effect on MCF‐10A as well as MCF‐7 cells. In addition, SC‐60 induced mild apoptosis‐inducing effect on MCF‐10A at 5 μm and MCF‐7 at 2 and 5 μm compared to that of TNBC cell lines (Figs 1B and S2B). Our results showed that SC‐60 had more cytotoxic effects on breast cancer cells than on normal breast MCF‐10A cells.
Figure 1.
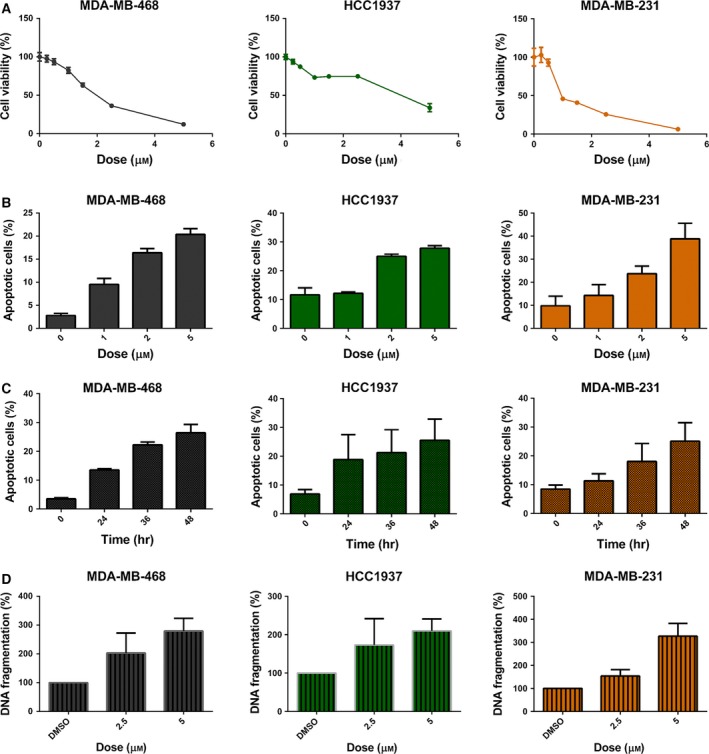
SC‐60 shows antiproliferative effects in human triple‐negative breast cancer cells. (A) Cells were exposed to SC‐60 at the indicated doses for 72 h, and cell viability was assessed by MTT assay. (B,C) Flow cytometry assay revealed that SC‐60 treatment resulted in increased percentage of apoptotic cells in the aforementioned cell lines in a dose (B)‐ and time (C)‐dependent manner. (D) SC‐60 treatment led to increased DNA fragmentation in TNBC cells in a dose‐dependent manner. Means of at least three independent experiments performed in triplicate are shown. Data are shown as mean ± SD.
3.2. SC‐60 enhanced cell apoptosis through p‐STAT3 inhibition
To investigate the mechanisms by which SC‐60 inhibited cell growth and induced cell apoptosis in TNBC, we analyzed the protein expressions of p‐STAT3 and its downstream proteins that have been demonstrated to be involved in cancer cell proliferation and survival. We found that SC‐60 inhibited the expression of p‐STAT3 and its downstream effectors including Mcl‐1, cyclin D1, and survivin in TNBC cells in a dose‐ and time‐dependent manner. Because STAT3 has been reported as a transcription factor, therefore, we also checked whether SC‐60 affected the mRNA levels of its downstream molecules. As shown in Fig. S3, SC‐60 indeed decreased the mRNA levels of STAT3 downstream target genes, cyclin D1, Mcl‐1, and survivin in MDA‐MB‐231 cells. Furthermore, the levels of apoptosis‐related proteins were also activated by SC‐60 in a dose‐ and time‐dependent manner in TNBC cells (Fig. 2A,B). To analyze whether the activity of STAT3 is affected by SC‐60, luciferase assays were performed using the Cignal STAT3 Reporter Assay Kit. TNBC cells were transfected with reference pCMV‐Renilla luciferase plasmid with a plasmid driven by the promoter region containing STAT3‐specific binding sites and treated with SC‐60. Results showed that SC‐60 dose dependently decreased the activity of STAT3 in TNBC cells (Fig. 2C). To further validate the role of STAT3 in SC‐60‐induced apoptosis in TNBC cells, we established stable STAT3‐overexpressing MDA‐MB‐468 cells. As shown in Fig. 2D, constitutively expressing STAT3 reversed SC‐60‐mediated p‐STAT3 inhibition and suppressed the apoptotic effect of SC‐60 on MDA‐MB‐468 cells, indicating that STAT3 mediates SC‐60‐induced apoptosis in TNBC cells.
Figure 2.
