

Abstract
An accurate blood‐based RAS mutation assay to determine eligibility of metastatic colorectal cancer (mCRC) patients for anti‐EGFR therapy would benefit clinical practice by better informing decisions to administer treatment independent of tissue availability. The objective of this study was to determine the level of concordance between plasma and tissue RAS mutation status in patients with mCRC to gauge whether blood‐based RAS mutation testing is a viable alternative to standard‐of‐care RAS tumor testing. RAS testing was performed on plasma samples from newly diagnosed metastatic patients, or from recurrent mCRC patients using the highly sensitive digital PCR technology, BEAMing (beads, emulsions, amplification, and magnetics), and compared with DNA sequencing data of respective FFPE (formalin‐fixed paraffin‐embedded) tumor samples. Discordant tissue RAS results were re‐examined by BEAMing, if possible. The prevalence of RAS mutations detected in plasma (51%) vs. tumor (53%) was similar, in accord with the known prevalence of RAS mutations observed in mCRC patient populations. The positive agreement between plasma and tumor RAS results was 90.4% (47/52), the negative agreement was 93.5% (43/46), and the overall agreement (concordance) was 91.8% (90/98). The high concordance of plasma and tissue results demonstrates that blood‐based RAS mutation testing is a viable alternative to tissue‐based RAS testing.
Keywords: anti‐EGFR therapy, CRC, ctDNA, plasma, RAS mutations
Abbreviations
- BEAMing
beads, emulsions, amplification, and magnetics
- CRC
colorectal cancer
- ctDNA
circulating tumor DNA
- EGFR
epidermal growth factor receptor
- FFPE
formalin‐fixed paraffin‐embedded
- LoB
limits of blank
- LOD
limits of detection
- MAF
mutant allelic fraction
- mCRC
metastatic colorectal cancer
- NPA
negative percent agreement
- PPA
positive percent agreement
- SOC
standard of care
- WT
wild‐type
1. Introduction
Colorectal cancer (CRC) is a major health problem with ~1.3 million new cases diagnosed annually (‘International Agency for Research on Cancer. GLOBOCAN 2012: estimated cancer incidence, mortality, and prevalence worldwide in 2012’, n.d.). The disease stage at diagnosis is a significant prognostic factor, and in spite of global screening efforts, ~50% of patients will either present with, or relapse with metastatic disease (Kievit, 2002; Siegel et al., 2015). A key priority for patients with metastatic colorectal cancer (mCRC) is the implementation of an appropriate first‐line treatment strategy. Numerous randomized controlled trials have demonstrated the benefit of treating mCRC patients with monoclonal antibodies targeting the epidermal growth factor receptor (EGFR), such as cetuximab and panitumumab (Jonker et al., 2007; Saltz et al., 2004; Tabernero et al., 2007; Van Cutsem et al., 2007). The identification of patients wild‐type (WT) for KRAS codons 12 and 13 mutations increased response rates to anti‐EGFR therapy by as much as 60% (Douillard et al., 2010; Van Cutsem et al., 2009a) and reinforced the approach of incorporating molecular diagnostics into clinical practice. Recent trials have demonstrated that a more comprehensive evaluation of RAS, so‐called expanded RAS, to include KRAS and NRAS codons 12, 13, 59, 61, 117, 146 can more precisely identify patients with mCRC for anti‐EGFR therapy than KRAS codons 12 and 13 testing alone (Bokemeyer et al., 2014; Douillard et al., 2013; Heinemann et al., 2014; Peeters et al., 2014).
Genotyping of tumor tissue can present challenges to even the most advanced clinical practice. Studies evaluating the genomic profiles of primary tumors and metastases have shown discordant results, attributed largely to molecular inter‐ and intratumor/metastasis heterogeneity (De Mattos‐Arruda et al., 2014; Gerlinger et al., 2012; Mao et al., 2015). Operationally, an optimal RAS testing procedure for biopsy and surgical resection specimens has yet to be uniformly established (Allegra et al., 2009; van Krieken et al., 2008; Tack et al., 2015). As an alternative and complement to tumor tissue genotyping, analysis of tumor DNA derived from plasma can provide a rapid genotype result, which accurately reflects the mutation status of tumor tissue (Bettegowda et al., 2014; Diehl et al., 2008). A unique feature of blood‐based genotyping is its potential to provide an integrative and gene mutation‐specific highly sensitive molecular analysis of an individual patient's tumor and/or metastases (Bettegowda et al., 2014; Diaz and Bardelli, 2014), thus eliminating sampling bias related to tissue heterogeneity.
Blood‐based tumor genotyping derives from observations that patients with cancer have markedly higher concentrations of circulating cell‐free DNA (cfDNA) than healthy individuals (Stroun et al., 1987). In patients with metastatic cancer, plasma‐derived ctDNA has been shown to be a reliable surrogate for genomic alterations in tumor tissue (Bettegowda et al., 2014; Diehl et al., 2008; Morelli et al., 2015). Among the various plasma ctDNA assays, BEAMing (beads, emulsions, amplification, and magnetics), based on emulsion digital PCR, has been shown to exhibit high sensitivity, enabling the detection of one mutant allele within a background of 10 000 wild‐type alleles (Diehl et al., 2006; Dressman et al., 2003; Li et al., 2006). In clinical trials, BEAMing has been extensively validated to assess tumor mutation status from the blood of patients with mCRC (Bettegowda et al., 2014; Morelli et al., 2015; Tabernero et al., 2015), with exemplary performance for the accurate assessment of expanded RAS (Bokemeyer et al., 2015; Van Cutsem et al., 2015; Venook et al., 2014). The objective of this study was to demonstrate the utility of a standardized blood‐based RAS genotyping system as an alternative to tissue‐based RAS genotyping prior to treatment with anti‐EGFR therapy.
