

Abstract
Novel agents, including the proteasome inhibitor bortezomib, have significantly improved the response and survival of patients with multiple myeloma (MM) over the last decade. Despite these advances, many patients relapse or do not benefit from the currently available therapies; thus, MM remains an incurable disease. Deacetylase inhibitors (DACi), including panobinostat and vorinostat, have recently emerged as novel agents being evaluated in the treatment of MM. Deacetylases are a group of enzymes with effects on various intracellular proteins including histones, transcription factors, and molecular chaperones. Although DACi inhibit cell growth and induce apoptosis in MM cells as a single agent, synergistic activity has been observed when they were used in combination with bortezomib. The mechanistic basis of synergy is multifactorial and includes disruption of protein degradation and inhibition of the interaction of MM cells with the tumor microenvironment. This review summarizes recent advancements in the understanding of the mechanism of action of proteasome inhibitors and DACi in MM and examines the biological basis of their synergistic effects. Data from the studies summarized here have been used as the rationale for the implementation of phase II and III clinical trials of DACi, alone and combined with bortezomib, in relapsed and refractory MM.
Keywords: proteasome inhibitors, bortezomib, deacetylase inhibitors, multiple myeloma, aggresome
INTRODUCTION TO MULTIPLE MYELOMA
Epidemiology and Treatment
Multiple myeloma (MM) is a plasma cell malignancy predominantly localized in the bone marrow and characterized clinically by paraproteinemia (M-protein), destructive bone disease, hypercalcemia, renal failure, and hematologic dysfunction. In 2010, it was estimated that 20,180 new myeloma cases would be diagnosed in the United States alone, accounting for 1.3% of all newly diagnosed cancer cases (1). Myeloma-related deaths accounted for an estimated 1.9% of all cancer deaths, with an estimated 10,650 in 2010 (1).
Treatments for MM have included corticosteroids (e.g., dexamethasone and prednisone) and cytotoxic drugs (e.g., melphalan, vincristine, cyclophosphamide, and doxorubicin) (2). In the past decade, there have been significant developments in the treatment of patients with MM, including the Food and Drug Administration approval of 3 novel agents: the immunomodulatory drugs thalidomide and lenalidomide and the proteasome inhibitor bortezomib. Randomized clinical trials with these agents have demonstrated significant benefit in patient response and outcome (3–5). The most compelling evidence for the impact of these therapies is the remarkable improvement in the survival of patients with MM diagnosed since the development of these novel agents (6). However, a significant unmet need remains for patients with acquired or intrinsic resistance to these therapies. A recent analysis demonstrated that patients who relapsed on and/or were refractory to prior bortezomib, thalidomide, or lenalidomide had poor outcomes, with an overall survival of 6 months and an event-free survival of 1 month (7). Therefore, despite the significant developments in MM treatment, there is a clear need for the development of new agents to improve long-term outcomes, particularly in patients who derive limited benefit from the currently available treatment options.
MM Disease Biology
Continued research on the tumor microenvironment has led to an increased understanding of the factors that affect MM cell growth and survival; this understanding has been integral to the development of novel agents. Cell adhesion molecules and cytokines play a key role in tumor cell localization, invasion, and spread of the disease (8). Within the bone marrow (BM), adhesion molecules facilitate the interaction of MM cells to both the BM stromal cells (BMSCs) and the extracellular matrix (ECM) (8). Binding of MM cells to BMSCs occurs, at least in part, through binding of very late antigen 4 to vascular cell adhesion molecule 1 and leukocyte function-associated antigen 1 to intracellular adhesion molecule 1 (ICAM1) (8). The interaction between MM cells and the ECM is mediated by the binding of syndecan 1 (CD138) to collagen and very late antigen 4 to fibronectin (8). Importantly, the interaction of MM cells to BMSCs activates the transcription and secretion of interleukin-6 (IL-6), which facilitates the paracrine-mediated growth and survival of MM cells (8). IL-6 also downregulates the expression of syndecan 1, leading to the spread of cells into the bloodstream, which may lead to the development of plasma cell leukemia (8).