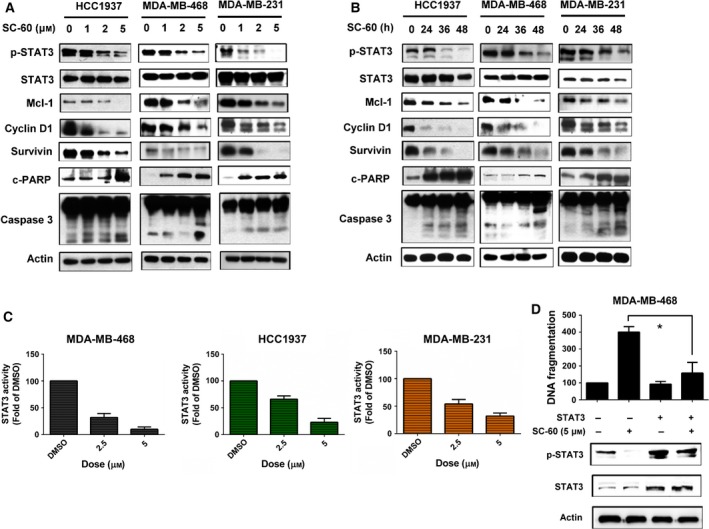
SC‐60 enhances apoptosis and reduces p‐STAT3 signaling in TNBC cells. (A,B) The effects of SC‐60 on p‐STAT3 and its downstream molecules (Mcl‐1, cyclin D1, and survivin) were analyzed by western blot. Cells were treated with SC‐60 at the (A) indicated doses for 48 h or (B) treated with SC‐60 (5 μm) at the indicated times. (C) Cells were transfected with inducible STAT3‐responsive firefly luciferase construct and constitutively expressing Renilla luciferase construct as internal control for 48 h; then, the cells were treated with SC‐60 at indicated dose for 6 h. The activities of STAT3 were measured by dual luciferase assay. The activities of STAT3 of tested samples were normalized with cells treated with DMSO. (D) Overexpression of STAT3 reversed the apoptotic effect of SC‐60. MDA‐MB‐468 cells were transfected with STAT3‐expressing vector with myc‐tag for 24 h and then treated with DMSO or SC‐60 at 5 μm for another 24 h. DNA fragmentation was measured, and the effect on p‐STAT3 was analyzed by western blot. Means of at least three independent experiments performed in triplicate are shown. *P < 0.05. Data are shown as mean ± SD.
As there are other molecular events that typically modulate STAT3 activity, such as VEGFR2, PDGFRβ, JAK1, JAK2, and ERK1/2 (Schreiner et al., 2002; Tian and An, 2004; Yu et al., 2014), we also examined the effects of SC‐60 on these molecules and found that SC‐60 did not affect VEGFR2, PDGFRβ, JAK1, JAK2, and ERK1/2 (Fig. S4).
3.3. SHP‐1 mediates effects of SC‐60 on p‐STAT3 inhibition in TNBC cells
To further determine the mechanism by which SC‐60 inhibits STAT3 phosphorylation, we analyzed the activity of its phosphatase, SHP‐1, in SC‐60‐treated cells. As shown in Fig. 3A, SC‐60 increased SHP‐1 activity significantly in three TNBC cell lines. We then added a specific SHP‐1 inhibitor, an α‐haloacetophenone derivative PTP inhibitor III that acts as a covalent inhibitor of PTPs and binds the catalytic domain of SHP‐1 (Arabaci et al., 1999), in MDA‐MB‐468 cells and found that it significantly reduced SC‐60‐induced downregulation of p‐STAT3 and apoptosis (Fig. 3B). Similarly, silencing SHP‐1 with small interfering RNA (siRNA) in MDA‐MB‐468 cells inhibited the effects of SC‐60 on p‐STAT3 inhibition and cell apoptosis induction (Figs 3C and S5). Previous studies have indicated that the autoinhibitory structure from the N‐SH2 domain to PTP domain is the major regulator of SHP‐1 activity (Wu et al., 2003; Yang et al., 1998, 2003). The mutant SHP‐1 constructs (DN1 and D61A) shown in Fig. 3D have been generated to mimic the open‐form structure of SHP‐1 (Tai et al., 2014b), which showed higher SHP‐1 activities than wild‐type SHP‐1. Therefore, these two mutants would show more potent inhibition on p‐STAT3 expression due to higher SHP‐1 activities (Fig. 3E,F, left). Moreover, it is expected that SC‐60 would interfere the open/close structures of SHP‐1, thereby increasing the SHP‐1 activity. Indeed, when the SHP‐1 was maintained as open structure (DN1 and D61A mutants), the effects of SC‐60 on p‐STAT3 inhibition (Fig. 3E) and apoptosis were reduced (Fig. 3F, right).
Figure 3.
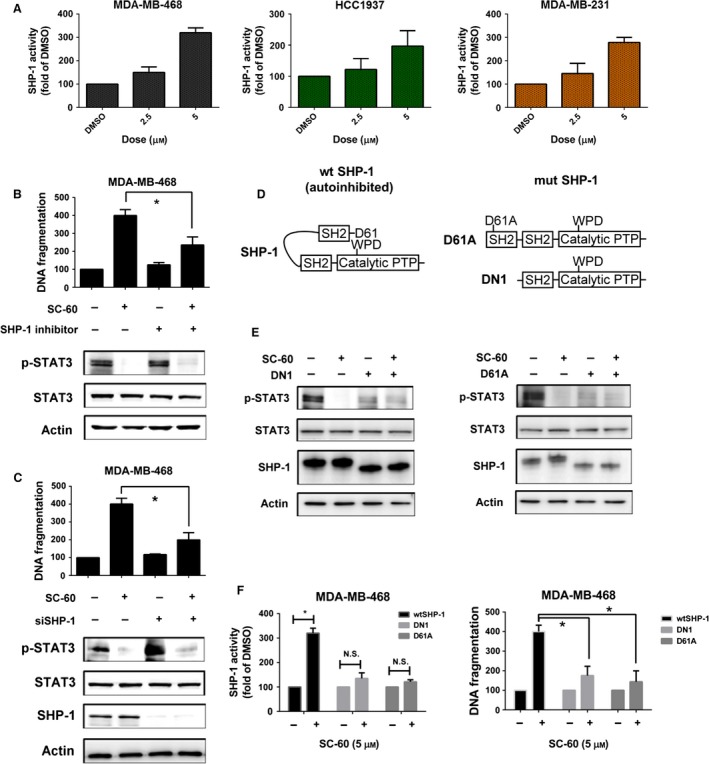
