

Abstract
Transforming growth‐interacting factor (TGIF) is a homeobox transcriptional repressor that has been implicated in holoprosencephaly and various types of cancer. TGIF is expressed in hematopoietic stem cells and modulates TGF‐β and retinoic acid (RA) signaling, both of which play an important role in hematopoiesis. We recently reported that TGIF's levels correlate inversely with survival in patients with acute myelogenous leukemia. Here we present the first direct evidence of a role for TGIF in myelopoiesis. We used short hairpin RNA interference to define the effects of TGIF knockdown on proliferation and differentiation of myeloid leukemia‐derived cell lines. Decreased TGIF expression resulted in reduced proliferation and differentiation and lower expression of CEBPβ, CEBPϵ, PU.1 and RUNX1, key myeloid transcription factors. Furthermore, TGF‐β signaling was increased and RA signaling was decreased. Further insights into the molecular basis of TGIF's effects were provided by a genome‐wide chromatin immunoprecipitation‐based elucidation of TGIF target genes. Together, these data suggest that TGIF has an important role myelopoiesis and may regulate the balance between proliferation and differentiation. Reduced TGIF expression could tip the balance toward quiescence thus providing progenitor as well as hematopoietic stem cells protection from anti‐cycle agents.
Keywords: TGIF, Leukemia, Acute myeloid leukemia, Hematopoiesis, HL60
1. Introduction
Transforming growth‐interacting factor (TGIF) is a transcriptional repressor and a member of the three amino acid loop extension (TALE) class of homeodomain proteins. TGIF was initially identified as a nuclear protein that binds a retinoid X receptor (RXR) response element in the retinol binding protein II promoter (Bertolino et al., 1995). Transcriptional repression by TGIF likely involves multiple mechanisms, including competition with RXR for RXR response elements, interaction with the ligand‐binding domain of RXR (Bartholin et al., 2006), and co‐repression in conjunction with Smad2 (Wotton et al., 2001, 1999, 1999).
Mutations and deletions of the TGIF gene are associated with holoprosencephaly (HPE), which is the most common structural abnormality of the forebrain in humans. HPE is an autosomal dominant disorder, and TGIF is one of several genes, including Sonic Hedgehog (Belloni et al., 1996), SIX3 (Wallis et al., 1999), ZIC2 (Brown et al., 1998), GLI2 (Roessler et al., 2003), PATCHED‐1 (Ming et al., 2002) and TDGF‐1 (de la Cruz et al., 2002), that have been associated with HPE. Although the role of TGIF has not been defined in specific hematopoietic lineages, TGIF transcripts have been detected in murine hematopoietic stem cells (HSCs) (Phillips et al., 2000). This transcription factor is also expressed in mouse and human embryonic stem cells and is represented on a short list of proteins proposed to mediate their “stemness” (Sato et al., 2003). Clinical relevance of a potential role for TGIF in hematopoiesis was provided by our observation that expression levels of TGIF are an independent predictor of overall survival in acute myelogenous leukemia (AML). AML patients with lower TGIF levels in their leukemia cells had a worse prognosis than patients with higher TGIF levels (Hamid et al., Submitted for publication).
While definitive evidence of TGIF's role in hematopoiesis may be lacking, several studies over the past decade have suggested that both retinoic acid (RA) and TGF‐β signaling play an important role in hematopoiesis and, in particular, myelopoiesis. TGF‐β is generally regarded as a negative regulator of all stages of hematopoiesis, but, depending upon the context, it can be pro‐ or anti‐proliferative, pro‐ or anti‐apoptotic, and pro‐ or anti‐differentiative (reviewed in (Kim and Letterio, 2003; Larsson and Karlsson, 2005)). Retinoids (all‐trans‐RA and 9‐cis‐RA) and their nuclear receptors, the retinoid alpha‐receptor (RAR) and RXR, control the growth and development of various cell types, including hematopoietic cells. In hematopoiesis, the best‐documented action of RA is the induction of differentiation in progenitor cells (reviewed in (Collins, 2002; Evans, 2005; Purton, 2007)).
Here, we provide the first direct evidence of a role for TGIF in hematopoiesis. We found that TGIF levels correlated in a linear fashion with myeloid cell proliferation. TGIF knockdown decreased and its over‐expression increased proliferation and differentiation of myeloid cell lines. This phenotype was not due to a block at a specific stage of the cell cycle or a significant increase in apoptosis. Our data suggest that TGIF knockdown enhanced TGF‐β and inhibited RA signaling. Furthermore, it also inhibited RA‐induced differentiation and altered the myeloid transcriptional program. Identification and analysis of direct TGIF targets provided additional clues about the molecular basis of TGIF's role in hematopoiesis.
2. Results
2.1. TGIF levels affect myeloid cell proliferation in a linear fashion
To determine a role for TGIF in myelopoiesis more directly, we used a lentiviral‐based shRNA approach to knockdown TGIF transcripts and protein levels and the vector pEFIRES‐P (P721) containing the strong elongation factor 1a promoter (Hobbs et al., 1998) to over‐express TGIF in HL60 cells. We confirmed the degree of knockdown (∼90%) and over‐expression (3‐fold) by Western blot and real‐time PCR analysis (Figure 1A). We isolated several individual clones and subjected them to proliferation analysis by manual counting with trypan blue staining. Equal numbers of cells were plated in triplicate, and the number of viable cells was counted at 24‐h intervals. As shown in Figure 1B, 120h of TGIF knockdown resulted in a nearly 6‐fold reduction in proliferation (p<0.0001) of shRNA‐transduced HL60 cells compared to cells transduced with a non‐mammalian (GFP) control shRNA sequence. Interestingly, TGIF over‐expression increased proliferation of HL60 cells by nearly 1.9‐fold (p<0.0001) (Figure 1C), although this effect was not as pronounced as that seen with TGIF knockdown. These data thus suggest that TGIF affects myeloid cells in a linear fashion, that is, knockdown results in decreased proliferation and over‐expression results in increased proliferation. To confirm that these effects were not HL60‐specific, we knocked down TGIF in two additional myeloid cell lines, TF‐1 and AML‐193, which showed a similar effect of TGIF depletion on cell proliferation (Supplementary Figures 1 and 2).
Figure 1.

TGIF has a linear effect on HL60 cell proliferation. (A) Lentiviral shRNA‐mediated TGIF knockdown in HL60 cells. Several clones were analyzed and the degree of knockdown was confirmed with both relative real‐time PCR and Western blot analysis (TGIF knockdown Western blot was exposure time was ∼60s while the TGIF over‐expression Western blot time was ∼10s; unequal times were used for better visualization of the respective bands). Data from one such clone is shown. The real‐time analysis was repeated at least 3 times and the results of one representative experiment are shown. Error bars represent SEM. Proliferation inhibition of HL60 cells with TGIF knockdown. (B) TGIF over‐expression (with plasmid p721) resulted in increased proliferation. (C) Proliferation analysis was done in triplicate with manual counting with trypan blue exclusion. Each sample of the triplicate was counted at least 3 times at each time point. X‐axis represents time in hours, Y‐axis shows the number of cells per ml. Several clones were analyzed and compared with HL60 cells and cells transduced with non‐mammalian shRNA. Proliferation analysis was repeated twice and the results of one such analysis is shown. Error bars represent SEM. P values were calculated using students t‐test with the help of Prism 5 statistical suite.
2.2. TGIF knockdown did not affect the cell cycle of HL60 cells but did slightly increase apoptosis
To determine if the observed effects of TGIF knockdown on cell proliferation were associated with alterations in the cell cycle, we evaluated the cell cycle distribution of HL60 clones by PI staining. As shown in Figure 2A, we did not observe a significant difference in the distribution of cells in cycle in HL60 cells transduced with a non‐mammalian control shRNA (GFP) or with TGIF shRNA. We did notice, however, that there was a small (p<0.0001) increase in the subG0 population in the TGIF knockdown clones. Since this population contains apoptotic cells, we further characterized this population with an Annexin V binding assay, a more sensitive method of detecting apoptosis. As shown in Figure 2B, TGIF knockdown resulted in a small increase in apoptosis, consistent with the cell cycle results. Whereas the proliferation analysis presented in Figure 1B showed that the TGIF knockdown resulted in a 6‐fold decrease in proliferation, the differences in early (Annexin V staining) and late stages of apoptosis (Annexin V and PI staining) between the clones and control were much smaller. This suggests that the proliferation inhibition observed in the TGIF knockdown clones was not solely due to increased apoptosis.
Figure 2.