Materials and methods
Study design
Two separate cohorts of advanced CRC patients from Australia and Germany were evaluated for concordance of RAS mutation status between plasma and tissue. A single blood sample from each patient was obtained immediately prior to biopsy or resection of tumors from either primary or metastatic sites. RAS mutation analysis of plasma was compared with the standard‐of‐care (SOC) tumor RAS testing performed on a primary or metastatic specimen (FFPE tumor tissue) from the same patient. In instances of discrepant RAS results between plasma and tissue, repeat RAS mutation testing was performed using BEAMing applied to the same FFPE tumor block as that used for SOC RAS testing. To determine concordance of plasma vs tissue RAS testing results, positive percent agreement (PPA), negative percent agreement (NPA), and overall percent agreement (OA) were calculated. In cases where SOC testing resulted in a WT determination and tissue BEAMing analysis revealed a RAS mutation, the BEAMing result was favored if the fraction of mutant alleles exceeded the SOC cutoff of 2%. Histopathology was performed and CEA levels were determined by the pathology and diagnostic laboratories at each hospital, respectively.
Patients and samples
The local ethical committees approved sample collection, and consent was obtained for plasma analysis prior to tumor biopsy or resection (ethical votes Australia: Melbourne 03/90, Newcastle 11/04/20/4.03; ethical votes Germany: Munich 1926/07; Bochum 16‐5683). Collected patient characteristics included age, gender, disease status, treatment history, CEA concentration if available, histopathology and tumor staging. Overall, 98 patients were included in the concordance analysis. Four patient cases were excluded, with three patient plasma samples exhibiting inadequate plasma‐derived DNA for analysis and one patient for whom a RAS mutation result could not be confirmed in the original FFPE specimen when re‐evaluated by DNA sequencing.
The Australian cohort was comprised of 32 CRC patients having advanced disease (stage IV, or stage III with multiple lymph nodes affected). All FFPE tissue and plasma samples originated from patients at the John Hunter Hospital in Newcastle, New South Wales, or the Peter MacCallum Cancer Centre in East Melbourne, Victoria, Australia. The majority of patients (n = 24) presented with recurrent metastatic disease for whom tissue was obtained from the metastatic lesion. Eight patients with stage III disease and involvement of multiple (>2) lymph nodes were also evaluated.
The German cohort (n = 66) was comprised of 61 newly diagnosed and five mCRC patients with recurrent disease. All FFPE samples and accompanying RAS testing results were provided by the Medical University of Bochum Hospital, Bochum, and the University Hospital Klinikum rechts der Isar, Munich, Germany. In contrast to those in the Australian cohort, tumor samples from Germany were comprised largely of primary tumors obtained at first diagnosis of mCRC.
For patients in both cohorts, plasma samples were prepared from blood collected in K2‐EDTA Vacutainer® tubes (Becton Dickinson, Franklin Lakes, NJ, USA) within 4 h of phlebotomy according to approved procedures for ctDNA analyses including a centrifugation step to pellet any cell debris (Sysmex Inostics GmbH, Hamburg Germany). All plasma samples were stored and shipped as 1 mL aliquots at −80 °C. Approximately 2 mL of plasma from each sample was thawed at room temperature for 10 min prior to ctDNA isolation. Purification of DNA from plasma was performed using the QIAamp DNA purification kit (Qiagen, Venlo, the Netherlands) according to the manufacturer's instructions. The total amount of human genomic DNA purified from plasma samples was quantified using a modified version of LINE‐1 real‐time PCR assay (Diehl et al., 2008). Any samples with total genome equivalents (GE) below 500 GE were deemed insufficient for mutational analysis, according to standard operating procedures (Sysmex Inostics GmbH).
Plasma RAS mutation testing using the BEAMing method
Plasma samples were analyzed for 33 mutations in KRAS and NRAS exons 2, 3, 4 by BEAMing at Sysmex Inostics. BEAMing utilizes emulsion digital PCR performed on magnetic beads to amplify single DNA molecules. Individual beads are then hybridized to allele‐specific fluorescently labeled probes complementary to the mutant and wild‐type DNA sequences. Finally, the bead population is analyzed by flow cytometry to count and sort wild‐type and mutant beads. The result is reported as the fractional abundance of mutant DNA alleles relative to wild‐type DNA alleles in a plasma sample. To generate the ratio of mutant to wild‐type DNA alleles (mutant allelic fraction, MAF), an average of 3 × 106 beads are interrogated in each BEAMing analysis (approximately 90 000 beads per mutation). The absolute number of RAS‐mutant alleles is not reported by BEAMing as the determination of mutant status is dependent on the total amount of DNA in an individual sample. Total circulating DNA levels (both wild‐type and mutant) are subject to interpatient variability, which may be directly related to tumor burden or other characteristics such as inflammation and immune response.
In this study, the cutoff for the BEAMing RAS assay was 0.02%. Although it has been shown that BEAMing can detect one mutant molecule in a background of 10 000 wild‐type molecules (0.01%), the setting of the cutoff to 0.02% ensured that the limits of detection (LODs) for each of the 33 RAS mutations in the BEAMing RAS assay were well above background signals or limits of blank (LoBs) for each analyte to be detected in clinical samples. LODs were determined by probit regression analyses by spiking wild‐type (non‐RAS mutation‐containing) plasma samples with each RAS analyte. Background signals (LoBs) were determined in DNA prepared from wild‐type plasma samples lacking RAS mutations at low, medium, and high concentrations of genomic DNA. Based on the results of these experiments, the cutoff of 0.02% was observed to be appropriate so as to obtain a 95% probability/confidence interval of reporting a ‘mutation detected’ result (Sysmex Inostics GmbH, internal validation).