SC‐60 induces cell apoptosis by SHP‐1/p‐STAT3 signaling in TNBC cells. (A) The activities of SHP‐1 were measured at the indicated doses for 48 h in MDA‐MB‐468, HCC1937, and MDA‐MB‐231 TNBC cell lines. (B) The protective effects of SHP‐1 inhibitor on SC‐60‐induced apoptosis in MDA‐MB‐468 cells. Cells were pretreated with 50 μm SHP‐1 inhibitor (PTP inhibitor III) for 1 h and then treated with SC‐60 at 5 μm for 36 h. DNA fragmentation was determined by the Cell Death Detection ELISAPLUS Kit. (C) Knockdown of SHP‐1 reversed the biological effects of SC‐60 on apoptosis (left) and p‐STAT3 inhibition (right). MDA‐MB‐468 cells were transfected with control siRNA (scrambled) or SHP‐1 siRNA for 24 h and then treated with SC‐60 at 5 μm for another 24 h. The protein levels of p‐STAT3, STAT3, SHP‐1, and actin (as loading control) were analyzed by western blot, and DNA fragmentation was measured by Cell Death Detection ELISAPLUS Kit. (D) Schematic representation of wild‐type SHP‐1 (autoinhibited), deletion and single mutants of SHP‐1 (mimic activated SHP‐1). (E,F) MDA‐MB‐468 cells were transfected with mutant SHP‐1 (DN1 or D61A) for 24 h and then treated with SC‐60 at 5 μm for another 24 h. (E) The protein levels of p‐STAT3, STAT3, SHP‐1, and actin (as loading control) were analyzed by western blot. (F) The SHP‐1 activity was assessed by SHP‐1 phosphatase activity (left) and DNA fragmentation (right) was measured by Cell Death Detection ELISAPLUS Kit as described in 2. Means of at least three independent experiments performed in triplicate are shown. *P < 0.05. Data are shown as mean ± SD.
3.4. SC‐60 inhibits MDA‐MB‐468 tumor growth in vivo
To investigate the clinical therapeutic options, we combined SC‐60 with docetaxel in MDA‐MB‐231 cells. This combination increased apoptosis (Fig. 4A, upper), DNA fragmentation (Fig. 4A, middle), and strongly reduced STAT3 phosphorylation (Fig. 4A, lower) in comparison with SC‐60 treatment alone. These results suggest that the combination of a lower dose of SC‐60 with docetaxel provides new therapeutic option in the treatment of patients with TNBC. To determine whether the effect of SC‐60 on TNBC cells is potentially clinically relevant, we tested the in vivo effect of SC‐60 on tumor growth in a xenograft mouse model. We found that SC‐60 treatment showed reduced tumor weight and suppressed tumor growth (Figs 4B and S6), increased SHP‐1 activity (Fig. 4C), and decreased p‐STAT3 and Mcl‐1 expressions in xenografted tumors (Figs 4D and S6). These results indicated that SC‐60 inhibited tumor growth through STAT3 inactivation.
Figure 4.
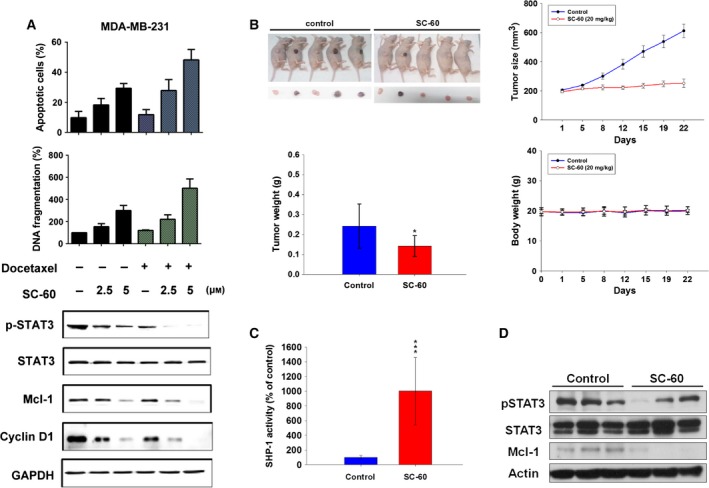
Combination of SC‐60 and docetaxel increases cell apoptosis by reducing p‐STAT3, and SC‐60 diminishes xenograft tumor growth of TNBC cells. (A) Cells were treated with docetaxel (0.1 μm) for 24 h, then treated with SC‐60 at the indicated doses (0, 2.5, and 5 μm) for another 24 h. Effect of docetaxel/SC‐60 combination on cell apoptosis (upper), DNA fragmentation (middle), p‐STAT3 and its downstream signaling (lower) were measured. Means of at least three independent experiments performed in triplicate are shown. Data are shown as mean ± SD. (B–D) MDA‐MB‐468‐bearing mice were treated with vehicle or SC‐60 orally at 20 mg·kg−1 three times a week. (B) Mice images (upper left), growth curves (lower left), tumor weight (upper right), body weight (lower right), (C) SHP‐1 activity, and (D) western blot analysis of p‐STAT3, STAT3, and Mcl‐1 were measured. Data of growth curve (n = 5) are shown as mean ± SE. Data of tumor weight, body weight, and SHP‐1 activity (n = 5) are shown as mean ± SD. *P < 0.05. ***P < 0.001.
4. Discussion
In the current study, we demonstrated the in vitro effectiveness of SC‐60 in three TNBC cell lines MDA‐MB‐468, HCC1937, and MDA‐MB‐231, and its in vivo antitumor efficacy in a MDA‐MB‐468 xenograft mouse model. We confirmed that SC‐60 exhibits its efficacy via the SHP‐1/p‐STAT3 pathway (Fig. 3). Moreover, the potential combination with docetaxel was shown by enhanced apoptosis in vitro (Fig. 4). Our study further reinforces the notion that targeting p‐STAT3 by enhancing SHP‐1 activity may be a useful anticancer therapeutic approach, and also provides a new chemical entity with pharmacological potential.