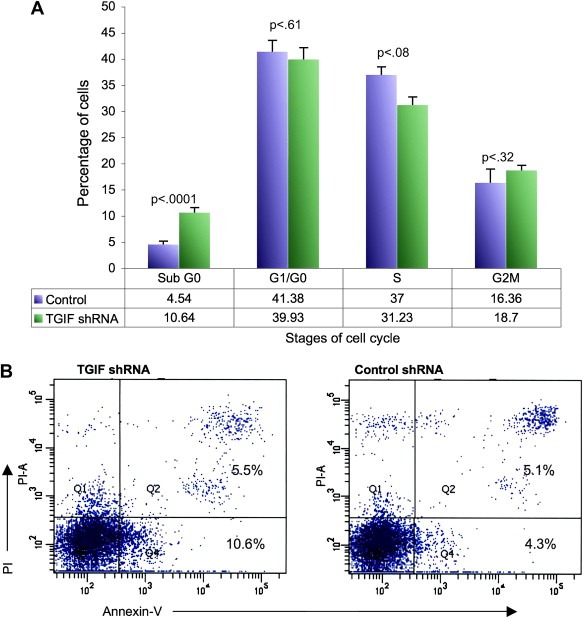
(A) Cell cycle analysis of exponentially growing TGIF knockdown HL60 cells. Cells were stained with PI and analyzed by flow‐cytometry. The histograms represent the mean of three separate experiments showing the cell cycle percentage of the isolated HL60 clones. SubG0 percentages likely represent apoptotic cells. Control cells were HL60 cells transduced with non‐mammalian shRNA. Error bars represent SEM. P values were calculated using students t‐test with the help of Prism 5 statistical suite. (B) Annexin V and PI staining of the TGIF knockdown HL60 cells. The x‐axis shows Annexin V‐FITC binding and the y‐axis staining of the vital dye PI. Cells in the lower left quadrant are viable, cells in the lower right are early apoptotic and those in the upper right are late stage apoptotic/dead cells. The numbers represent the percentage of cells present in each quadrant. Results from one typical experiment are shown. Each experiment was repeated three times.
2.3. TGIF knockdown inhibits differentiation of myeloid cells lines
Since TGIF levels have a linear effect on HL60 cell proliferation, we investigated whether TGIF levels would also affect the differentiation potential of these cells. As shown in Figure 3A, after five days of exposure to all‐trans‐retinoic acid (ATRA), 88.0% of control HL60 cells (transduced with a non‐mammalian shRNA) differentiated into CD11b‐expressing terminal myeloid cells compared to 11.1% of HL60 cells with TGIF knockdown. Just as TGIF over‐expression increased HL60 proliferation, TGIF over‐expression also resulted in the increased differentiation of HL60 cells into terminal myeloid cells (Figure 3B). These data thus support the conclusion that TGIF knockdown inhibits terminal myeloid differentiation of HL60 cells. TGIF's effects on differentiation were not ATRA‐specific, since similar decreased differentiation was also seen with TPA stimulation (data not shown) as well as cell line specific since TGIF knockdown also inhibited differentiation of TF‐1 and AML‐193 cells (Supplementary Figures 1 and 2). Furthermore, the block in differentiation was not absolute and could be overcome by a 5‐fold higher dose of TPA or ATRA (data not shown).
Figure 3.

TGIF knockdown resulted in decreased differentiation (A) and its over‐expression (with plasmid p721) resulted in increased differentiation (B). For HL60 cells with TGIF knockdown, ATRA stimulation continued for 5 days while for cells with TGIF over‐expression ATRA stimulation was stopped after 2 days. Cells were assayed for the expression of terminal myeloid marker CD11b with a PE‐conjugated antibody. The numbers represent the percentage of cells that differentiated following induction with ATRA. Experiment was repeated three times and the results of one representative experiment are shown. P values were calculated using students t‐test with the help of Prism 5 statistical suite.
2.4. TGIF knockdown results in increased TGF‐β and decreased RA signaling
Since TGIF has been shown to modulate TGF‐β and RA signaling, both of which have pivotal roles in hematopoiesis, we initially hypothesized that the observed effects of TGIF knockdown on HL60 cell growth and differentiation were mediated by an effect on endogenous TGF‐β or RA signaling or both. We analyzed the expression levels of several TGF‐β and RA pathway genes, by real‐time PCR, to provide a read‐out of pathway activity. The results showed that TGF‐β target gene expression, in particular the expression of the prototypic target gene PAI1, was higher in TGIF knockdown HL60 cells compared to controls (Figure 4A). On the other hand, reduced expression of RARα and a number of RA target genes, HOXB1 and STRA6 in particular, indicated that RA signaling was less active in TGIF knockdown clones compared to the controls (Figure 4B). Since increased TGF‐β signaling can inhibit myeloid cell proliferation (Kim and Letterio, 2003) and RA signaling can lead to their differentiation (Gaines and Berliner, 2003; Kastner and Chan, 2001), our data suggest that TGIF may affect myeloid cell proliferation and differentiation through modulation of both TGF‐β and RA signaling.
Figure 4.
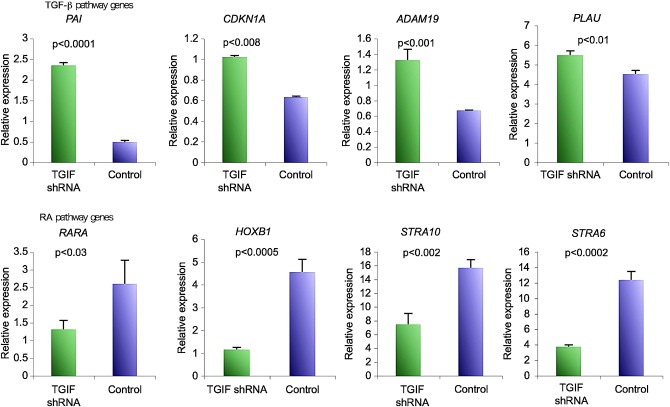
Expression levels of TGF‐β and RA target genes in TGIF knockdown HL60 cells as measured by relative real‐time PCR. TGF‐β target genes are up‐regulated while RA target genes are down regulated as compared to control shRNA‐transduced HL60 cells. Experiment was done in triplicate and repeated twice; the results of one representative experiment are shown. Error bars represent SEM. P values were calculated using students t‐test with the help of Prism 5 statistical suite.
2.5. TGIF knockdown disrupts the myeloid transcription program
The transcriptional program associated with myeloid differentiation is well described and includes a limited number of transcription factors that are expressed at specific developmental stages to facilitate proper myeloid differentiation (Iwasaki and Akashi, 2007; Rosenbauer and Tenen, 2007). Since TGIF knockdown resulted in decreased differentiation of HL60 cells, we hypothesized that TGIF knockdown disrupted the myeloid transcription program. To that end, we determined the abundance of mRNA encoding 16 transcription factors, by real‐time PCR, before and after induction of differentiation of the TGIF knockdown clonal HL60 cells. At baseline, the levels of only three transcription factors, CEBPβ, EFNA2, and c‐MYB, were significantly different between TGIF knockdown and control HL60 cells (data not shown). We then compared the expression levels of these transcription factors by real‐time PCR analysis after induction of differentiation with ATRA. As shown in Figure 5, upon induction of differentiation, HL60 cells with TGIF knockdown had lower transcript levels of five key myeloid transcription factors, CEBPβ, CEBPϵ, PU.1, RUNX1, ZFPM1 and increased levels of c‐MYB as compared to the control cells. Since increased expression of CEBPβ, CEBPε, PU.1, RUNX1 and ZFPM1 and decreased expression of c‐MYB are required for proper myeloid differentiation (Rosenbauer and Tenen, 2007), these data suggest that alteration in this transcriptional program may be, in part, responsible for the decreased differentiation seen in the TGIF knockdown clones. Additionally, since CEBPε, PU.1 and ZFPM1 (FOG1) have all been shown to be positively regulated by RA signaling, these data provide additional support for our hypothesis that reduced RA‐directed transcription may play a role in the observed decrease in differentiation (Evans, 2005; Kastner and Chan, 2001; Lawson and Berliner, 1999; Mueller et al., 2006; Verbeek et al., 1999; Zhuang et al., 2003).
Figure 5.