Numerous cytokines, in addition to IL-6, play an important role in MM cell proliferation, survival, migration, and drug resistance in the tumor microenvironment. Besides the direct effects of these cytokines/chemokines on MM cells, several studies have examined the role of VEGF in neovascularization in the BM microenvironment and disease progression in MM (9). Myeloma cells secrete VEGF, which contributes to new blood vessel formation in vitro (10). Moreover, VEGF-mediated stimulation of microvascular endothelial cells results in increased secretion of IL-6, with continued MM cell growth (11, 12). In addition to its proangiogenic effects in the BM, VEGF has also been shown to directly induce tumor cell proliferation through the Raf/mitogen-activated protein kinase extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase pathway, as well as tumor cell migration through a protein kinase C–dependent pathway (12).
Tumor necrosis factor (TNF)-α plays an important role in the biology of MM cells within the tumor microenvironment. Although TNF-α has modest effects on MM cell proliferation, it does lead to increased expression of adhesion molecules, including ICAM1, on both MM cells and BMSCs. The cytokine also leads to increased heterotypic adhesion, thereby triggering NF-κB–dependent upregulation of transcription and secretion of IL-6 and resulting in paracrine MM cell growth by BMSCs (13). Insulin-like growth factor 1 (IGF1) is another cytokine that supports MM cell proliferation and survival. IGF1 is present in both the BM microenvironment and peripheral blood and stimulates MM cell proliferation and survival (14) via activation of NF-κB and Akt, as well as increased expression of several antiapoptotic proteins including FADD-like IL-1β-converting enzyme inhibitory protein, X-linked inhibitor of apoptosis, and survivin (14). Although IGF1 induces more prominent Akt activation than IL-6, it does not activate the Janus kinase 2/signal transducers and activators of transcription 3 pathway, which is commonly activated by gp130 family cytokines including IL-6.
The proliferation and survival of MM cells within the tumor microenvironment is therefore dependent on their interaction with the BMSC and the ECM. These factors supporting MM cell survival are complex and provide many mechanisms for the development of resistance to commonly used agents. Novel strategies to target or disrupt these pathways are urgently needed.
MECHANISTIC BASIS OF ANTIMYELOMA ACTIVITY OF PROTEASOME INHIBITORS IN MM
Proteasomes are abundant multienzyme complexes that provide the main pathway for degradation of intracellular proteins and contribute to the maintenance of protein homeostasis and clearance of misfolded/unfolded and cytotoxic proteins (15). The 26S proteasome is a large 2.4-MDa ATP-dependent proteolytic complex located in both the cytoplasm and nucleus. This proteasome consists of a 20S core catalytic cylindrical complex capped at both ends by 19S regulatory subunits (15). Polyubiquitination is an essential event for proteins targeted for proteasomal degradation (15). Proteins degraded by the proteasome include mediators of cell cycle progression and apoptosis, such as the cyclins, caspases, B-cell lymphoma 2 (BCL2), and NF-κB activation (15).
It has been hypothesized that cancer cells are more dependent on the proteasome for clearance of abnormal or mutant proteins (15). In fact, several preclinical studies have demonstrated that malignant cells are more sensitive to proteasome inhibition than normal cells.(16–20) The proteasome inhibitor bortezomib is a dipeptide boronic acid analog that reversibly inhibits the chymotryptic activity of the 20S subunit of the proteasome (19). Bortezomib has been demonstrated to directly inhibit proliferation and induce apoptosis in MM cell lines and patient tumor cells resistant to conventional therapies (20). Furthermore, bortezomib demonstrated enhanced anti-MM activity with dexamethasone and overcame resistance to apoptosis conferred by IL-6 or adhesion to BMSCs (20). Significant tumor growth inhibition and increased host survival was also observed in vivo using a mouse-human MM cell xenograft model (19).