At present, there are no clinically approved STAT3‐targeted agents. One possible hindrance may be that direct inhibition of STAT3 dimerization requires total inhibition of STAT3 molecules in the cell, which may need high drug concentrations (Chai et al., 2016; Furtek et al., 2016). In contrast, kinase inhibitors or enzyme activators may be feasible approaches (Zhang et al., 2015; Zhong et al., 2015). JAK kinase inhibitors, the so‐called jakinibs, were originally designated as therapy for myeloproliferative diseases (Kontzias et al., 2012). Recently, the therapeutic implication of these jakinibs has moved toward cancer and autoimmune diseases such as rheumatoid arthritis (Kontzias et al., 2012). Indeed, the JAK inhibitor ruxolitinib is currently in clinical trials for solid cancers (Hurwitz et al., 2014) and is closer to clinical approval for cancer therapy compared with other STAT3‐targeting agents. As mentioned earlier, agents aiming to inhibit STAT3 phosphorylation other than JAK inhibitors are also an interesting field of drug development. Previously, we demonstrated that a SHP‐1 enhancer, SC‐43, is effective for TNBC cells (Liu et al., 2013). A number of compounds or drugs have been reported to be capable of enhancing SHP‐1 activity (Fig. 5). Sorafenib analogues with a urea‐based structure (SC agents such as SC‐1, SC‐49, SC‐43, SC‐60, and SC‐78) have been tested as SHP‐1 enhancers for anticancer activity in preclinical settings (Chen et al., 2012c; Fan et al., 2014; Liu et al., 2013; Su et al., 2016; Tai et al., 2011). Among these agents, SC‐60 has been found to form the hydrogen bonding with N280 of the PTP domain through a docking model (Fan et al., 2014). Another class of SHP‐1 enhancer is obatoclax analogue, SC‐2001, increasing SHP‐1 expression through transcription factor RFX‐1 (Chen et al., 2012b; Liu et al., 2014; Su et al., 2012, 2014a,b). The core structure of obatoclax and SC‐2001 includes a pyrrole and indole ring. Nintedanib has been found to elevate SHP‐1 activity through interacting with SHP‐1 at Glu524 through hydrogen bonding in a docking model (Tai et al., 2014c). Although many SHP‐1 activators have been identified (Chen et al., 2012a,b,c; Liu et al., 2014; Su et al., 2014a,b, 2016; Tai et al., 2012), the structure and activity relationship still needs to be further investigated.
Figure 5.
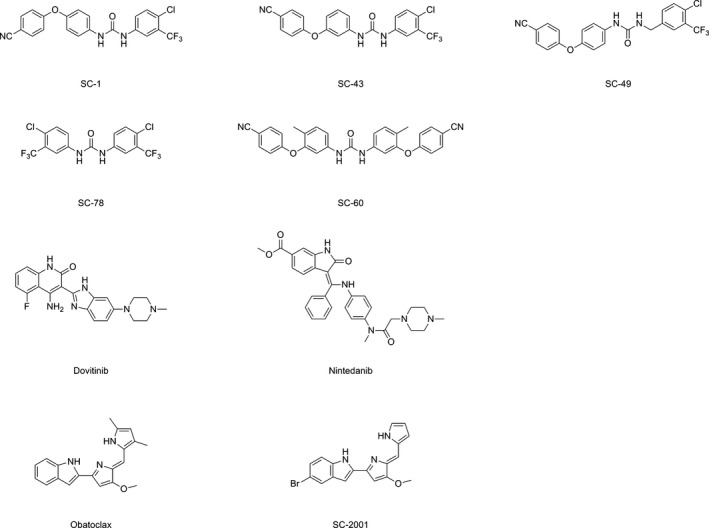
Chemical structures of compounds or drugs that have been reported to enhance SHP‐1 activity.
It is clear that many solid cancers have aberrant and activated JAK/STAT signaling (Roxburgh and McMillan, 2016; Sansone and Bromberg, 2012). Despite STAT3 signaling not being specific for TNBC, targeting STAT3 activation in TNBC is feasible for several reasons: First, currently there are no single pathways specific for therapy for all subtypes of TNBC (Mayer et al., 2014). Given the heterogeneity of TNBC revealed by molecular profiling (Lehmann et al., 2011; Mayer et al., 2014), the general applicability of STAT3 signaling in cancer cells provides an alternative strategy to traditional chemotherapy. Moreover, we also demonstrated the potential combination of chemotherapeutics with SHP‐1 agonists (Fig. 4), suggesting the possibility of combining p‐STAT3 inhibitors with chemotherapy in the future. Furthermore, we recently discovered a VEGF‐A‐dependent autocrine/paracrine loop in TNBC which could be disrupted by SHP‐1 enhancers, suggesting that the SHP‐1/p‐STAT3/VEGF‐A axis is a potential therapeutic target for metastatic TNBC (Su et al., 2016). Interestingly, our results showed that SC‐60 had more cytotoxic effects on breast cancer cells than on normal breast MCF10A cells, and it seemed that TNBC cells might be more sensitive than hormone receptor‐positive MCF‐7 cells in terms of apoptotic effects (Figs 1 and S2). Estrogen is a growth factor for hormone receptor‐positive breast cancer cells and may contribute to chemoresistance (Jiang et al., 2012). Moreover, estrogen receptor (ERα) can bind to STAT3 and JAK2, resulting in enhanced JAK2 activity upstream of STAT3 in response to stimulation which might lead to an increased (ERα)‐dependent cell viability (Binai et al., 2010). However, it remains speculative and more studies are necessary to address possible mechanisms for the differential apoptosis sensitivities among TNBC and hormone receptor‐positive breast cancer cell lines.