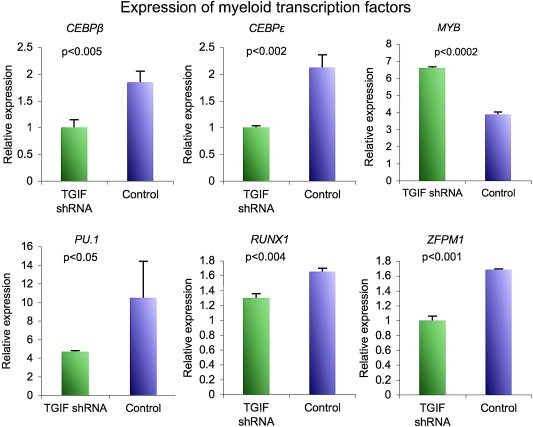
Comparison of expression levels of myeloid specific transcription factors in HL60 cells with TGIF knockdown after the induction of differentiation as measured by real‐time PCR. Out of the 16 transcription factors analyzed, six (CEBPε, CEBPβ, RUNX1, PU.1, ZFPM1, MYB) showed significant differences in expression, between HL60 cells with TGIF knockout and the control HL60cells. Experiment was done in triplicate and repeated twice; the results of one representative experiment are shown. Error bars represent SEM. P values were calculated using students t‐test with the help of Prism 5 statistical suite.
2.6. TGIF target genes have key functions in hematopoiesis
Our data show that TGIF knockdown alters TGF‐β and RA signaling, suggesting that its effects on HL60 proliferation and differentiation may be secondary to its modulation of these pathways. However, transcriptional repression by TGIF also involves its binding to target genes, and thus a thorough understanding of the molecular mechanisms that mediate its actions requires that the role of these target genes should also be taken into account. In practice this proved difficult, since no information existed as to the identity of direct TGIF target genes.
We therefore decided to use a genome‐wide chromatin immunoprecipitation (ChIP) sequencing approach (Brynczka et al., 2007) to identify potential TGIF target genes. This analysis identified nearly 1655 direct targets. We then applied Ingenuity Pathway Analysis (IPA) to these data to obtain further insight into potential cellular pathways that may be modified/regulated by TGIF and its direct targets. This analysis showed that 35% of TGIF target genes regulate cellular proliferation, differentiation and apoptosis, 18% had a previously documented role in hematopoiesis and 15% were involved in cancer (Figure 6). We then used IPA to generate networks of connectivity based on the identified TGIF target genes. Seventy‐two networks were generated; out of these, 34 were considered statistically significant insofar as they exceeded the IPA algorithm cutoff. The top eight are listed in Table 1. The most common functions associated with these networks were related to cancer, hematopoietic development, differentiation, apoptosis and the cell cycle. This was not surprising since, as noted above, 18% of the genes identified as TGIF targets have a defined role in hematopoiesis. Further analyses showed that TGIF binding sites were present in the upstream regions (between −1 and −1000bp) of a majority of genes shown in Table 2.
Figure 6.
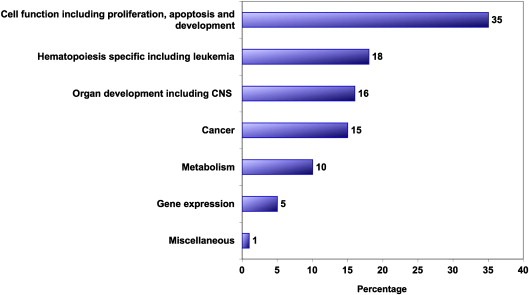
Ingenuity Pathways Analysis (IPA) of the downstream TGIF targets. Numbers represents the percentage of genes in each category.
Table 1.
Biological networks generated by Ingenuity Pathways Analysis. Direct TGIF target genes in bold.
| Molecules in networks | Functions |
|---|---|
| Ap1, BCL10, CD3, CD38, CDX1, CREB3, FKBP5, FPRL1 (includes EG:2358), GAB3, GNA15, HCLS1, IFNAR2, Ikb, IKBKG, IKK, IL1, LTA, MAP3K14, MAP4K1, NF‐κB, Nfat, NFkB, NOD2, PTAFR, PTPN11, RELT, RHOH, SDC4, SIGLEC7, SIGLEC9, TCR, Tnf receptor, TNFRSF1A, TRAF3, TREML1 | Hematopoietic System Development and Function, Organ Morphology, Cancer |
| Ahr‐aryl hydrocarbon‐Arnt, BAX, BBC3, Caspase, CBX5, CCL2, CCL5, CD44, CDKN1A, Creb, Cyclin A, DTL, E2f, E2F3, ETV5, FGF2, Histone h3, HNRNPA1, JAK, LDL, Mek, MLL, Mmp, NFE2, P38 MAPK, PI3K, PIK3CD, Rb, RNA polymerase II, TASP1, Tgf beta, TGFBR2, TRIP11, UCN2, Vegf | Cellular Growth and Proliferation, Cancer, Cell Death |
| AMN , beta‐estradiol, BICD1, BMI1, CBX6, CDC14B, CHMP4A, COL4A5, CUBN, FUCA1, GLIPR1, HDAC1 (includes EG:3065), HIST2H2BE, HOXA9, HOXC5, HOXC12, HS3ST1, IFNA14, MLLT10, MYOG, PBXIP1, PDCD6IP, PHC3, PTPRU, RAD54B, RCBTB2, RECQL4, retinoic acid, SCMH1, SIX5, SMARCB1, TGIF2, THSD4, TP53, TSC22D1 | Hematological Disease, Cell Cycle, Cancer |
| Akt, ANXA11, ATYPICAL PROTEIN KINASE C, CBLB, COL13A1, Cyclin D, Gsk3, IL8RB, Insulin, Mapk, Mek1/2, MYB, N‐cor, Pdgf, PDGF BB, Pdgfr, PDGFRB, PDPK1, Pkc(s), Pld, PP2A, PRKCD, RAB2A, RARA, RARG, Ras, Rsk, Rxr, S100A8, Sos, STAT, STAT5a/b, TBL1XR1, VitaminD3‐VDR‐RXR | Embryonic Development, Nervous System Development and Function |
| ARL6IP5 , BAG3, BMP1, C5, CAPRIN1, CD46, EGF, ETV3, FOXH1, G3BP1, GNPAT, GPR77, HMMR, HOXB5, HRH2, JARID1B, MBOAT5, MIA3, MTSS1, MYC, NOP5/NOP58, PDE6H, PHLDA2, PLC gamma, PLS3, PRPS1, PTPRD, RALGPS1, SRC, SRM, STOML2, TGFB1, TOM1L1, UNC119, ZFP161 | Cellular Movement, Cell Morphology, Cellular Assembly and Organization |
| ARHGAP26 , ASGR1, CABIN1, Calmodulin, Calmodulin‐Hsp90‐Nos3, Calpain, CDC37L1, Dynein, EIF3K, F Actin, FGD4, FKBP4, FKBP51‐TEBP‐GR‐HSP90‐HSP70, FKBP52‐Dyenin‐Glucocorticoid‐GR‐HSP90, Hsp70, Hsp90, IFNZ, IL29, IL27RA, Jnk, MAP6, MAP4K3, Pka, PLC, RAB19, Rac, Ras homolog, RASGRP2, SMC3, SOCS4, SRP9, STAT3, STAT5A, STAT5B, USP6 | Hematological Disease, Cell Death, Cellular Growth and Proliferation |
| B4GALT1 , BCL10, CASP8AP2, CD3, CD38, CDX1, CREB3, DDX58, ECSIT, FKBP4, FKBP5, FPRL1 (includes EG:2358), GNA15, Ikb, IKBKG, IKK, ITCH, LTA, MAP3K14, MUC2 (includes EG:4583), NF‐kappa;B, NFkB, NIBP, NKIRAS1 (includes EG:28512), NOD2, Peptidylprolyl isomerase, PPIF, PRKDC, PTAFR, RHOH, SDC4, SLC11A2, SLIT2, Tnf receptor, TXLNA | Cell signaling, Cellular Movement |
| CBLB , CNN1, DDEF1, DUSP3, ENaC, FCHSD2, GAB3, IL7, JAK, KLHL21, LYN, NEDD4L, Pdgf, PDGF BB, Pdgfr, PDGFRB, PI3K, PIK3CD, PIK3R5, PLC gamma, PLK2, PTPN11, SCNN1A, SND1, Sos, STAT, STAT5A, STAT5a/b, STAT5B, SV2A, TP53I11, TREML1, VAV, VAV3, WNK4 | Gene Expression, Hematological System Development and Function, Immune and Lymphatic System Development and Function |
Table 2.