Bortezomib demonstrated remarkable clinical activity in patients with MM and was rapidly approved by the Food and Drug Administration in 2003 to treat relapsed and refractory MM. Clinical activity resulting in high response rates and increased progression-free and overall survival has also been observed in multiple phase III clinical trials with bortezomib. Of note, bortezomib has shown increased activity in combination with melphalan and prednisone compared with melphalan and prednisone alone in newly diagnosed patients with MM (4), as well as increased activity as a single agent compared with dexamethasone in patients with relapsed or refractory disease (21).
The mechanism of action and target of bortezomib, leading to disruption of intracellular protein metabolism, are well characterized. The downstream biological effects of proteasome inhibition are multifactorial, with direct effects on both MM cells and the MM cell microenvironment, including inhibition of cytokine secretion, suppression of adhesion molecule expression, and inhibition of angiogenesis. The initial rationale to use bortezomib in cancer was its inhibitory effect on NF-κB activity, thereby modulating transcription. Specifically, the NF-κB canonical pathway is regulated by inhibitor protein IκB, which blocks nuclear translocation of the p50 (NFκB1)/p65 (RelA) heterodimer. Importantly, IκB is a substrate of the proteasome, and proteasome inhibition by bortezomib can therefore lead to an increase in cytoplasmic level of IκB, resulting in a blockade of NF-κB translocation to the nucleus and DNA-binding activity (15). NF-κB has also been identified as a mediator of paracrine signaling between MM cells and BMSCs within the BM microenvironment. For example, NF-κB–dependent upregulation of IL-6 in BMSCs is induced by adhesion to MM cells or via TNF-α secretion by MM cells (13, 22). TNF-α–induced upregulation of NF-κB leads to increased expression of the adhesion molecules ICAM1 and vascular cell adhesion molecule 1 on MM cells and BMSCs, thus enhancing intercellular binding (13). Bortezomib blocks the TNF-α–induced upregulation of NF-κB, leading to decreased binding of MM cells to BMSCs and related decreased IL-6 secretion (13, 20). Of note, the specific IκB kinase inhibitors PS-1145 and MLN120B also inhibit secretion of IL-6 and adhesion of MM cells and BMSCs; however, these agents only lead to partial inhibition of MM cell growth (23). Therefore, bortezomib-triggered anti-MM activities are not solely mediated by NF-κB inhibition.
Bortezomib inhibits angiogenesis in the BM microenvironment, which plays an important role in both MM pathogenesis and disease progression. In preclinical models using MM patient–derived endothelial cells, bortezomib inhibited cell proliferation, chemotaxis, adhesion, and capillary formation, thus supporting its angiogenic inhibitory activity in vivo (24). Bortezomib also inhibited the expression and secretion of several proangiogenic factors, including VEGF (24). In addition, VEGF-mediated migration of MM cells was inhibited by bortezomib (12).
Inhibition of the proteasome induces accumulation of intracellular misfolded/unfolded proteins (20), which triggers the unfolded protein response (UPR) signaling pathway to protect cells against cellular stress (25, 26). Because MM cells produce large amounts of immunoglobulin, a functional UPR is required for their survival (25, 26). Treatment of MM cells with bortezomib leads to induction of proapoptotic UPR components, including growth arrest– and DNA damage–inducible gene 153 (26). Proteasome inhibition also interferes with the stability of mRNA transcripts of X box–binding protein 1, a downstream transcription factor of IRE1α, regulating the UPR (25). Taken together, these data demonstrate the reliance of MM cells on the UPR for survival and identify disruption of protein metabolism as a potential therapeutic target in MM.