Notwithstanding, the prognostic role of p‐STAT3 in cancer patient outcome seems to be conflicting among various solid cancers (Thomas et al., 2015). A recent review summarizing studies on the relationship between JAK/STAT activation and prognosis suggested that most studies have utilized immunohistochemistry to determine p‐STAT3 signaling (Thomas et al., 2015). In several cancers such as prostate, non‐small‐cell lung cancers, cervical cancers, renal cell carcinoma, and glioblastoma, activation of STAT3 or STAT5 is associated with a worse prognosis; conversely, STAT3 is associated with favorable prognosis in breast cancer and in some studies in colorectal cancer and head and neck squamous cell carcinoma (Thomas et al., 2015). This difference in prognosis prediction may be partly due to different tumor biology among cancers, and by the various regulatory mechanisms upstream of p‐STAT3 signaling, for example, endogenous negative regulators such as the suppressor of cytokine signaling family, protein inhibitor of activated STAT (PIAS) proteins and the PTP family, or post‐translational modifications (Chai et al., 2016).
In this study, we showed that SC‐60 acts as a SHP‐1 agonist. SC‐60 increased SHP‐1 activity, thereby decreased p‐STAT3, and subsequently decreased cyclin D1 expression. However, SC‐60 seemed to affect cyclin D1 expression more prominently compared to p‐STAT3 expression (Fig. 2). Cyclin D1 can be regulated by numerous effectors, such as STAT3 (Leslie et al., 2006), ERK1/2 (Balmanno and Cook, 1999), and SRC/PI3K/AKT (Xing et al., 2008) pathways. This may indicate that p‐STAT3 is not the only effector in SC‐60‐mediated cyclin D1 suppression. There might be other STAT3‐independent effects of SC‐60 on cyclin D1. Moreover, previous studies have shown that SHP‐1 also regulates MAPK/ERK pathway by dephosphorylating ERK (Cai et al., 2006) and that lack of SHP‐1 prevents phosphorylation of the active site (Tyr416) on Src (Okenwa et al., 2013). In contrast, our data showed that SC‐60 did not significantly alter the phosphorylation of ERK1/2 (Fig. S4), whereas the phosphorylation of Src on Try416 was found to be mildly decreased with SC‐60 treatment (Fig. S5). However, why SC‐60 (as a SHP‐1 agonist) could affect p‐Src (Try416) is not clearly understood; we suggest that SC‐60 may exert other than SHP‐1 agonist activity (off‐target effect) that decreases p‐Src (Try416) or may through other effectors to decrease the expression of cyclin D1. Nevertheless, further studies are necessary.
In summary, our study demonstrates the preclinical activity of a SHP‐1 agonist SC‐60 in TNBC, the therapeutic implication of targeting SHP‐1/p‐STAT3, and the potential combination of a SHP‐1 agonist with chemotherapeutic docetaxel as a feasible therapeutic strategy for TNBC.
Author contributions
CYL, JCS, and TTH drafted the manuscript. CYL, JCS, TTH, CHL, KYL, WCT, HPY, and CTH conducted in vitro experiments. PYC, WLW, TTH, and JCS conducted animal experiments. CWS, LMT, and KFC helped in data interpretation and statistical analysis. CYL, JCS, TTH, PYC, CTH, and WLW prepared the figures. CWS and KFC designed and synthesized SC‐60. All authors made substantial contributions to the conception or design of the work. All authors have read, revised critically for intellectual content, and approved the final manuscript. All authors agreed with the accuracy and integrity of all parts of the work.
Supporting information
Fig. S1. The chemical structure and solubility of SC‐60.
Fig. S2. Cytotoxicity effect of SC‐60 on MCF‐10A normal human breast epithelial cells and MCF‐7 luminal breast cancer cells.
Fig. S3. SC‐60 decreased the mRNA levels of STAT3 downstream target genes.
Fig. S4. SC‐60 had no obvious effects on VEGFR2, PDGFRβ, JAK1, JAK2 and ERK1/2 in MDA‐MB‐231 cells.
Fig. S5. The effects of SC‐60 on SHP1‐depleted MDA‐MB‐468 cells.
Fig. S6. SC‐60 diminishes xenograft tumor growth of MDA‐MB‐468 cells.
Acknowledgements
This work was supported by grants from the Taiwan Clinical Oncology Research Foundation; the Yen Tjing Ling Medical Foundation (CI‐104‐07); the Ministry of Science and Technology, Taiwan (MOST 103‐2811‐B‐002‐157, MOST 103‐2325‐B‐075‐002, MOST 104‐2628‐B‐075‐001‐MY3, MOST 104‐2811‐B‐002‐030, MOST 104‐2321‐B‐010‐017 and 105‐2314‐B‐002‐190‐MY2); the National Health Research Institutes, Taiwan (NHRI‐EX106‐10608BI); Yang‐Ming Branch of Taipei City Hospital (10601‐62‐020); Taipei Veterans General Hospital (V103C‐141, V104C‐151, V105C‐067 and V106C‐101); the TVGH‐NTUH Joint Research Program (VN103‐08, VN106‐07, and VN105‐09) from Taipei Veterans General Hospital and National Taiwan‐University Hospital, and from the Ministry of Health and Welfare, Executive Yuan, Taiwan (MOHW105‐TDU‐B‐211‐134‐003 and MOHW106‐TDU‐B‐211‐144‐003 for the Center of Excellence and MOHW103TDU‐212‐114‐002 for Cancer Research at Taipei Veterans General). This study was also partially supported by the Chong Hin Loon Memorial Cancer and the Biotherapy Research Center of National Yang‐Ming University, Taipei, Taiwan.