Tissue RAS mutation testing
Formalin‐fixed paraffin‐embedded specimens were evaluated for RAS mutations in KRAS and NRAS exons 2, 3, and 4 using pyrosequencing, Sanger sequencing, or next‐generation sequencing according to the procedures established in routine clinical use at participating institutions. The cutoff threshold RAS MAF for calling a specimen mutant as validated at the local institutions was 2% and 5% for Australian and German cohorts, respectively, with pyrosequencing utilized for all German specimens. For cases where only KRAS exon 2 was evaluated by the SOC method, tissue specimens were re‐examined for expanded RAS either by DNA sequencing at the provider's institution or by BEAMing. BEAMing of tissue samples was also used to re‐evaluate the result provided by the standard‐of‐care assay for cases where the BEAMing plasma and SOC tissue results were discordant. The cutoff for tissue BEAMing was set at 1.0% MAF as demonstrated in CRYSTAL and OPUS studies (Bokemeyer et al., 2015; Van Cutsem et al., 2015).
Statistical analyses
Concordance of RAS mutation status was determined by calculating the agreement of RAS‐mutant and WT cases. Fisher's exact test was used to assess the significance of relationships for plasma and tissue RAS results. All statistical tests were two‐sided; the threshold for statistical significance was P < 0.05. MAF values for newly diagnosed vs recurrent mCRC patients were evaluated by calculating mean MAF values with standard errors and compared with p values derived using Welch's unequal variances t‐test. Correlation between ctDNA and CEA levels was assessed using Pearson's rank correlation.
Results
Patient characteristics
In total, 98 patients with histologically confirmed metastatic or stage III CRC with multiple lymph node involvement (54 males and 44 females having a median age of 66 years) were evaluated. Staging was based on postoperative histopathology and imaging diagnoses. The baseline characteristics of patients are shown in Table 1. At the time of study inclusion, the majority of patients (91.8%) had stage IV colorectal adenocarcinoma. Most patients (70/98) had newly diagnosed disease and were naïve to treatment (71.4%). With respect to treatment, ~ one‐third of the patients had recurrent disease and received at least one line of prior chemotherapy. The colorectum was the predominant site of tissue biopsy (78%). In patients for whom a metastatic site was submitted for RAS mutation analysis (22%), the most frequent site was the liver (77%) followed by the lung (18%).
Table 1.
Patient characteristics
| Patients | 98 |
| Median age (range) | 66 (21–92) |
| Gender | |
| Female, n (%) | 44 (44.9%) |
| Male, n (%) | 54 (55.1%) |
| Disease status at time of biopsy, n (%) | |
| Stage III, newly diagnosed | 8 (8.2%) |
| Stage IV, newly diagnosed | 62 (63.3%) |
| Stage IV, recurrent disease | 28 (28.6%) |
| Site of tissue biopsy, n (%) | |
| Primary tumor | 76 (78%) |
| Metastases | 22 (22%) |
| Liver | 18 (82%) |
| Lung | 4 (18%) |
| Therapeutic history, n (%) | |
| Treatment naive | 70 (71.4%) |
| ≥first line of therapy | 28 (28.6%) |
| Baseline serum CEA, median (IQR) (n = 65) | 18.4 (6.25–60.25) |
Levels of CEA in blood samples were available for 65 of 98 patients in the concordance analysis. The median CEA concentration from these 65 patients was 18.40 ng·mL−1 (range: 2–4069; mean ± SD 193.6 ± 624.6). A comparison of CEA concentrations in newly diagnosed stage IV patients versus those diagnosed with metastatic disease at recurrence was then made. The median CEA concentrations in newly diagnosed stage IV patients and those with recurrent disease were 19.40 ng·mL−1 (mean ± SD 236.3 ± 700.0) and 9.90 ng·mL−1 (mean ± SD 37.86 ± 52.26), respectively. A two‐tailed t‐test of CEA levels indicated that differences in CEA levels between these two patient groups were at the threshold of statistical significance (P = 0.0504).
RAS mutation status analysis from plasma and tissue
RAS mutation analysis in plasma was performed using the BEAMing expanded RAS mutation panel (Table 2), which detects 33 mutations encoding pathogenic variants of KRAS and NRAS proteins. RAS mutation status was evaluable in both plasma and tissue of all 94 patients. Overall, RAS mutations were detected in 53% of tumor tissue samples and in 51% of plasma samples (Table 3). The frequency of RAS mutations in patients investigated in this study was in agreement with the results of other groups performing expanded RAS analysis (Sorich et al., 2015). The vast majority of mutations detected by both plasma and tissue methods were KRAS codons 12 and 13 (Table 3). RAS codon 61 mutations were detected in only four cases.
Table 2.
Individual KRAS and NRAS mutations detected by BEAMing
| KRAS | NRAS | ||
|---|---|---|---|
| Exon | Mutation | Exon | Mutation |
| 2 |
G12S G12R G12C G12D G12A G12V G13D |
2 |
G12S G12R G12C G12D G12A G12V G13D G13R G13V |
| 3 |
A59T Q61L Q61Ha Q61Ha |
3 |
A59T Q61L Q61Ha Q61Ha Q61K Q61R |
| 4 |
K117Na
K117Na A146T A146V |
4 |
K117Na
K117Na A146T |
Denotes two separate mutations detected for each of these codons.
Table 3.