MECHANISTIC BASIS OF ANTIMYELOMA ACTIVITY OF DACi IN MM
Histone deacetylases (HDACs) have emerged as a relevant clinical target in MM. HDACs and histone acetyl transferases regulate the acetylation of target proteins (27). Specifically, HDACs remove acetyl groups from target proteins that regulate their activity (27). Eighteen HDACs have been identified in humans and subsequently divided into 4 classes based on their homology to yeast HDACs: class I (HDAC1, HDAC2, HDAC3, and HDAC8), class IIa (HDAC4, HDAC5, HDAC7, and HDAC9), class IIb (HDAC6 and HDAC10), class III (SIRT family), and class IV (HDAC11) (27). The different classes of enzymes also differ in their subcellular localization, with class I HDACs found in the nucleus and class II enzymes found in both nucleus and cytoplasm, and their intracellular targets (27). The name HDAC is based on the identification of histone proteins as the initial target of HDACs; however, a recent study in cancer cell lines identified 3,600 acetylation sites on 1,750 proteins associated with various intracellular functions including gene expression, DNA replication and repair, cell cycle progression, cytoskeletal reorganization, and protein chaperone activity (28). Therefore, the term deacetylase (DAC) may be more appropriate when referring to these enzymes.
Although there are several DACi in various stages of clinical development, only 2 are approved, vorinostat (suberoylanilide hydroxamic acid) and romidepsin (FK228 or FR901228), for treatment of cutaneous T-cell lymphoma (29, 30). DACi differ in their structure and potency toward the HDAC enzymes. Romidepsin is a cyclic tetrapeptide with DAC inhibitory activity primarily toward class I HDACs (27). Other DACi include the benzamide class, which includes mocetinostat (MGCD103) and entinostat (MS-275) class I specific inhibitors (27). The hydroxamic acid–based DACi, vorinostat, panobinostat (LBH589), and belinostat (PXD101), are pan-DACi, with inhibitory activity against class I, II, and IV HDACs (27) (Fig. 1). Panobinostat is among the most potent DACi, with nanomolar DAC inhibitory activity (31).
Figure 1.
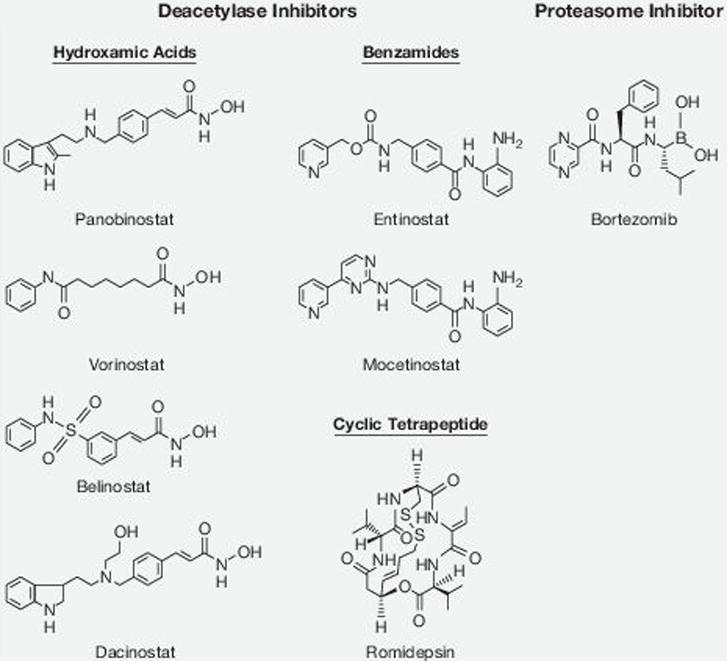
Molecular structures of deacetylase inhibitors and the proteasome inhibitor bortezomib.
Preclinical studies demonstrated that DACi have potent antimyeloma activity. The class I specific DACi, romidepsin, along with the pan-DACi dacinostat (LAQ824), vorinostat, and panobinostat have been shown to inhibit proliferation and induce apoptosis in MM cell lines in vitro and in vivo in mouse xenograft models (32–36). In many of these preclinical studies, additive or synergistic effects were observed when DACi were combined with other agents, including corticosteroids and proteasome inhibitors, establishing the rationale for clinical studies of DACi in combination with these agents (32–36).