Contributor Information
Ling‐Ming Tseng, Email: lmtseng@vghtpe.gov.tw.
Kuen‐Feng Chen, Email: kfchen1970@ntu.edu.tw.
References
- Arabaci G, Guo XC, Beebe KD, Coggeshall KM and Pei D (1999) alpha‐Haloacetophenone derivatives as photoreversible covalent inhibitors of protein tyrosine phosphatases. J Am Chem Soc 121, 5085–5086. [Google Scholar]
- Balmanno K and Cook SJ (1999) Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP‐1 proteins in CCL39 cells. Oncogene 18, 3085–3097. [DOI] [PubMed] [Google Scholar]
- Banerjee K and Resat H (2015) Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer 138, 2570–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berishaj M, Gao SP, Ahmed S, Leslie K, Al‐Ahmadie H, Gerald WL, Bornmann W and Bromberg JF (2007) Stat3 is tyrosine‐phosphorylated through the interleukin‐6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res 9, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binai NA, Damert A, Carra G, Steckelbroeck S, Lower J, Lower R and Wessler S (2010) Expression of estrogen receptor alpha increases leptin‐induced STAT3 activity in breast cancer cells. Int J Cancer 127, 55–66. [DOI] [PubMed] [Google Scholar]
- Cai J, Jiang WG, Ahmed A and Boulton M (2006) Vascular endothelial growth factor‐induced endothelial cell proliferation is regulated by interaction between VEGFR‐2, SH‐PTP1 and eNOS. Microvasc Res 71, 20–31. [DOI] [PubMed] [Google Scholar]
- Chai EZ, Shanmugam MK, Arfuso F, Dharmarajan A, Wang C, Kumar AP, Samy RP, Lim LH, Wang L, Goh BC et al (2016) Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther 162, 86–97. [DOI] [PubMed] [Google Scholar]
- Chen KF, Chen HL, Liu CY, Tai WT, Ichikawa K, Chen PJ and Cheng AL (2012a) Dovitinib sensitizes hepatocellular carcinoma cells to TRAIL and tigatuzumab, a novel anti‐DR5 antibody, through SHP‐1‐dependent inhibition of STAT3. Biochem Pharmacol 83, 769–777. [DOI] [PubMed] [Google Scholar]
- Chen KF, Su JC, Liu CY, Huang JW, Chen KC, Chen WL, Tai WT and Shiau CW (2012b) A novel obatoclax derivative, SC‐2001, induces apoptosis in hepatocellular carcinoma cells through SHP‐1‐dependent STAT3 inactivation. Cancer Lett 321, 27–35. [DOI] [PubMed] [Google Scholar]
- Chen KF, Tai WT, Hsu CY, Huang JW, Liu CY, Chen PJ, Kim I and Shiau CW (2012c) Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP‐1 phosphatase activity. Eur J Med Chem 55, 220–227. [DOI] [PubMed] [Google Scholar]
- Chen KF, Tai WT, Huang JW, Hsu CY, Chen WL, Cheng AL, Chen PJ and Shiau CW (2011) Sorafenib derivatives induce apoptosis through inhibition of STAT3 independent of Raf. Eur J Med Chem 46, 2845–2851. [DOI] [PubMed] [Google Scholar]
- D'Anello L, Sansone P, Storci G, Mitrugno V, D'Uva G, Chieco P and Bonafe M (2010) Epigenetic control of the basal‐like gene expression profile via Interleukin‐6 in breast cancer cells. Mol Cancer 9, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE Jr, Kerr IM and Stark GR (1994) Jak‐STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Diaz N, Minton S, Cox C, Bowman T, Gritsko T, Garcia R, Eweis I, Wloch M, Livingston S, Seijo E et al (2006) Activation of stat3 in primary tumors from high‐risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res 12, 20–28. [DOI] [PubMed] [Google Scholar]
- Fan LC, Teng HW, Shiau CW, Lin H, Hung MH, Chen YL, Huang JW, Tai WT, Yu HC and Chen KF (2014) SHP‐1 is a target of regorafenib in colorectal cancer. Oncotarget 5, 6243–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada T, Hibi M, Yamanaka Y, Takahashi‐Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K and Hirano T (1996) Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti‐apoptosis. Immunity 5, 449–460. [DOI] [PubMed] [Google Scholar]
- Furtek SL, Backos DS, Matheson CJ and Reigan P (2016) Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol 11, 308–318. [DOI] [PubMed] [Google Scholar]
- Gariboldi MB, Ravizza R, Molteni R, Osella D, Gabano E and Monti E (2007) Inhibition of Stat3 increases doxorubicin sensitivity in a human metastatic breast cancer cell line. Cancer Lett 258, 181–188. [DOI] [PubMed] [Google Scholar]
- Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, Nam S, Eweis I, Diaz N, Sullivan D et al (2006) Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res 12, 11–19. [DOI] [PubMed] [Google Scholar]
- Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, Mazumdar A, Hilsenbeck SG, Mills GB et al (2013) Growth of triple‐negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL‐6 and IL‐8. Cancer Res 73, 3470–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz H, Uppal N, Wagner SA, Bendell JC, Thaddeus Beck JT, Wade S, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA et al (2014) A randomized double‐blind phase 2 study of ruxolitinib (RUX) or placebo (PBO) with capecitabine (CAPE) as second‐line therapy in patients (pts) with metastatic pancreatic cancer (mPC). J Clin Oncol 32, 55 (suppl; abstr 4000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Guo J, Shen J, Jin M, Xie S and Wang L (2012) The role of estrogen receptor alpha in mediating chemoresistance in breast cancer cells. J Exp Clin Cancer Res 31, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 3, 422–433. [DOI] [PubMed] [Google Scholar]