Selected direct TGIF target genes with functions in hematopoiesis and myelopoiesis.
| Gene ID | Gene symbol | Gene name |
|---|---|---|
| GeneID:5914 | RARA | Retinoic acid receptor, alpha |
| GeneID:6776 | STAT5A | Signal transducer and activator of transcription 5A |
| GeneID:6777 | STAT5B | Signal transducer and activator of transcription 5B |
| GeneID:6774 | STAT3 | Signal transducer and activator of transcription 3 (acute‐phase response factor) |
| GeneID:5914 | RARA | Retinoic acid receptor, alpha |
| GeneID:8915 | BCL10 | B‐cell CLL/lymphoma 10 |
| GeneID:10488 | CREB3 | cAMP responsive element binding protein 3 |
| GeneID:4297 | MLL | Myeloid/lymphoid or mixed‐lineage leukemia (trithorax homolog, Drosophila) |
| GeneID:649 | BMP1 | Bone morphogenetic protein 1 |
| GeneID:4602 | MYB | v‐myb myeloblastosis viral oncogene homolog (avian) |
| GeneID:8928 | FOXH1 | Forkhead box H1 |
| GeneID:8028 | MLLT10 | Myeloid/lymphoid or mixed‐lineage leukemia (trithorax homolog, Drosophila); translocated to, 10 |
| GeneID:3228 | HOXC12 | Homeobox C12 |
| GeneID:60436 | TGIF2 | TGFB‐induced factor 2 (TALE family homeobox) |
| GeneID:2299 | FOXI1 | Forkhead box I1 |
| GeneID:9020 | MAP3K14 | Mitogen‐activated protein kinase kinase kinase 14 |
| GeneID:147912 | SIX5 | Sine oculis homeobox homolog 5 (Drosophila) |
| GeneID:55193 | PB1 | Polybromo 1 |
| GeneID:50485 | SMARCAL1 | SWI/SNF related, matrix‐associated, actin dependent regulator of chromatin, subfamily a‐like 1 |
| GeneID:11244 | ZHX1 | Zinc fingers and homeoboxes 1 |
| GeneID:51130 | ASB3 | Ankyrin repeat and SOCS box‐containing 3 |
| GeneID:114907 | FBXO32 | F‐box protein 32 |
| GeneID:142 | PARP1 | Poly (ADP‐ribose) polymerase 1 |
| GeneID:29109 | FHOD1 | Formin homology 2 domain containing 1 |
| GeneID:3228 | HOXC12 | Homeobox C12 |
| GeneID:144699 | FBXL14 | F‐box and leucine‐rich repeat protein 14 |
| GeneID:114907 | FBXO32 | F‐box protein 32 |
| GeneID:3274 | HRH2 | Histamine receptor H2 |
| GeneID:7048 | TGFBR2 | Transforming growth factor, beta receptor II (70/80kDa) |
| GeneID:80790 | CMIP | c‐Maf‐inducing protein |
| GeneID:23523 | CABIN1 | Calcineurin binding protein 1 |
| GeneID:81693 | AMN | Amnionless homolog (mouse) |
| GeneID:5159 | PDGFRB | Platelet‐derived growth factor receptor, beta polypeptide |
| GeneID:3274 | HRH2 | Histamine receptor H2 |
| GeneID:6352 | CCL5 | Chemokine (C–C motif) ligand 5 |
| GeneID:3695 | ITGB7 | Integrin, beta 7 |
| GeneID:868 | CBLB | Cas‐Br‐M (murine) ecotropic retroviral transforming sequence b |
| GeneID:4049 | LTA | Lymphotoxin alpha (TNF superfamily, member 1) |
| GeneID:56899 | EB‐1 | E2a‐Pbx1‐associated protein |
| GeneID:8928 | FOXH1 | Forkhead box H1 |
| GeneID:5170 | PDPK1 | 3‐Phosphoinositide dependent protein kinase‐1 |
| GeneID:10550 | JWA | Cytoskeleton related vitamin A responsive protein |
| GeneID:27335 | eIF3k | Eukaryotic translation initiation factor 3 subunit k |
| GeneID:148423 | gm117 | gm117 |
| GeneID:4602 | MYB | v‐myb myeloblastosis viral oncogene homolog (avian) |
| GeneID:79718 | IRA1 | Likely ortholog of mouse IRA1 protein |
| GeneID:952 | CD38 | CD38 antigen (p45) |
| GeneID:285852 | TLT4 | Triggering receptor expressed on myeloid cells‐like 4 |
| GeneID:5781 | PTPN11 | Protein tyrosine phosphatase, non‐receptor type 11 (Noonan syndrome 1) |
| GeneID:7187 | TRAF3 | TNF receptor‐associated factor 3 |
| GeneID:399 | RHOH | ras homolog gene family, member H |
| GeneID:9321 | TRIP11 | Thyroid hormone receptor interactor 11 |
| GeneID:5293 | PIK3CD | Phosphoinositide‐3‐kinase, catalytic, delta polypeptide |
| GeneID:5580 | PRKCD | Protein kinase C, delta |
| GeneID:4297 | MLL | Myeloid/lymphoid or mixed‐lineage leukemia (trithorax homolog, Drosophila) |
| GeneID:8778 | SIGLEC5 | Sialic acid binding Ig‐like lectin 5 |
| GeneID:6385 | SDC4 | Syndecan 4 (amphiglycan, ryudocan) |
| GeneID:2247 | FGF2 | Fibroblast growth factor 2 (basic) |
| GeneID:51514 | RAMP | RA‐regulated nuclear matrix‐associated protein |
| GeneID:6347 | CCL2 | Chemokine (C–C motif) ligand 2 |
We then reasoned that if TGIF directly binds to the proximal regulatory regions of these genes then its knockdown would affect their transcript levels positively and vice versa. We thus analyzed the transcripts' levels of a number of the genes listed in Table 2 by real‐time PCR, in the HL60 cells with TGIF knockdown and over‐expression. As shown in Table 3, TGIF knockdown resulted in increased transcription and its over‐expression in decreased transcription of 13 of the 23 genes analyzed. These data represent the first experimental identification of downstream TGIF targets. It is interesting to note that several of these genes have key functions in hematopoiesis and hematopoietic stem cell biology, including MLL, MLLT10 and the Forkhead transcription factor family members.
Table 3.
Real‐time PCR confirmation of the ChIP identified TGIF targets. Plus (+) signifies genes that were up‐regulated with TGIF knockdown and down regulated with TGIF over‐expression. Negative (−) represent genes whose expression did not increase with TGIF knockdown and decrease with TGIF over‐expression *HOXC12 and BCL10 were down regulated by TGIF knockdown and unaffected by TGIF over‐expression.
| Gene | Up‐regulated by TGIF knockdown & down regulated by TGIF over‐expression | P value |
|---|---|---|
| FOXI1 | + | 0.0004 |
| MLL | + | 0.04 |
| MLLT10 | + | 0.0024 |
| FHOD1 | + | 0.014 |
| FOXH1 | + | 0.0001 |
| FBXL14 | + | 0.0008 |
| TGFBR2 | + | 0.0001 |
| PARP1 | + | 0.0011 |
| ASB3 | + | 0.0001 |
| PBRM1 | + | 0.034 |
| TGIF2 | + | 0.02 |
| STAT5B | + | 0.0107 |
| MYB | + | 0.02 |
| BMP | − | 0.39 |
| CREB | − | 0.36 |
| SMARCAL1 | − | 0.66 |
| HOXC12* | − | 0.0001 |
| BCL10* | − | 0.0002 |
| FBOX32 | − | 0.34 |
| RARA | − | 0.2 |
| STAT5A | − | 0.07 |
| MAP3K14 | − | 0.3228 |
| SIX5 | − | 0.879 |
3. Discussion
In this study, we present the first in vitro evidence of a role for TGIF in myelopoiesis. Based on TGIF's membership in a small group of genes whose expression constitutes a molecular signature for HSC proliferation and our finding that its abundance correlated with survival in AML, we proceeded to determine whether TGIF has a direct role in myelopoiesis. We used HL60 cells, a well‐characterized cell line model of leukemic myeloid cell differentiation, to study the effects of TGIF knockdown on hematopoietic myeloid cell function. We show that TGIF has a linear effect on HL60 growth and differentiation, that is, its knockdown resulted in decreased proliferation and differentiation and its over‐expression had the opposite effect. Our data also suggest that the molecular mechanisms behind the observed effects are complex and likely mediated through modulations of both RA and TGF‐β signaling and further complemented by TGIF's interactions with its direct downstream target genes.