RAS mutation by exon/codon: frequency and prevalence
| RAS mutation | Tissue | Plasma | ||
|---|---|---|---|---|
| N | % | N | % | |
| KRAS Exon 2 Codon 12 | 40 | 77 | 35 | 70 |
| KRAS Exon 2 Codon 13 | 10 | 19 | 10 | 20 |
| KRAS Exon 3 Codon 61 | 1 | 2 | 1 | 2 |
| NRAS Exon 3 Codon 61 | 1 | 2 | 3 | 6 |
| KRAS Exon 4 Codon 146 | 1 | 2 | 1 | 2 |
| RAS prevalence | 52/98 = 53.1% | 50/98 = 51.0% | ||
| WT prevalence | 46/98 = 46.9% | 48/98 = 49.0% | ||
The concordance of RAS status between matched plasma and tissue from each patient is summarized in Table 4. The RAS mutation status determined by BEAMing from plasma versus the reference method was concordant in 90 of 98 cases examined (91.8% OPA). RAS mutations were found in 47 of 52 cases tested (90.4% PPA). Of 46 patients identified as WT by tissue testing, 43 were also found to be WT in plasma (93.5% NPA). When considering only patients with mCRC, plasma RAS mutations were found in 45 of 49 cases (91.8% PPA), no RAS mutations were found in 38 of 41 cases (92.7% NPA), and RAS mutation status was concordant in 83 of 90 cases (92.2% OPA). Initially, nine discrepant RAS mutation results were found among the 98 cases. Five samples evaluated by tissue testing were RAS mutation+, whereas the corresponding plasma samples were WT. Conversely, plasma analysis revealed RAS mutations in four patients whose tumors were determined WT by tissue testing.
Table 4.
Concordance of plasma and tissue RAS mutation results
| Tumor tissue RAS result | |||||||
|---|---|---|---|---|---|---|---|
| RAS | Mutant | WT | Total | PPA (95% CI) | NPA (95% CI) | OPA (95% CI) | |
| Plasma ctDNA RAS result | Mutant | 47 | 3 | 50 | 100 × 47/52 = 90.4% (79%, 96%) | 100 × 43/46 = 93.5% (82%, 98%) | 100 × 90/98 = 91.8% (85%, 96%) |
| WT | 5 | 43 | 48 | ||||
| Total | 52 | 46 | 98 | ||||
Discordance analysis
To evaluate discrepancies between the results of matched tumor and plasma samples, tissue BEAMing was employed as an orthogonal assay. Previous studies have shown that BEAMing is an accurate technique for the mutational analysis of archival FFPE tumor tissue (Bokemeyer et al., 2015; Morelli et al., 2015; Van Cutsem et al., 2015). Whenever possible, FFPE samples matched to the date of surgery and tissue site for cases having discrepant results were re‐analyzed by BEAMing. All but three FFPE specimens were either unavailable or exhibited severely degraded DNA and could not be tested. Of the three samples adequate for re‐analysis, tissue BEAMing confirmed the results of SOC tissue testing in two cases and the results of plasma testing in one case (Table 5). In the two cases in which SOC tissue results were confirmed, BEAMing detected a result of WT in one and a KRAS codon 12 mutation in the other. However, in one case, re‐examination with tissue BEAMing confirmed a KRAS G12D mutation (2.86% MAF) also detected in the plasma, but not detected by SOC tissue testing.
Table 5.
Discordant analysis. In cases where no tissue re‐evaluation was possible, the final call remained discordant
| Sample ID | Stage | Site of tissue biopsy | Tissue result | Plasma result | Plasma MAF% | Tissue re‐evaluation | Final call | CEA (ng·mL−1) |
|---|---|---|---|---|---|---|---|---|
| AUS007 | IV | MET (lung) |
KRAS
G12C |
WT |
KRAS
G12C (12.1%) |
P‐FN | 2.4 | |
| AUS030 | IIIB | Primary |
KRAS G12D |
WT | Low DNA/NA | Discordant | Not available | |
| GER010 | IV | Primary | WT |
NRAS Q61R |
0.258% | NTA | Discordant | 1452 |
| GER016 | IV | Primary |
KRAS G12D |
WT | NTA | Discordant | 18.4 | |
| GER024 | IV | Primary | WT |
NRAS Q61R |
0.237% | NTA | Discordant | 7.1 |
| GER028 | IV | Primary | WT |
KRAS
G12D |
0.425% |
KRAS
G12D (2.9%) |
Concordant | 27.5 |
| GER029 | IV | Primary |
KRAS G12D |
WT | NTA | Discordant | 41.4 | |
| GER051 | IV | Primary |
KRAS G12V |
WT | NTA | Discordant | 42.6 | |
| GER056 | IV | Primary | WT |
KRAS G12A |
0.111% | WT | P‐FP | 4.52 |
WT, wild‐type; NA, nonanalyzable; NTA, no tissue available; P‐FN, plasma false‐negative; P‐FP, plasma false‐positive.
Comparisons of RAS mutation results obtained by BEAMing analyses of tissue and those obtained by the SOC tissue test are designated in boldface type.
As a patient's CEA concentration is used as a prognostic indicator of disease status, we examined whether CEA concentration may be related to the likelihood of detecting a plasma mutation that might explain discordance in the eight stage IV patients (e.g., low CEA levels correspond to undetectable ctDNA). As shown in Table 5, one patient with a KRAS codon 12 mutation in the tumor but showing no detectable RAS mutation in plasma had a normal CEA concentration (2.4 ng·mL−1). The median CEA concentration in patients for whom a mutation was detected in plasma, but not in the tissue, was 17.3 ng·mL−1. In patients for whom a mutation was present in the tissue, but not detected in plasma, the median CEA concentration was 29.9 ng·mL−1. Based on the available samples, we did not observe any correlation between CEA levels and the ability to detect RAS mutations.
Plasma mutant allele frequency and correlation with tumor burden
A unique feature of BEAMing ctDNA analysis is the ability to determine the fraction of cell‐free mutant alleles as a proportion of the overall cell‐free DNA content in circulation at the time of sampling. For the 50 patients with detectable plasma RAS mutations, the average MAF was 6.82% (Fig. 1). Given the possibility that the frequency of circulating mutant alleles is related to overall tumor burden or extent of metastatic invasion, MAF values in plasma were compared between newly diagnosed mCRC patients and those having metastatic recurrence. A statistically significant relationship was observed between the patient clinical diagnosis status and mean proportion of mutant RAS alleles in circulation. In stage IV newly diagnosed patients with intact primary tumors of the colorectum, the MAF was 6.5‐fold higher (9.63%) compared with those patients who presented with recurrent disease after removal of their primary tumors (1.49%) (P = 0.0055, Fig. 1). In patients presenting with stage III newly diagnosed disease, the average MAF was also lower (0.55%); this is in line with results of previous reports that earlier‐stage tumors tend to release lower amounts of tumor DNA into circulation (Bettegowda et al., 2014). There appeared to be no correlation between CEA concentration and MAF among those patients whose CEA levels were available at the time of blood collection for ctDNA analysis (Pearson's r = 0.231, P = 0.1822).