Clinical studies in patients with MM have demonstrated limited single-agent activity of DACi (37, 38). In a phase I trial of single-agent vorinostat(37, 38){{268 Richardson,P. 2008; 269 Niesvizky,R. 2010}}(37,38){{268 Richardson,P. 2008; 269 Niesvizky,R. 2010}} in 13 patients with relapsed and/or refractory MM, only 1 patient demonstrated a minimal response and 9 patients demonstrated disease stabilization (37). In a phase II trial of single-agent romidepsin in 13 patients with refractory MM, no objective responses were observed; however, there was evidence of disease stabilization and resolution of disease-related symptoms (38). Overall, the activity of DACi as single agents has been limited, and a clearer understanding of the biological activity of these agents will help determine the ideal combination therapies for clinical development.
Because DACi act on many intracellular targets, the biological basis of their antimyeloma activity is due to a number of effects on MM cells and their interaction with the tumor microenvironment. For example, LAQ824 induces upregulation of CDK inhibitor p21, leading to cell cycle arrest followed by apoptosis through activation of caspases 8, 9, and 3 (33). Vorinostat also induced p21 expression, leading to cell cycle arrest and apoptosis; however, no significant caspase 8, 9, or 3 cleavage was observed, suggesting caspase-independent apoptosis (39). Romidepsin has been shown to induce MM cell apoptosis through downregulation of the antiapoptotic proteins BCL2, BCLXL, and myeloid cell leukemia sequence 1 (35).
DACi also modulate interaction of MM cells with cellular components in the BM microenvironment. LAQ824 inhibited MM cell proliferation even in the presence of exogenous IL-6 or BMSC coculture (33). Vorinostat suppresses the stimulation of IL-6 secretion in BMSCs by MM cell adhesion, with no effect on BMSC viability (39). Vorinostat also suppresses autocrine IGF1 production, directly interrupting the IGF1/IGF1R/Akt signaling pathway critical for antiapoptosis and survival of MM cells (40). DACi therefore can induce direct MM cell cycle arrest and apoptosis, as well as disruption of signaling between MM cells and BMSCs.
Recent studies have shown that aggresomes represent an alternative pathway for catabolism of misfolded proteins and develop when production of misfolded ubiquitinated proteins exceeds the capacity of proteasomes to degrade them (41). Misfolded proteins can form aggregates that are transported by microtubules via dynein motor complexes to the autophagosome, where they are degraded by lysosomes. HDAC6 belongs to the class IIb HDACs and is broadly expressed in different types of cells. HDAC6 regulates acetylation of α-tubulin and facilitates the transport of the aggresome to the lysosome (42). DACi that target HDAC6, such as the pan-DACi panobinostat and HDAC6-specific inhibitor tubacin, lead to hyperacetylation of α-tubulin, disruption of the interaction between HDAC6 and dynein, and resultant increases in ubiquitinated proteins (41, 43). Therefore, inhibition of protein degradation through targeting of the aggresome by DACi, represents an attractive model for the treatment of cancers, such as MM, that are reliant on efficient protein metabolism.
MECHANISMS OF SYNERGY BETWEEN PROTEASOME INHIBITORS AND DACi IN MM
The molecular sequelae of proteasome inhibitors and DACi in MM are associated with key pathways vital to the proliferation and survival of MM cells. Although there are some common targets and pathways affected by each agent, some of the pathways targeted are complementary and may underlie the synergistic effects. In fact, synergistic anti-tumor activities between DACi and bortezomib have been observed in several preclinical studies (36, 41, 43–45). Either the pan-DACi vorinostat or panobinostat with bortezomib have synergistic effects on inhibition of cell growth and increasing apoptosis in MM cells (41, 44). Similar effects were observed with the HDAC6-specific inhibitor tubacin combined with bortezomib, associated with a marked increase in polyubiquitinated proteins (43). These effects were observed in both MM cell lines and primary tumor cells isolated from MM patients. Importantly, class I–specific inhibitor romidepsin and the pan-DACi panobinostat have also demonstrated antitumor effects in vivo in human MM cell mouse xenograft models (36, 45).