Since TGIF is so intimately involved in the regulation of TGF‐β and RA signaling pathways and may play a role in cross‐talk between these pathways (Zhang et al., 2009), we first sought to determine whether our findings could be explained by altered activity of either of these pathways. TGF‐β has well described inhibitory actions on cell proliferation, including hematopoietic cells (Batard et al., 2000; Garbe et al., 1997; Sitnicka et al., 1996), and can also function as an inducer of apoptosis (Lee and Bae, 2002). TGF‐β can also affect cell cycle progression through alterations in expression of cytokine receptors and/or certain cyclin‐dependent kinase (cdk) inhibitors, such as CDKN1A (p21). Our data showed that TGIF knockdown increased TGF‐β signaling, providing support to our hypothesis that the observed growth inhibition may be due to increased TGF‐β signaling. It is unlikely that the concomitantly decreased RA signaling is responsible for the observed slow proliferation, since it is increased RA activity that leads to growth inhibition, not decreased activity. These data are also consistent with the previous report that knockdown of TGIF results in increased TGF‐β‐induced expression of PAI1 in Mv1Lu cells (Seo et al., 2006), increased TGF‐β activity in mesenchyme micomass cultures (Zhang et al., 2009) and that murine embryonic fibroblasts isolated from Tgif−/− mice do not proliferate as well as those isolated from wild‐type mice in vitro (Mar and Hoodless, 2006). However, there remain caveats to this hypothesis, one of the most important being that HL60 cells over‐express c‐Myc due to extra chromosomal amplification, and c‐Myc over‐expression has been shown to block the growth inhibitory responses of TGF‐β (Ruegemer et al., 1990). In addition, even though a TGF‐β‐mediated reduction in overall progression through cell cycle has been reported (Stoeck et al., 1989), the most common effect of TGF‐β on the cell cycle is G1 arrest (Ewen et al., 1993). Thus, significant growth inhibition in the presence of c‐Myc gene amplification and overall slow progression through the cell cycle without an obvious block at a particular stage (specifically G1) suggest that other pathways or genes may mediate TGIF's effects. Our TGIF target data provide intriguing clues to this end. For example, TGIF binds to several Forkhead and Forkhead binding factor genes. TGIF knockdown led to increased FOXH1 expression, which has been shown have an inhibitory effect on the cell cycle through its interactions with p21 (van der Vos and Coffer, 2008). Similarly, TGIF binds to MLL regulatory regions and its knockdown increases MLL expression, which can decrease cell cycle progression (Milne et al., 2005).
The effects of TGIF on differentiation functions of HL60 cells, on the other hand, are likely secondary to altered RA signaling. In hematopoiesis, the best‐documented action of RA is the induction of differentiation of progenitor cells (reviewed in (Collins, 2002; Evans, 2005; Purton, 2007)). Our data show that TGIF knockdown resulted not only in down‐regulation of several known RA target genes, but also in altered expression of several RA‐regulated myeloid transcription factors, including CEBPβ, CEBPε, PU.1, RUNX1, c‐MYB and ZFPM1, supporting the notion that the decreased differentiation observed with TGIF depletion resulted in part from down‐regulation of RA signaling. The inability of the TGIF knockdown HL60 cells to up‐regulate CEBPε is particularly intriguing in that it is consistent with a recent report demonstrating that shRNA‐mediated TGIF knockdown in pre‐adipocytes inhibited their differentiation into mature adipocytes through a decrease in CEBPε expression (Horie et al., 2008). CEBPϵ is known to be important in myeloid differentiation (Yamanaka et al., 1997).
Our data thus indicate that TGIF's regulation of myelopoiesis is complex, involving modulation of TGF‐β and RA signaling, alteration of the myeloid transcription program, and interaction with downstream TGIF targets. Based on the data reported in this study and our recent finding that TGIF levels predict survival in AML, it was not surprising that a significant number of TGIF targets have a documented role in hematopoiesis. These hematopoiesis‐specific targets include master regulators of transcriptional programs such as Forkhead box (FOX) proteins; STAT5B and MLL, which regulate histone methylation; cell cycle regulators such as p21; and important receptors such as TGFBR2.
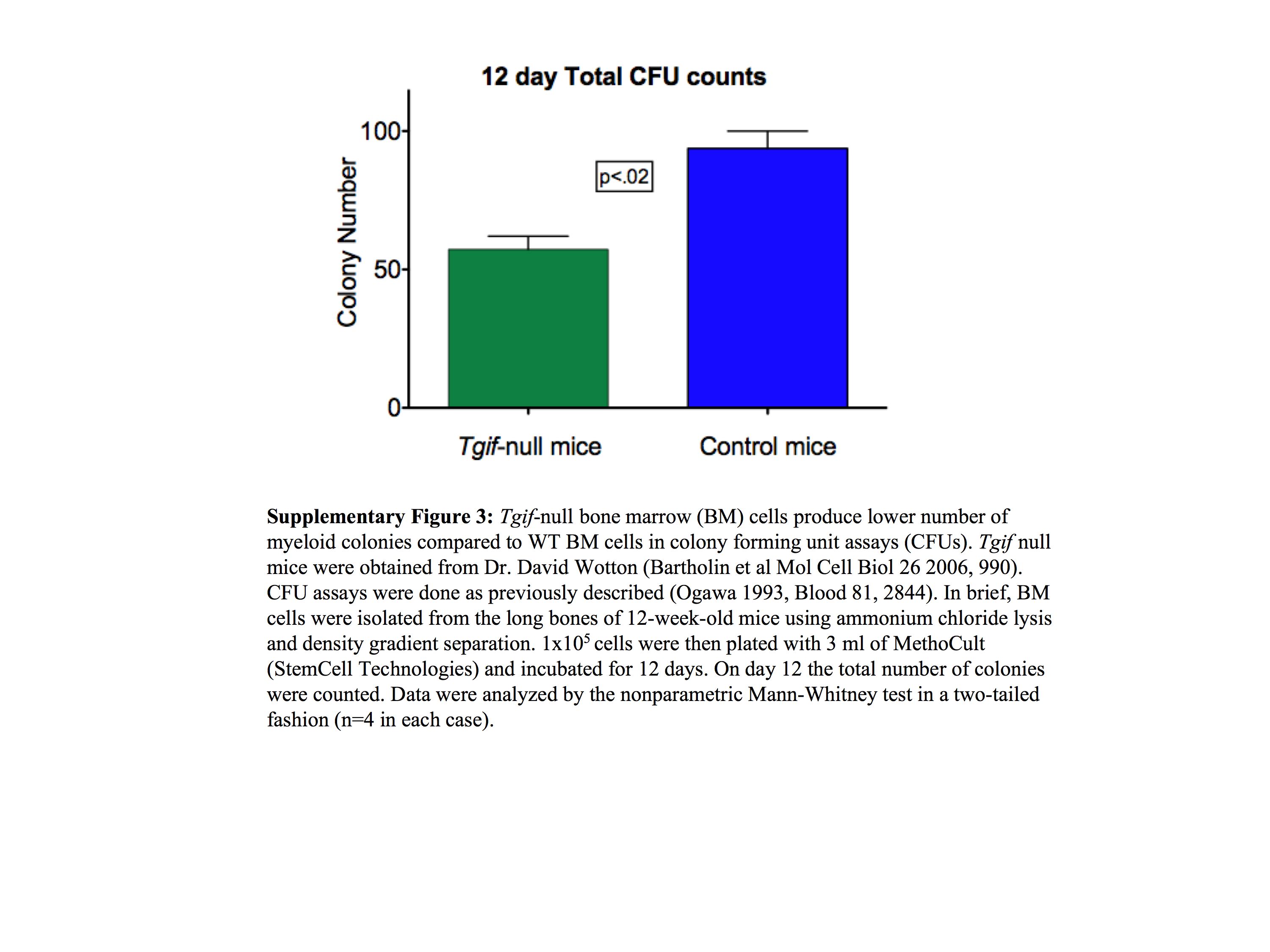
The data presented in this report are only the first steps toward understanding how TGIF regulates myelopoiesis. Further studies will require the elucidation of the complete TGIF transcriptional network, which would include careful validation of all of the candidate target genes as well as identification of genes that regulate TGIF – both active areas of research in our laboratory. One caveat to our findings is that our data were collected using an in vitro model of myelopoiesis and thus would require detailed analyses using an in vivo model system. Preliminary studies from a knockout model, under validation in our laboratory, show that bone marrow cells from Tgif‐null mice produce fewer myeloid colonies in colony forming units assays suggesting that, as was the case with the myeloid cell lines, Tgif knockout may also produce proliferation inhibition in primary bone marrow cells (Supplementary Figure 3).