Figure 1.
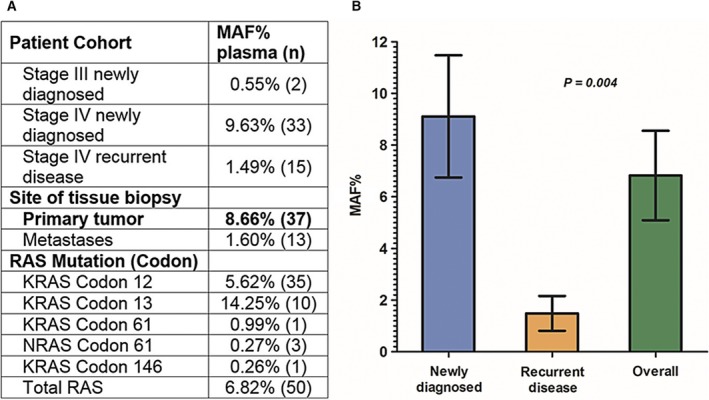
(A) Bar chart showing the average plasma DNA‐mutant fractions detected in ctDNA of patients with newly diagnosed compared to recurrent disease as well as the overall cohort of patients with RAS mutations detected in the plasma by BEAMing. (B) Average value and standard error for all patients (stage III and stage IV), those with newly diagnosed and recurrent disease are 6.82 ± 1.73, 9.11 ± 2.36, and 1.49 ± 0.67, respectively. P = 0.004 for MAF% in newly diagnosed patients compared to those with recurrent disease. P values were derived from a Welch's unequal variances t‐test.
Discussion
The accurate prescription of anti‐EGFR therapy is of high clinical importance for patients with mCRC. Retrospective analyses of data from randomized controlled clinical trials consistently demonstrate that KRAS exon 2 mutation is a contraindication for the administration of anti‐EGFR therapy, regardless of the chemotherapy backbone (Amado et al., 2008; Bokemeyer et al., 2011; Douillard et al., 2010; Karapetis et al., 2008; Peeters et al., 2010; Van Cutsem et al., 2011). In 2009, KRAS testing was thus established in both European and US clinical practice guidelines as a predictive marker of response to anti‐EGFR therapy (Allegra et al., 2009; Benson et al., 2014; Van Cutsem et al., 2009a, b). With KRAS exon 2 mutation status widely accepted as a predictor for a lack of response to anti‐EGFR therapy, it became clear that not all KRAS exon 2 wild‐type patients responded to treatment. Further refinement in biomarker testing was pursued to improve patient outcomes and avoid unnecessary treatment‐related side effects and costs, with a focus on KRAS exon 3 and 4 mutations—shown to confer resistance to EGFR antibodies similar to KRAS exon 2 mutations (Janakiraman et al., 2010; Smith et al., 2010). Subsequent evaluation of NRAS mutations revealed their occurrence in CRC tumors with persistent GTPase activity similar to alterations in KRAS (Irahara et al., 2010). Incorporation of expanded RAS testing into practice is expected to increase the proportion of patients ineligible for anti‐EGFR therapy from ~45% to ~55% (Sorich et al., 2015). Current clinical practice guidelines now recommend expanded RAS analysis be performed to more precisely identify patients for anti‐EGFR therapy (Allegra et al., 2016; Benson et al., 2014; Van Cutsem et al., 2014).
Phase III clinical trials utilizing the BEAMing expanded RAS mutation panel have shown superior overall survival for RAS WT vs. RAS‐mutant mCRC patients when treated in first line with EGFR antibodies (Bokemeyer et al., 2015; Van Cutsem et al., 2015; Venook et al., 2014). Notably, specimens from 548 patients with mCRC previously defined as KRAS exon 2 WT in CRYSTAL and OPUS studies re‐examined with BEAMing detected other RAS mutations in 14.7% and 26% of patients, respectively. These results validated the use of BEAMing to evaluate expanded RAS testing in order to select patients for anti‐EGFR therapy. Although great strides have been made, the standardization of RAS testing has been difficult to achieve, largely due to variability of testing methodologies and DNA quality and quantity of FFPE specimens. Implementation of reliable tumor tissue genotyping programs has also been challenging. For instance, a recent external quality assessment (EQA) revealed significant interlaboratory variability within routine approaches to testing of RAS in Europe (Tack et al., 2015). Moreover, obtaining suitable tumor tissue samples can pose a challenge—particularly in recurrent mCRC patients with distant metastases (Wang et al., 2010). At initial diagnosis, the timing of molecular testing results is of critical importance for first‐line treatment. However, in a recent study of patients with mCRC evaluated for first‐line therapy, ~25% of patients did not have RAS testing requested at or at least one month after their initial diagnosis of metastatic disease (Longin, 2015). These findings are supported by a survey of European physicians, which revealed that RAS testing turnaround times and the unavailability of tissue were the most frequent factors cited for treating mCRC patients with unknown KRAS status (Trojan et al., 2015). Moreover, a recent EQA survey found that half of all participating laboratories exceeded the required turnaround time of 14 days for RAS testing (Tack et al., 2015). This presents a challenge to the broad realization of individualizing therapy and providing an accurate blood‐based RAS mutation assay with rapid turnaroundtime would help circumvent these issues.