Despite the observation that synergistic effects were observed with bortezomib and a variety of DACi across several studies, the conclusions made regarding the biological basis of the synergy observed are varied. This can be partially explained by the differential potency and targets of the various DACi tested in these studies. In addition, the pleiotropic effects that these agents elicit in MM cells, along with the experimental design of the individual studies, may have led the investigators to focus on the most relevant biological effects observed. As the data from preclinical studies have demonstrated, there are numerous genes affected by bortezomib or DACi (39, 40, 46), and it is therefore most likely that a combination of these effects leads to the synergy observed between the 2 classes of agents.
The most well-characterized models of synergy between proteasome inhibitors and DACi is through dual inhibition of the proteasome and aggresome pathways (41, 43) (Fig. 2). Targeting both the proteasome with bortezomib and the aggresome with HDAC6 inhibitors in tumor cells induces greater accumulation of polyubiquitinated proteins, resulting in increased cellular stress and apoptosis (41, 43). More specifically, proteasome inhibition drives the formation of aggresomes, which are dependent on the interaction of HDAC6 with tubulin and dynein complex (41). Moreover, the proteasome inhibitor (bortezomib) and HDAC6 inhibitors (tubacin or panobinostat) lead to increased hyperacetylation of tubulin and generation of polyubiquitinated proteins, thus increasing cellular stress response (i.e., c-Jun N-terminal protein kinase activation) and leading to apoptosis, which is in part dependent on caspase activity (41–43).
Figure 2.
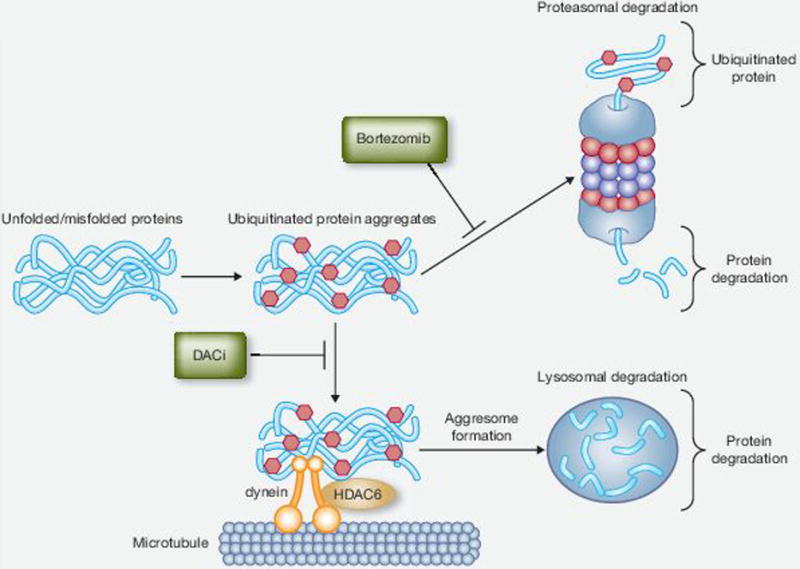
Inhibitions of the proteasome and aggresome pathways by bortezomib and deacetylase inhibitors (DACi). Unfolded/misfolded proteins are targeted by ubiquitin for degradation by the proteasome and aggresome pathways. The proteasome inhibitor bortezomib leads to the accumulation of ubiquitin protein aggregates. These aggregates are shuttled to the lysosome, where they are degraded via the aggresome pathway. Aggresome formation involves the shuttling of the protein aggregates along microtubules by dynein motor proteins. The interaction of the unfolded/misfolded protein complexes is facilitated by histone deacetylase 6 (HDAC6). Conversely, DACi with inhibitory activity toward HDAC6 block this process. The combination of proteasome inhibitors and DACi lead to increased cellular stress and apoptosis.
Although disruption of protein degradation represents a major contributor to the synergistic antitumor activity observed between proteasome inhibitors and DACi, other studies have also identified additional mechanisms. For example, the combination of bortezomib and vorinostat results in enhanced cytochrome-c release, caspase and poly-ADP-ribose polymerase cleavage, and inactivation of NF-κB, followed by apoptosis (44). Conversely, antioxidative agents including N-acetyl-L-cysteine block these effects (44).