Lastly, if TGIF knockdown results in growth inhibition and a relative differentiation block, as our data clearly indicate, how might this explain our previously reported finding that TGIF levels correlate inversely with clinical outcome in AML? One explanation may be that TGIF's effect on leukemic cells work within the framework of the “two‐hit” hypothesis of leukemogenesis. In this scenario, decreased TGIF would provide the first hit, leading to a differentiation block; an additional hit would endow the cell with a proliferative/anti‐apoptotic advantage, thus leading to accumulation of immature blasts. Another possible explanation could be that TGIF's actions in myeloid cells are quite different than in hematopoietic or leukemic stem cells. Clinically, growth inhibitory effects of TGIF knockdown may result in decreased HSC cycling, offering them added protection against chemotherapeutic agents, since these agents primarily affect more rapidly growing cells. These growth inhibitory effects are likely due to increased TGF‐β signaling, based on the data presented here and the known effects of TGF‐β signaling in HSC function. TGIF's known interactions with cPML provide further support for this hypothesis, since they result in the nuclear sequestration of cPML and the disruption of the cPML‐SARA complex with resulting decreased TGF‐β signaling. Knockdown of TGIF by siRNA caused increased association of cPML with SARA and cytoplasmic accumulation of cPML with resultant increased TGF‐β signaling (Faresse et al., 2008; Seo et al., 2006).
The association of TGIF with cPML is particularly intriguing since PML is the target of reciprocal translocation with the RAR locus in acute promyelocytic leukemia. PML likely regulates the activity of key tumor suppressive pathways such as p53 and retinoblastoma and has been shown regulate cell cycle, apoptosis and induction of quiescence (Bernardi et al., 2008). However, TGIF interacts with the cytoplasmic rather than the nuclear isoforms of PML, and the function of these isoforms in leukemia or other types of cancer remains to be fully elucidated. Thus, based on TGIF's role in TGF‐β signaling through its cPML‐mediated and cPML‐independent interactions with Smad proteins, low TGIF‐expressing leukemic HSCs from an AML patient would be expected to survive induction chemotherapy, increasing the likelihood of relapse. We plan to test this hypothesis with the help of a Tgif knockout mouse model under validation.
In summary, this is the first report that identifies a role for the stem cell‐expressed homeobox gene, TGIF, in myelopoiesis. In light of our earlier observation that the level of TGIF expression in blast cells at diagnosis appears to be a powerful predictor of disease relapse and patient survival in AML, our findings indicate that TGIF may also have a role in myeloid leukemia. Further in vivo work is necessary to confirm these findings and fully elucidate TGIF's role in normal and abnormal myelopoiesis.
4. Experimental procedures
4.1. shRNA‐mediated silencing of TGIF expression
Four pLKO.1‐based lentiviral vectors that contain stem‐loop cassettes encoding shRNAs targeted to human TGIF mRNA (GenBank accession number NM_003244) were obtained from Open Biosystems, cat number RHS4533‐NM_003244 (Huntsville, AL). pLKO.1 TRC control (Addgene plasmid 10879) (Moffat et al., 2006), a non‐mammalian silencing control against GFP (Addgene plasmid 12273) (Orimo et al., 2005), and packaging plasmids pMD2.G (Addgene plasmid 12259) (Klages et al., 2000) and pCMV‐dR8.2 dvpr (Addgene plasmid 8455) (Stewart et al., 2003) were obtained from Addgene Inc. (Cambridge, MA). Lentiviral particles were produced by co‐transfection of 293‐T cells with pLKO.1 constructs and the compatible packaging plasmids as previously described (Naldini et al., 1996; Zufferey et al., 1997).
1ml (high multiplicity of infection) of the harvested lentiviral supernatants were used to transduce HL60 cells at a density of 0.3×103 in a 10ml volume containing 8μg/ml Polybrene (American Bioanalytical) using the RNAi Consortium lentiviral protocol (http://www.broad.mit.edu/node/563). Infected cells were selected with 0.4μg/ml puromycin (Sigma‐Aldrich). After the selection was complete, the TGIF shRNA‐lentiviral infected cells, as well as the cells infected with the three control lentiviral‐shRNA constructs, GFP, TRC and Scramble, were tested for TGIF knockdown by real‐time PCR and Western blot analysis (see below) to determine the degree of knockdown for each of the four lentiviruses tested. We then used limiting dilution in 96‐well plates to isolate individual clones and quantify the extent of TGIF knockdown by real‐time PCR and Western blot analysis.
4.2. Western blot analysis
Cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), transferred to polyvinylidenefluoride (PVDF) membranes and probed with a rabbit anti‐TGIF polyclonal antibody, sc‐9084, and an anti‐actin polyclonal antibody, sc‐1615‐R (Santa Cruz Biotechnology, Santa Cruz, CA). Proteins were detected with horseradish peroxidase‐labeled secondary antibodies and the Immobilon Chemiluminescent HRP substrate (Millipore, Billerica, Massachusetts).
4.3. Proliferation assays
Logarithmically growing HL60 cells were counted with a hematocytometer and seeded in triplicate at a density of 2×104cells/ml in full growth media without puromycin. The cell number and viability were determined every 24h by manual counting with a hematocytometer using trypan blue (0.03%).
4.4. TGIF over‐expression
TGIF over‐expression vector was prepared by inserting TGIF cDNA into the vector pEFIRES‐P at its EcoR1 cloning site. The vector contains the strong elongation factor 1a promoter (Hobbs et al., 1998). This plasmid is referred to as p721 in the text.
4.5. Apoptosis assay and cell cycle analysis
In order to measure apoptosis we used an Annexin‐V/Propidium iodide (PI) double staining according to the manufacturer's instructions (Invitrogen Carlsbad, CA). For cell cycle analysis, exponentially growing cells were fixed with 70% ethanol for 24h and then treated with 300μl of DNA staining solution (10ml of 0.1% Triton X‐100 in PBS, 2mg DNAse‐free RNAse A and 0.40ml of 500μg/ml PI) for 15min. Cells were analyzed by flow‐cytometry (BD Bioscience, San Jose CA) using FACSDiva software (BD Bioscience, San Jose CA). Sample size was set at 20,000 events.
4.6. Differentiation assay
HL60 cells were induced to differentiate as previously described (Gaines and Berliner, 2005; Le Cabec et al., 1997). In brief, exponentially growing cells were seeded at a density of 0.5×106 per ml and then induced to differentiate with 0.5μM all‐trans‐retinoic acid (ATRA) or 50nM TPA for 2 days. Differentiation was monitored by the expression of the antigen marker CD11b, which is only expressed on the terminally differentiated granulocytes/monocytes, by a PE‐conjugated anti‐CD11b antibody. Data were acquired by flow‐cytometry (BD Bioscience, San Jose CA) using FACSDiva software (BD Bioscience, San Jose CA). Sample size was set at 20,000 events.
4.7. Real‐time PCR analysis
Real‐time PCR analysis for TGIF abundance was done as previously described (Hamid et al., 2008). For analysis of RNA encoded by the TGF‐β pathway and myeloid transcription factor genes and TGIF target genes we used predesigned QuantiTect Primer assays (Qiagen, Valencia, CA). Real‐time PCR was carried out with SybrGreen (Applied Biosystems, Foster City, CA) and consisted of 95°C for 30s, 55°C for 30s, and 72°C for 30s for 40 cycles.
4.8. Chromatin immunoprecipitation (ChIP) sequencing analysis
This analysis was done using Genpathway's FactorPath Discovery approach, as described previously (Brynczka et al., 2007). Briefly, 10 million HL60 cells were cross‐linked by 1% formaldehyde and lysed by Dounce homogenization. Chromatin was sheared to an average length of 500bp and TGIF–DNA complexes were immunoprecipitated using an anti‐TGIF polyclonal antibody (sc‐9084, Santa Cruz Biotech, Santa Cruz, CA). To assay for TGIF binding sites in purified ChIP DNA, target‐specific primers were used to measure amounts of target sequence in immunoprecipitated samples by real‐time PCR.
Enriched ChIP DNA was ligated to a DNA adapter and amplified by PCR. The amplified ChIP DNA was cloned into the pCR2.1‐TOPO vector (Invitrogen) and sequenced. Individual tag sequences were aligned to NCBI human genome build v35.1 using Megablast 2.2.13. Tags producing low scoring or multiple alignments were eliminated from consideration. Clusters were generated by grouping of tags mapping to within 2kb of each other. Alignments were associated with annotated or predicted genes mapping to within 10kb of each tag. To confirm candidate TGIF binding sites, PCR primers targeting a region within 200bp of each selected alignment or cluster were used to measure the amount of sequence in immunoprecipitated samples by real‐time PCR.