- Kontzias A, Kotlyar A, Laurence A, Changelian P and O'Shea JJ (2012) Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol 12, 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Seo NJ, Jeong SJ, Park Y, Jung DB, Koh W, Lee EO, Ahn KS, Lu J and Kim SH (2011) Oral administration of penta‐O‐galloyl‐beta‐D‐glucose suppresses triple‐negative breast cancer xenograft growth and metastasis in strong association with JAK1‐STAT3 inhibition. Carcinogenesis 32, 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y and Pietenpol JA (2011) Identification of human triple‐negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Pietenpol JA and Tan AR. (2015) Triple‐negative breast cancer: molecular subtypes and new targets for therapy. Am Soc Clin Oncol Educ Book e31–e39. [DOI] [PubMed] [Google Scholar]
- Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C et al (2006) Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res 66, 2544–2552. [DOI] [PubMed] [Google Scholar]
- Liu CY, Su JC, Ni MH, Tseng LM, Chu PY, Wang DS, Tai WT, Kao YP, Hung MH, Shiau CW et al (2014) Obatoclax analog SC‐2001 inhibits STAT3 phosphorylation through enhancing SHP‐1 expression and induces apoptosis in human breast cancer cells. Breast Cancer Res Treat 146, 71–84. [DOI] [PubMed] [Google Scholar]
- Liu CY, Tseng LM, Su JC, Chang KC, Chu PY, Tai WT, Shiau CW and Chen KF (2013) Novel sorafenib analogues induce apoptosis through SHP‐1 dependent STAT3 inactivation in human breast cancer cells. Breast Cancer Res 15, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Ruiz P, Rodriguez‐Ubreva J, Cariaga AE, Cortes MA and Colas B (2011) SHP‐1 in cell‐cycle regulation. Anticancer Agents Med Chem 11, 89–98. [DOI] [PubMed] [Google Scholar]
- Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain‐Qimron N, Kim JJ, Choudhury SA, Maruyama R et al (2011) The JAK2/STAT3 signaling pathway is required for growth of CD44+CD24– stem cell‐like breast cancer cells in human tumors. J Clin Invest 121, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer IA, Abramson VG, Lehmann BD and Pietenpol JA (2014) New strategies for triple‐negative breast cancer–deciphering the heterogeneity. Clin Cancer Res 20, 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okenwa C, Kumar A, Rego D, Konarski Y, Nilchi L, Wright K and Kozlowski M (2013) SHP‐1‐Pyk2‐Src protein complex and p38 MAPK pathways independently regulate IL‐10 production in lipopolysaccharide‐stimulated macrophages. J Immunol 191, 2589–2603. [DOI] [PubMed] [Google Scholar]
- Qin C, Wavreille AS and Pei D (2005) Alternative mode of binding to phosphotyrosyl peptides by Src homology‐2 domains. Biochemistry 44, 12196–12202. [DOI] [PubMed] [Google Scholar]
- Rajendran P, Li F, Shanmugam MK, Kannaiyan R, Goh JN, Wong KF, Wang W, Khin E, Tergaonkar V, Kumar AP et al (2012) Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res 5, 631–643. [DOI] [PubMed] [Google Scholar]
- Real PJ, Sierra A, De Juan A, Segovia JC, Lopez‐Vega JM and Fernandez‐Luna JL (2002) Resistance to chemotherapy via Stat3‐dependent overexpression of Bcl‐2 in metastatic breast cancer cells. Oncogene 21, 7611–7618. [DOI] [PubMed] [Google Scholar]
- Roxburgh CS and McMillan DC (2016) Therapeutics targeting innate immune/inflammatory responses through the interleukin‐6/JAK/STAT signal transduction pathway in patients with cancer. Transl Res 167, 61–66. [DOI] [PubMed] [Google Scholar]
- Sansone P and Bromberg J (2012) Targeting the interleukin‐6/Jak/stat pathway in human malignancies. J Clin Oncol 30, 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner SJ, Schiavone AP and Smithgall TE (2002) Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem 277, 45680–45687. [DOI] [PubMed] [Google Scholar]
- Song JI and Grandis JR (2000) STAT signaling in head and neck cancer. Oncogene 19, 2489–2495. [DOI] [PubMed] [Google Scholar]
- Su JC, Chen KF, Chen WL, Liu CY, Huang JW, Tai WT, Chen PJ, Kim I and Shiau CW (2012) Synthesis and biological activity of obatoclax derivatives as novel and potent SHP‐1 agonists. Eur J Med Chem 56, 127–133. [DOI] [PubMed] [Google Scholar]
- Su JC, Mar AC, Wu SH, Tai WT, Chu PY, Wu CY, Tseng LM, Lee TC, Chen KF, Liu CY et al (2016) Disrupting VEGF‐A paracrine and autocrine loops by targeting SHP‐1 suppresses triple negative breast cancer metastasis. Sci Rep 6, 28888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JC, Tseng PH, Hsu CY, Tai WT, Huang JW, Ko CH, Lin MW, Liu CY, Chen KF and Shiau CW (2014a) RFX1‐dependent activation of SHP‐1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget 5, 4909–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JC, Tseng PH, Wu SH, Hsu CY, Tai WT, Li YS, Chen IT, Liu CY, Chen KF and Shiau CW (2014b) SC‐2001 overcomes STAT3‐mediated sorafenib resistance through RFX‐1/SHP‐1 activation in hepatocellular carcinoma. Neoplasia 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai WT, Cheng AL, Shiau CW, Huang HP, Huang JW, Chen PJ and Chen KF (2011) Signal transducer and activator of transcription 3 is a major kinase‐independent target of sorafenib in hepatocellular carcinoma. J Hepatol 55, 1041–1048. [DOI] [PubMed] [Google Scholar]
- Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH, Lin MW, Chen PJ and Chen KF (2012) Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP‐1‐mediated inhibition of STAT3. Mol Cancer Ther 11, 452–463. [DOI] [PubMed] [Google Scholar]
- Tai WT, Chu PY, Shiau CW, Chen YL, Li YS, Hung MH, Chen LJ, Chen PL, Su JC, Lin PY et al (2014a) STAT3 mediates regorafenib‐induced apoptosis in hepatocellular carcinoma. Clin Cancer Res 20, 5768–5776. [DOI] [PubMed] [Google Scholar]
- Tai WT, Shiau CW, Chen PJ, Chu PY, Huang HP, Liu CY, Huang JW and Chen KF (2014b) Discovery of novel Src homology region 2 domain‐containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology 59, 190–201. [DOI] [PubMed] [Google Scholar]
- Tai WT, Shiau CW, Li YS, Chang CW, Huang JW, Hsueh TT, Yu HC and Chen KF (2014c) Nintedanib (BIBF‐1120) inhibits hepatocellular carcinoma growth independent of angiokinase activity. J Hepatol 61, 89–97. [DOI] [PubMed] [Google Scholar]
- Tan FH, Putoczki TL, Stylli SS and Luwor RB (2014) The role of STAT3 signaling in mediating tumor resistance to cancer therapy. Curr Drug Targets 15, 1341–1353. [DOI] [PubMed] [Google Scholar]
- Thomas SJ, Snowden JA, Zeidler MP and Danson SJ (2015) The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer 113, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian ZJ and An W (2004) ERK1/2 contributes negative regulation to STAT3 activity in HSS‐transfected HepG2 cells. Cell Res 14, 141–147. [DOI] [PubMed] [Google Scholar]
- Wang W, Liu L, Song X, Mo Y, Komma C, Bellamy HD, Zhao ZJ and Zhou GW (2011) Crystal structure of human protein tyrosine phosphatase SHP‐1 in the open conformation. J Cell Biochem 112, 2062–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Tweardy DJ, Zhang M, Zhang X, Landua J, Petrovic I, Bu W, Roarty K, Hilsenbeck SG, Rosen JM et al (2014) STAT3 signaling is activated preferentially in tumor‐initiating cells in claudin‐low models of human breast cancer. Stem Cells 32, 2571–2582. [DOI] [PubMed] [Google Scholar]
- Wu C, Guan Q, Wang Y, Zhao ZJ and Zhou GW (2003) SHP‐1 suppresses cancer cell growth by promoting degradation of JAK kinases. J Cell Biochem 90, 1026–1037. [DOI] [PubMed] [Google Scholar]
- Xing J, Zhang Z, Mao H, Schnellmann RG and Zhuang S (2008) Src regulates cell cycle protein expression and renal epithelial cell proliferation via PI3K/Akt signaling‐dependent and ‐independent mechanisms. Am J Physiol Renal Physiol 295, F145–F152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liang X, Niu T, Meng W, Zhao Z and Zhou GW (1998) Crystal structure of the catalytic domain of protein‐tyrosine phosphatase SHP‐1. J Biol Chem 273, 28199–28207. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ and Zhou GW (2003) Crystal structure of human protein‐tyrosine phosphatase SHP‐1. J Biol Chem 278, 6516–6520. [DOI] [PubMed] [Google Scholar]
- Yao X, Zhu F, Zhao Z, Liu C, Luo L and Yin Z (2011) Arctigenin enhances chemosensitivity of cancer cells to cisplatin through inhibition of the STAT3 signaling pathway. J Cell Biochem 112, 2837–2849. [DOI] [PubMed] [Google Scholar]
- Yu H, Lee H, Herrmann A, Buettner R and Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14, 736–746. [DOI] [PubMed] [Google Scholar]
- Zhang SS, Liu MG, Kano A, Zhang C, Fu XY and Barnstable CJ (2005) STAT3 activation in response to growth factors or cytokines participates in retina precursor proliferation. Exp Eye Res 81, 103–115. [DOI] [PubMed] [Google Scholar]
- Zhang M, Xu C, Ma L, Shamiyeh E, Yin J, von Moltke LL and Smith WB (2015) Effect of food on the bioavailability and tolerability of the JAK2‐selective inhibitor fedratinib (SAR302503): results from two phase I studies in healthy volunteers. Clin Pharmacol Drug Dev 4, 315–321. [DOI] [PubMed] [Google Scholar]
- Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, Tan X, Sun X, Jia W and Zhou Z (2015) Pharmacological activation of aldehyde dehydrogenase 2 by Alda‐1 reverses alcohol‐induced hepatic steatosis and cell death in mice. J Hepatol 62, 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The chemical structure and solubility of SC‐60.
Fig. S2. Cytotoxicity effect of SC‐60 on MCF‐10A normal human breast epithelial cells and MCF‐7 luminal breast cancer cells.
Fig. S3. SC‐60 decreased the mRNA levels of STAT3 downstream target genes.
Fig. S4. SC‐60 had no obvious effects on VEGFR2, PDGFRβ, JAK1, JAK2 and ERK1/2 in MDA‐MB‐231 cells.
Fig. S5. The effects of SC‐60 on SHP1‐depleted MDA‐MB‐468 cells.
Fig. S6. SC‐60 diminishes xenograft tumor growth of MDA‐MB‐468 cells.