The primary objective of this study was to validate blood‐based RAS mutation analysis as an alternative to tissue‐based RAS mutation analysis prior to anti‐EGFR therapy. Two geographically isolated cohorts of patients with CRC from Australia and Germany were examined for concordance of RAS mutations in plasma and tissue. The Australian cohort mainly comprised patients with metastatic recurrences and was representative of the clinical practice of assessing eligibility for anti‐EGFR therapy following first‐line chemotherapy. In contrast, the German cohort was comprised predominantly of newly diagnosed, treatment‐naïve mCRC patients, a cohort eligible for first‐line anti‐EGFR therapy. The entire patient group in this study represented a reasonable cross‐section of the anti‐EGFR intended use population.
Previous concordance studies of SOC FFPE KRAS mutation detection assays showed minimal variation in the presence or absence of KRAS mutations, most likely due to differential tumor cell selection from formalin‐fixed tumor tissue (Whitehall et al., 2009). Overall, the concordance between plasma and tissue RAS mutation status determined in this study was 93% and suggests that plasma RAS mutation detection is as good as tissue‐based detection strategies, but has the advantage that it does not rely on the testing of DNA isolated from fixed tissue. The most notable difference was the threshold cutoffs for RAS positivity between the two sites. Due to this variability, any tissue samples exhibiting RAS mutation status that differed from plasma were, if available, re‐examined by BEAMing. For one such case, BEAMing identified the same KRAS mutation in tissue that was identified in plasma, contrary to the original SOC RAS WT result. Interestingly, the MAF obtained by tissue BEAMing was 2.86%, falling between the 2% and 5% cutoffs of the SOC methods. This circumstance highlights the variability in SOC RAS testing techniques, and with the possibility to apply an orthogonal assay, this case was determined to be concordant. These comparisons also suggest that further improvements in the agreement of plasma and tissue RAS testing results may be achieved when both the methods of plasma preparation and FFPE RAS testing are standardized.
A systemic assessment of mutation status in a patient with metastatic CRC represents a key advantage of blood‐based testing. However, in a patient with widely metastatic disease, blood‐based mutation testing does not yet have the ability to discern the site of ctDNA origin. In our study for example, three patients had a RAS mutation that was detected in plasma, but not in the primary colorectal tumor. All three patients were newly diagnosed, treatment naïve with intact primary tumors, and presented with hepatic metastases; one patient had additional pulmonary metastases. As all three patients presented with distant metastases at the time of blood draw, a reasonable explanation of discordance may be attributed to heterogeneity of the genotype between metastatic and primary tumors. Indeed, several studies evaluating intertumor molecular heterogeneity between primary tumors and metastases in the same patient have shown mutational discordance in 3.6–32.4% of cases (Artale et al., 2008; Italiano et al., 2010; Kim et al., 2012; Knijn et al., 2011; Tie et al., 2011). As we have observed a lower plasma RAS MAF in patients with recurrent metastatic disease (Fig. 1), a tenable hypothesis is that in these three newly diagnosed patients lacking RAS mutations in their primary tumor, but having low plasma RAS MAF (0.111–0.258%), the RAS ctDNA in these three patients may be contributed by a single or few metastatic sites.
Conclusions
Determination of a patient's RAS mutational status from plasma may provide distinct advantages compared to RAS testing of FFPE samples. A specific application with future clinical utility may be the routine surveillance of plasma mutation status to assess RAS‐mediated resistance in patients receiving anti‐EGFR therapy. This approach has been shown to provide a more precise gauging of the efficacy/failure of anti‐EGFR therapy (Diaz et al., 2012; Morelli et al., 2015). Indeed, BEAMing has already revealed substantial differences in RAS mutation status between baseline mCRC tumor samples as compared with current plasma mutation status. This advantage was demonstrated in a clinical trial of 503 patients with mCRC that investigated whether plasma RAS mutation status was associated with response to regorafenib (Tabernero et al., 2015). In a cohort of patients whose archival tumor was KRAS WT and consequently received anti‐EGFR therapy, KRAS mutations were detected by BEAMing in the plasma of 48% of patients at disease progression. This exemplifies the use of plasma ctDNA testing to provide a real‐time assessment of mutation status. Blood‐based mutation assessment may therefore help define critical decision points for the individualized management of patients with CRC, providing a finer resolution of molecular cues to signal optimal timing for treatment hiatus and re‐initiation of targeted therapy.
A necessary first step toward implementing blood‐based RAS testing in clinical practice is to demonstrate concordance between ctDNA and tissue RAS mutation testing. The results presented herein provide a high level of confidence that the clinical performance of plasma RAS testing using BEAMing is comparable to FFPE tissue testing and can be useful in a clinical setting to select patients with mCRC for anti‐EGFR therapy.
Author contributions
WS, RJS, PR, DLE, FSJ, SH, and SBF conceived and designed the project. SD, WL, CJM, PP, BD, SS, CH, HP, AL, ES, KN, TC, DA, SPC, AT, ARS, WU, CT, HW, JS, RV, HF, KPJ, UN, JSH, MP, DV, AB, BDD, SHB, FSJ, and SH acquired the data. WS, RJS, AT, DLE, FSJ, SH, and SBF analyzed and interpreted the data. WS, RJS, DLE, FSJ, SH, and SBF wrote the manuscript.
Conflict of interest
P.R. is employee of the Merck KGAa; D.L.E. and F.S.J. are employees of the Sysmex Inostics Inc. W.S. has a nonprofit cooperation with Sysmex Inostics to use the BEAMing technology.
Acknowledgements
Funding for the collection of samples in Australia was made possible by grants from the CSIRO, the Laby Foundation, and Therapeutic Innovation Australia. Financial support for this study in Germany was provided by a grant (PURE) from the Ministry of Science, North Rhine‐Westphalia, Germany. This study was cooperatively funded by Sysmex Inostics and Merck KGaA. Drs Wolff Schmiegel and Rodney Scott are the guarantors of this work and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Contributor Information
Wolff Schmiegel, Email: Wolff.schmiegel@rub.de.