4.9. In silico functional analysis of the TGIF targets using ingenuity pathways analysis
We used Ingenuity Pathways Analysis (IPA) (Ingenuity® Systems, www.ingenuity.com) to generate networks and conduct TGIF target functional analyses. A data set containing gene identifiers was uploaded into the application. These genes, called focus genes, were overlaid onto a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. IPA generates models of gene interactions called networks that are presented graphically to show relationships between genes and the pathways they regulate. These networks have a maximum of 35 genes enriched for the input proteins and are ranked according to a score calculated via a right‐tailed Fisher's exact test. In the network representations, genes are represented as nodes and their relationships as edges (lines). All edges are supported by references from the literature. The functional analysis of a network identified the biological functions and/or diseases that were most common to the genes in the network. The network genes associated with biological functions and/or diseases in the Ingenuity Pathways Knowledge Base were considered for the analysis. Fischer's exact test was used to calculate a p‐value determining the probability that each biological function and/or disease assigned to that network is due to chance alone.
4.10. Cell culture
Human HL60, TF‐1, AML‐193 and 293‐T cells were purchased from American Type Culture Collection (ATCC, Rockville, MD). HL60, AML‐193 and TF‐1 are human acute myeloid leukemia cell lines and were maintained in 80% IMDM+20% FBS, 70% IMDM+10% FBS+2ηg/ml recombinant GM‐CSF, and 70% RPMI 1640+20% FBS+5ηg/ml recombinant GM‐CSF, respectively. 293‐T cells were maintained in 90% DMEM and 10% FBS.
Supporting information
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgments
The work presented in this study was supported in part by an institutional grant from the American Cancer Society (to R.H.) and the Vanderbilt Physician Scientist Program (to R.H.). We also received editorial assistance from Melissa Stauffer, PhD, of Scientific Editing Solutions (scientificeditingsolutions.com).
Supplementary data 1.
Supplementary data associated with this article can be found in the online version, at doi:10.1016/j.molonc.2009.07.004.
Hamid Rizwan, Brandt Stephen J., (2009), Transforming growth‐interacting factor (TGIF) regulates proliferation and differentiation of human myeloid leukemia cells, Molecular Oncology, 3, doi: 10.1016/j.molonc.2009.07.004.
References
- Bartholin, L. , Powers, S.E. , Melhuish, T.A. , Lasse, S. , Weinstein, M. , Wotton, D. , 2006. TGIF inhibits retinoid signaling. Mol. Cell. Biol.. 26, (3) 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batard, P. , Monier, M.N. , Fortunel, N. , Ducos, K. , Sansilvestri-Morel, P. , Phan, T. , Hatzfeld, A. , Hatzfeld, J.A. , 2000. TGF-(beta)1 maintains hematopoietic immaturity by a reversible negative control of cell cycle and induces CD34 antigen up-modulation. J. Cell. Sci.. 113, (Pt 3) 383–390. [DOI] [PubMed] [Google Scholar]
- Belloni, E. , Muenke, M. , Roessler, E. , Traverso, G. , Siegel-Bartelt, J. , Frumkin, A. , Mitchell, H.F. , Donis-Keller, H. , Helms, C. , Hing, A.V. , 1996. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet.. 14, (3) 353–356. [DOI] [PubMed] [Google Scholar]
- Bernardi, R. , Papa, A. , Pandolfi, P.P. , 2008. Regulation of apoptosis by PML and the PML-NBs. Oncogene. 27, (48) 6299–6312. [DOI] [PubMed] [Google Scholar]
- Bertolino, E. , Reimund, B. , Wildt-Perinic, D. , Clerc, R.G. , 1995. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J. Biol. Chem.. 270, (52) 31178–31188. [DOI] [PubMed] [Google Scholar]
- Brown, S.A. , Warburton, D. , Brown, L.Y. , Yu, C.Y. , Roeder, E.R. , Stengel-Rutkowski, S. , Hennekam, R.C. , Muenke, M. , 1998. Holoprosencephaly due to mutations in ZIC2, a homologue of drosophila odd-paired. Nat. Genet.. 20, (2) 180–183. [DOI] [PubMed] [Google Scholar]
- Brynczka, C. , Labhart, P. , Merrick, B.A. , 2007. NGF-mediated transcriptional targets of p53 in PC12 neuronal differentiation. BMC. Genomics.. 8, 139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S.J. , 2002. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia. 16, (10) 1896–1905. [DOI] [PubMed] [Google Scholar]
- de la Cruz, J.M. , Bamford, R.N. , Burdine, R.D. , Roessler, E. , Barkovich, A.J. , Donnai, D. , Schier, A.F. , Muenke, M. , 2002. A loss-of-function mutation in the CFC domain of TDGF1 is associated with human forebrain defects. Hum. Genet.. 110, (5) 422–428. [DOI] [PubMed] [Google Scholar]
- Evans, T. , 2005. Regulation of hematopoiesis by retinoid signaling. Exp. Hematol.. 33, (9) 1055–1061. [DOI] [PubMed] [Google Scholar]
- Ewen, M.E. , Sluss, H.K. , Whitehouse, L.L. , Livingston, D.M. , 1993. TGF beta inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell. 74, (6) 1009–1020. [DOI] [PubMed] [Google Scholar]
- Faresse, N. , Colland, F. , Ferrand, N. , Prunier, C. , Bourgeade, M.F. , Atfi, A. , 2008. Identification of PCTA, a TGIF antagonist that promotes PML function in TGF-beta signalling. Embo. J.. 27, (13) 1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaines, P. , Berliner, N. , 2003. Retinoids in myelopoiesis. J. Biol. Regul. Homeost. Agents. 17, (1) 46–65. [PubMed] [Google Scholar]
- Gaines, P. , Berliner, N. , 2005. Differentiation and characterization of myeloid cells. Cur. Protocols Immunol. Suppl.. 67, [DOI] [PubMed] [Google Scholar]
- Garbe, A. , Spyridonidis, A. , Mobest, D. , Schmoor, C. , Mertelsmann, R. , Henschler, R. , 1997. Transforming growth factor-beta 1 delays formation of granulocyte-macrophage colony-forming cells, but spares more primitive progenitors during ex vivo expansion of CD34+ haemopoietic progenitor cells. Br. J. Haematol.. 99, (4) 951–958. [DOI] [PubMed] [Google Scholar]
- Hamid R, Means-Powell J, Martincic D, Kravtsov VD, Shyr Y, Greer JP, Byrne DW, Koury MJ, Brandt SJ. Expression of the TG-Interacting Factor Gene in Blast Cells at Diagnosis Correlates with Patient Survival in Acute Myeloid Leukemia. Blood, Submitted for publication.
- Hamid, R. , Patterson, J. , Brandt, S.J. , 2008. Genomic structure, alternative splicing and expression of TG-interacting factor, in human myeloid leukemia blasts and cell lines. Biochimica et Biophysica Acta-Gene Regulatory Mechanisms. 1779, 347–355. [DOI] [PubMed] [Google Scholar]
- Hobbs, S. , Jitrapakdee, S. , Wallace, J.C. , 1998. Development of a bicistronic vector driven by the human polypeptide chain elongation factor 1alpha promoter for creation of stable mammalian cell lines that express very high levels of recombinant proteins. Biochem. Biophys. Res. Commun.. 252, (2) 368–372. [DOI] [PubMed] [Google Scholar]
- Horie, T. , Ono, K. , Kinoshita, M. , Nishi, H. , Nagao, K. , Kawamura, T. , Abe, Y. , Wada, H. , Shimatsu, A. , Kita, T. , 2008. TG-interacting factor is required for the differentiation of preadipocytes. J. Lipid Res. [DOI] [PubMed] [Google Scholar]
- Iwasaki, H. , Akashi, K. , 2007. Myeloid lineage commitment from the hematopoietic stem cell. Immunity. 26, (6) 726–740. [DOI] [PubMed] [Google Scholar]
- Kastner, P. , Chan, S. , 2001. Function of RARalpha during the maturation of neutrophils. Oncogene. 20, (49) 7178–7185. [DOI] [PubMed] [Google Scholar]
- Kim, S.J. , Letterio, J. , 2003. Transforming growth factor-beta signaling in normal and malignant hematopoiesis. Leukemia. 17, (9) 1731–1737. [DOI] [PubMed] [Google Scholar]
- Klages, N. , Zufferey, R. , Trono, D. , 2000. A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol. Ther.. 2, (2) 170–176. [DOI] [PubMed] [Google Scholar]
- Larsson, J. , Karlsson, S. , 2005. The role of Smad signaling in hematopoiesis. Oncogene. 24, (37) 5676–5692. [DOI] [PubMed] [Google Scholar]
- Lawson, N.D. , Berliner, N. , 1999. Neutrophil maturation and the role of retinoic acid. Exp. Hematol.. 27, (9) 1355–1367. [DOI] [PubMed] [Google Scholar]
- Le Cabec, V. , Calafat, J. , Borregaard, N. , 1997. Sorting of the specific granule protein, NGAL, during granulocytic maturation of HL-60 cells. Blood. 89, (6) 2113–2121. [PubMed] [Google Scholar]
- Lee, K.Y. , Bae, S.C. , 2002. TGF-beta-dependent cell growth arrest and apoptosis. J. Biochem. Mol. Biol.. 35, (1) 47–53. [DOI] [PubMed] [Google Scholar]
- Mar, L. , Hoodless, P.A. , 2006. Embryonic fibroblasts from mice lacking TGIF were defective in cell cycling. Mol. Cell. Biol.. 26, (11) 4302–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, T.A. , Hughes, C.M. , Lloyd, R. , Yang, Z. , Rozenblatt-Rosen, O. , Dou, Y. , Schnepp, R.W. , Krankel, C. , Livolsi, V.A. , Gibbs, D. , 2005. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U S A. 102, (3) 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming, J.E. , Kaupas, M.E. , Roessler, E. , Brunner, H.G. , Golabi, M. , Tekin, M. , Stratton, R.F. , Sujansky, E. , Bale, S.J. , Muenke, M. , 2002. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum. Genet.. 110, (4) 297–301. [DOI] [PubMed] [Google Scholar]
- Moffat, J. , Grueneberg, D.A. , Yang, X. , Kim, S.Y. , Kloepfer, A.M. , Hinkle, G. , Piqani, B. , Eisenhaure, T.M. , Luo, B. , Grenier, J.K. , 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 124, (6) 1283–1298. [DOI] [PubMed] [Google Scholar]
- Mueller, B.U. , Pabst, T. , Fos, J. , Petkovic, V. , Fey, M.F. , Asou, N. , Buergi, U. , Tenen, D.G. , 2006. ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU.1 expression. Blood. 107, (8) 3330–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini, L. , Blomer, U. , Gallay, P. , Ory, D. , Mulligan, R. , Gage, F.H. , Verma, I.M. , Trono, D. , 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 272, (5259) 263–267. [DOI] [PubMed] [Google Scholar]
- Orimo, A. , Gupta, P.B. , Sgroi, D.C. , Arenzana-Seisdedos, F. , Delaunay, T. , Naeem, R. , Carey, V.J. , Richardson, A.L. , Weinberg, R.A. , 2005. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 121, (3) 335–348. [DOI] [PubMed] [Google Scholar]
- Phillips, R.L. , Ernst, R.E. , Brunk, B. , Ivanova, N. , Mahan, M.A. , Deanehan, J.K. , Moore, K.A. , Overton, G.C. , Lemischka, I.R. , 2000. The genetic program of hematopoietic stem cells. Science. 288, (5471) 1635–1640. [DOI] [PubMed] [Google Scholar]
- Purton, E.L. , 2007. Roles of retinoids and retinoic Acid receptors in the regulation of hematopoietic stem cell self-renewal and differentiation. PPAR Res. 2007, 87934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler, E. , Du, Y.Z. , Mullor, J.L. , Casas, E. , Allen, W.P. , Gillessen-Kaesbach, G. , Roeder, E.R. , Ming, J.E. , Ruiz i Altaba, A. , Muenke, M. , 2003. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. U S A. 100, (23) 13424–13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer, F. , Tenen, D.G. , 2007. Transcription factors in myeloid development: balancing differentiation with transformation. Nat. Rev. Immunol.. 7, (2) 105–117. [DOI] [PubMed] [Google Scholar]
- Ruegemer, J.J. , Ho, S.N. , Augustine, J.A. , Schlager, J.W. , Bell, M.P. , McKean, D.J. , Abraham, R.T. , 1990. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J. Immunol.. 144, (5) 1767–1776. [PubMed] [Google Scholar]
- Sato, N. , Sanjuan, I.M. , Heke, M. , Uchida, M. , Naef, F. , Brivanlou, A.H. , 2003. Molecular signature of human embryonic stem cells and its comparison with the mouse. Dev. Biol.. 260, (2) 404–413. [DOI] [PubMed] [Google Scholar]
- Seo, S.R. , Ferrand, N. , Faresse, N. , Prunier, C. , Abecassis, L. , Pessah, M. , Bourgeade, M.F. , Atfi, A. , 2006. Nuclear retention of the tumor suppressor cPML by the homeodomain protein TGIF restricts TGF-beta signaling. Mol. Cell.. 23, (4) 547–559. [DOI] [PubMed] [Google Scholar]
- Sitnicka, E. , Ruscetti, F.W. , Priestley, G.V. , Wolf, N.S. , Bartelmez, S.H. , 1996. Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood. 88, (1) 82–88. [PubMed] [Google Scholar]
- Stewart, S.A. , Dykxhoorn, D.M. , Palliser, D. , Mizuno, H. , Yu, E.Y. , An, D.S. , Sabatini, D.M. , Chen, I.S. , Hahn, W.C. , Sharp, P.A. , 2003. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 9, (4) 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeck, M. , Miescher, S. , MacDonald, H.R. , Von Fliedner, V. , 1989. Transforming growth factors beta slow down cell-cycle progression in a murine interleukin-2 dependent T-cell line. J. Cell. Physiol.. 141, (1) 65–73. [DOI] [PubMed] [Google Scholar]
- van der Vos, K.E. , Coffer, P.J. , 2008. FOXO-binding partners: it takes two to tango. Oncogene. 27, (16) 2289–2299. [DOI] [PubMed] [Google Scholar]
- Verbeek, W. , Lekstrom-Himes, J. , Park, D.J. , Dang, P.M. , Vuong, P.T. , Kawano, S. , Babior, B.M. , Xanthopoulos, K. , Koeffler, H.P. , 1999. Myeloid transcription factor C/EBPepsilon is involved in the positive regulation of lactoferrin gene expression in neutrophils. Blood. 94, (9) 3141–3150. [PubMed] [Google Scholar]
- Wallis, D.E. , Roessler, E. , Hehr, U. , Nanni, L. , Wiltshire, T. , Richieri-Costa, A. , Gillessen-Kaesbach, G. , Zackai, E.H. , Rommens, J. , Muenke, M. , 1999. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat. Genet.. 22, (2) 196–198. [DOI] [PubMed] [Google Scholar]
- Wotton, D. , Knoepfler, P.S. , Laherty, C.D. , Eisenman, R.N. , Massague, J. , 2001. The Smad transcriptional corepressor TGIF recruits mSin3. Cell. Growth. Differ.. 12, (9) 457–463. [PubMed] [Google Scholar]
- Wotton, D. , Lo, R.S. , Lee, S. , Massague, J. , 1999. A Smad transcriptional corepressor. Cell. 97, (1) 29–39. [DOI] [PubMed] [Google Scholar]
- Wotton, D. , Lo, R.S. , Swaby, L.A. , Massague, J. , 1999. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J. Biol. Chem.. 274, (52) 37105–37110. [DOI] [PubMed] [Google Scholar]
- Yamanaka, R. , Barlow, C. , Lekstrom-Himes, J. , Castilla, L.H. , Liu, P.P. , Eckhaus, M. , Decker, T. , Wynshaw-Boris, A. , Xanthopoulos, K.G. , 1997. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc. Natl. Acad. Sci. U S A. 94, (24) 13187–13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Li, N. , Tang, Y. , Wu, W. , Zhang, Q. , Yu, Z. , 2009. Negative functional interaction of retinoic acid and TGF-beta signaling mediated by TG-interacting factor during chondrogenesis. Cell. Physiol. Biochem.. 23, (1–3) 157–164. [DOI] [PubMed] [Google Scholar]
- Zhuang, Y. , Faria, T.N. , Chambon, P. , Gudas, L.J. , 2003. Identification and characterization of retinoic acid receptor beta2 target genes in F9 teratocarcinoma cells. Mol. Cancer Res.. 1, (8) 619–630. [PubMed] [Google Scholar]
- Zufferey, R. , Nagy, D. , Mandel, R.J. , Naldini, L. , Trono, D. , 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol.. 15, (9) 871–875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data