Rodney J. Scott, Email: Rodney.scott@newcastle.edu.au
References
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF and Schilsky RL (2009) American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti‐epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 27, 2091–2096. [DOI] [PubMed] [Google Scholar]
- Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A and Schilsky RL (2016) Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti‐epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Oncol Pract 34, 179–185. [DOI] [PubMed] [Google Scholar]
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R et al (2008) Wild‐type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26, 1626–1634. [DOI] [PubMed] [Google Scholar]
- Artale S, Sartore‐Bianchi A, Veronese SM, Gambi V, Sarnataro CS, Gambacorta M, Lauricella C and Siena S (2008) Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol 26, 4217–4219. [DOI] [PubMed] [Google Scholar]
- Benson AB, Venook AP, Bekaii‐Saab T, Chan E, Chen Y‐J, Cooper HS, Engstrom PF, Enzinger PC, Fenton MJ, Fuchs CS et al (2014) Colon cancer, version 3.2014. J Natl Compr Cancer Netw 12, 1028–1059. [DOI] [PubMed] [Google Scholar]
- Bettegowda C, Sausen M, Leary R.J, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM et al (2014) Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 6, 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M and Koralewski P (2011) Efficacy according to biomarker status of cetuximab plus FOLFOX‐4 as first‐line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol 22, 1535–1546. [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Kohne C‐H, Ciardiello F, Lenz H‐J, Heinemann V, Klinkhardt U, Beier F, Duecker K and Tejpar S (2014) Treatment outcome according to tumor RAS mutation status in OPUS study patients with metastatic colorectal cancer (mCRC) randomized to FOLFOX4 with/without cetuximab. J Clin Oncol 32, 5s. [Google Scholar]
- Bokemeyer C, Köhne C‐H, Ciardiello F, Lenz H‐J, Heinemann V, Klinkhardt U, Beier F, Duecker K, van Krieken JH and Tejpar S (2015) FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer Oxf Engl 51, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mattos‐Arruda L, Weigelt B, Cortes J, Won HH, Ng CKY, Nuciforo P, Bidard F‐C, Aura C, Saura C, Peg V et al (2014) Capturing intra‐tumor genetic heterogeneity by de novo mutation profiling of circulating cell‐free tumor DNA: a proof‐of‐principle. Ann Oncol 25, 1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA and Bardelli A (2014) Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 32, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA et al (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl F, Li M, He Y, Kinzler KW, Vogelstein B and Dressman D (2006) BEAMing: single‐molecule PCR on microparticles in water‐in‐oil emulsions. Nat Methods 3, 551–559. [DOI] [PubMed] [Google Scholar]
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA et al (2008) Circulating mutant DNA to assess tumor dynamics. Nat Med 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douillard J‐Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J et al (2013) Panitumumab‐FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Douillard J‐Y, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J et al (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first‐line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28, 4697–4705. [DOI] [PubMed] [Google Scholar]
- Dressman D, Yan H, Traverso G, Kinzler KW and Vogelstein B (2003) Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci USA 100, 8817–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P et al (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366, 883‐892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling‐Kaiser U, Al‐Batran S‐E, Heintges T, Lerchenmüller C, Kahl C, Seipelt G et al (2014) FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment for patients with metastatic colorectal cancer (FIRE‐3): a randomised, open‐label, phase 3 trial. Lancet Oncol 15, 1065–1075. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer . GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence worldwide in 2012 [WWW Document], n.d. URL http://globocan.iarc.fr/Pages/fact_sheets_population.aspx (accessed 14 December 15).
- Irahara N, Baba Y, Nosho K, Shima K, Yan L, Dias‐Santagata D, Iafrate AJ, Fuchs CS, Haigis KM and Ogino S (2010) NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol 19, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano A, Hostein I, Soubeyran I, Fabas T, Benchimol D, Evrard S, Gugenheim J, Becouarn Y, Brunet R, Fonck M et al (2010) KRAS and BRAF mutational status in primary colorectal tumors and related metastatic sites: biological and clinical implications. Ann Surg Oncol 17, 1429–1434. [DOI] [PubMed] [Google Scholar]
- Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K, Ricarte Filho JC et al (2010) Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res 70, 5901–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au H‐J, Berry SR, Krahn M, Price T, Simes RJ et al (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357, 2040–2048. [DOI] [PubMed] [Google Scholar]
- Karapetis CS, Khambata‐Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S et al (2008) K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359, 1757–1765. [DOI] [PubMed] [Google Scholar]
- Kievit J (2002) Follow‐up of patients with colorectal cancer: numbers needed to test and treat. Eur J Cancer 1990, 986–999. [DOI] [PubMed] [Google Scholar]
- Kim M‐J, Lee HS, Kim JH, Kim YJ, Kwon JH, Lee J‐O, Bang S‐M, Park KU, Kim D‐W, Kang S‐B et al (2012) Different metastatic pattern according to the KRAS mutational status and site‐specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer 12, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijn N, Mekenkamp LJM, Klomp M, Vink‐Börger ME, Tol J, Teerenstra S, Meijer JWR, Tebar M, Riemersma S, van Krieken JHJM et al (2011) KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer 104, 1020–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Krieken JHJM, Jung A, Kirchner T, Carneiro F, Seruca R, Bosman FT, Quirke P, Fléjou JF, Plato Hansen T, de Hertogh G et al (2008) KRAS mutation testing for predicting response to anti‐EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch Int J Pathol 453, 417–431. [DOI] [PubMed] [Google Scholar]
- Li M, Diehl F, Dressman D, Vogelstein B and Kinzler KW (2006) BEAMing up for detection and quantification of rare sequence variants. Nat Methods 3, 95–97. [DOI] [PubMed] [Google Scholar]
- Longin J (2015) Review of the current status of Ras mutation testing in patients with metastatic colorectal cancer (Mcrc): flash‐Ras study. Value Health J Int Soc Pharmacoeconomics Outcomes Res 18, A491. [Google Scholar]
- Mao C, Yuan J‐Q, Yang Z‐Y, Fu X‐H, Wu X‐Y and Tang J‐L (2015) Blood as a substitute for tumor tissue in detecting EGFR mutations for guiding EGFR TKIs treatment of nonsmall cell lung cancer: a systematic review and meta‐analysis. Medicine (Baltimore) 94, e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli MP, Overman MJ, Dasari A, Kazmi SMA, Mazard T, Vilar E, Morris VK, Lee MS, Herron D, Eng C et al (2015) Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti‐EGFR treatment. Ann Oncol 26, 731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, Andre T, Chan E, Lordick F et al (2014) Analysis of KRAS/NRAS mutations in phase 3 study 20050181 of panitumumab (pmab) plus FOLFIRI versus FOLFIRI for second‐line treatment (tx) of metastatic colorectal cancer (mCRC). [WWW Document]. Journal of Clinical Oncology. URL http://meetinglibrary.asco.org/content/122548-143 (accessed 7 January 2016). [Google Scholar]
- Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJA et al (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second‐line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28, 4706–4713. [DOI] [PubMed] [Google Scholar]
- Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J and Mayer RJ (2004) Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol 22, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD and Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65, 5–29. [DOI] [PubMed] [Google Scholar]
- Smith G, Bounds R, Wolf H, Steele RJC, Carey FA and Wolf CR (2010) Activating K‐Ras mutations outwith “hotspot” codons in sporadic colorectal tumours ‐ implications for personalised cancer medicine. Br J Cancer 102, 693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA and Karapetis CS (2015) Extended RAS mutations and anti‐EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta‐analysis of randomized, controlled trials. Ann Oncol 26, 13–21. [DOI] [PubMed] [Google Scholar]
- Stroun M, Anker P, Lyautey J, Lederrey C and Maurice PA (1987) Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol 23, 707–712. [DOI] [PubMed] [Google Scholar]
- Tabernero J, Lenz H‐J, Siena S, Sobrero A, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C et al (2015) Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol 16, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero J, Van Cutsem E, Díaz‐Rubio E, Cervantes A, Humblet Y, André T, Van Laethem J‐L, Soulié P, Casado E, Verslype C et al (2007) Phase II trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first‐line treatment of metastatic colorectal cancer. J Clin Oncol 25, 5225–5232. [DOI] [PubMed] [Google Scholar]
- Tack V, Ligtenberg MJL, Tembuyser L, Normanno N, Vander Borght S, Han van Krieken J and Dequeker EMC (2015) External quality assessment unravels interlaboratory differences in quality of RAS testing for anti‐EGFR therapy in colorectal cancer. Oncologist 20, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J, Lipton L, Desai J, Gibbs P, Jorissen RN, Christie M, Drummond KJ, Thomson BNJ, Usatoff V, Evans PM et al (2011) KRAS mutation is associated with lung metastasis in patients with curatively resected colorectal cancer. Clin Cancer Res 17, 1122–1130. [DOI] [PubMed] [Google Scholar]
- Trojan J, Mineur L, Tomášek J, Rouleau E, Fabian P, de Maglio G, García‐Alfonso P, Aprile G, Taylor A, Kafatos G et al (2015) Panitumumab use in metastatic colorectal cancer and patterns of KRAS testing: results from a Europe‐Wide Physician Survey and Medical Records Review. PLoS One 10, e0140717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E, Cervantes A, Nordlinger B, Arnold D, ESMO Guidelines Working Group (2014) Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 25 (Suppl. 3), iii1–iii9. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Köhne C‐H, Hitre E, Zaluski J, Chang Chien C‐R, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G et al (2009a) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360, 1408–1417. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Köhne C‐H, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S et al (2011) Cetuximab plus irinotecan, fluorouracil, and leucovorin as first‐line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 29, 2011–2019. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Lenz H‐J, Köhne C‐H, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH et al (2015) Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 33, 692–700. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Oliveira J, ESMO Guidelines Working Group (2009b) Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow‐up. Ann Oncol 20 (Suppl. 4), 61–63. [DOI] [PubMed] [Google Scholar]
- Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon J‐L, Van Laethem J‐L, Maurel J, Richardson G et al (2007) Open‐label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy‐refractory metastatic colorectal cancer. J Clin Oncol 25, 1658–1664. [DOI] [PubMed] [Google Scholar]
- Venook A, Niedzwiecki D, Lenz HJ, Innocenti F, Mahoney MR, O'Neil B, Shaw J, Polite B, Hochster H, Atkins J et al (2014) O‐0019calgb/Swog 80405: Phase III trial of Irinotecan/5‐Fu/Leucovorin (FOLFIRI) or Oxaliplatin/5‐Fu/Leucovorin (MFOLFOX6) with Bevacizumab (BV) or Cetuximab (CET) for patients (PTS) with KRAS wild‐type (WT) untreated metastatic adenocarcinoma of the colon. Ann Oncol 25, ii112–ii113. [Google Scholar]
- Wang S, An T, Wang J, Zhao J, Wang Z, Zhuo M, Bai H, Yang L, Zhang Y, Wang X et al (2010) Potential clinical significance of a plasma‐based KRAS mutation analysis in patients with advanced non‐small cell lung cancer. Clin Cancer Res 16, 1324–1330. [DOI] [PubMed] [Google Scholar]
- Whitehall V, Tran K, Umapathy A, Grieu F, Hewitt C, Evans T‐J, Ismail T, Li WQ, Collins P, Ravetto P et al (2009) A multicenter blinded study to evaluate KRAS mutation testing methodologies in the clinical setting. J Mol Diagn 11, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]