In addition to the synergistic effects observed when these agents are combined, it is plausible that each agent affects complementary pathways in MM cells, thereby leading to synergistic effects on growth inhibition and apoptosis. As summarized in the preceding sections, bortezomib and DACi both affect pathways associated with the interaction of MM cells and the microenvironment, including cytokine signaling and cell adhesion (Fig. 3) In addition, overexpression of proto-oncogenes/oncogenic genes is a common mechanism of resistance in cancer, and a recent study demonstrated that bortezomib specifically downregulates the expression of class I HDACs, leading to histone hyperacetylation (45). It was also noted that exogenous overexpression of HDAC1 caused resistance to bortezomib both in vitro and in vivo, which was reversed by the class I DACi romidepsin (45). In addition, pan-DACi LAQ824 has been shown to decrease the activity of the 20S proteasome, as determined by reduced proteasome chymotrypsin-like activity (33). The ability of proteasome inhibitors to downregulate HDACs, along with the observation that DACi can decrease proteasome activity, may also contribute to the synergistic antitumor activities. Taken together, proteasome inhibitors and DACi target several relevant mechanisms in MM biology. Further research will delineate uncover additional mechanisms that contribute to the synergistic anti-tumor activities and potential avenues of resistance.
Figure 3.
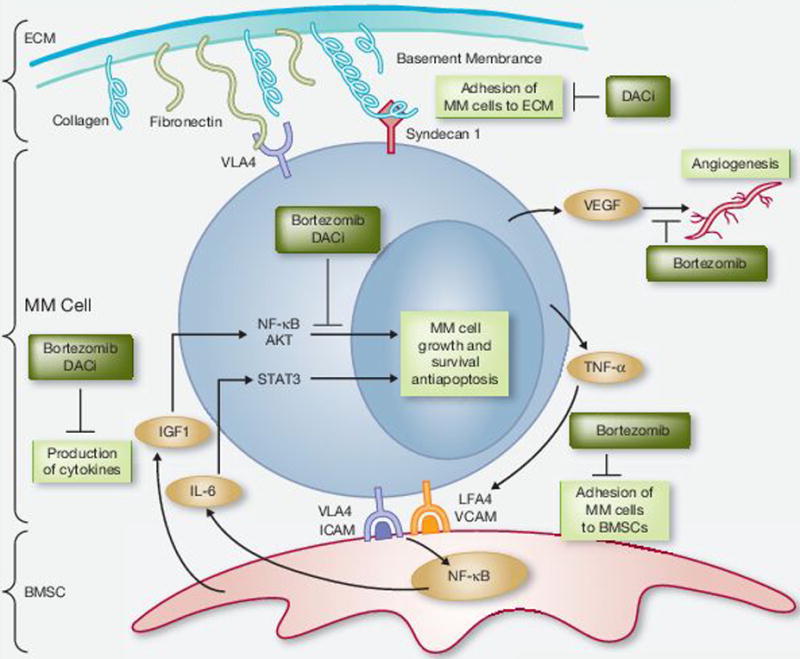
Bortezomib and deacetylase inhibitors (DACi) inhibit key pathways associated with multiple myeloma (MM) cell growth and survival. Growth and survival of MM cells are dependent on functioning intracellular pathways that drive proliferation of and the interaction with the extracellular matrix (ECM) and bone marrow stromal cells (BMSC). The combination of bortezomib and DACi leads to inactivation of NF-κB and MM cell apoptosis. Both DACi and bortezomib suppress the production of cytokines including interleukin-6 (IL-6) and insulin-like growth factor 1 (IGF1). Bortezomib also suppresses tumor necrosis factor α (TNF-α), leading to inhibition of the interaction of MM cells and BMSCs. Bortezomib has been shown to decrease the secretion of VEGF, leading to inhibition of angiogenesis. The cell surface proteoglycan syndecan 1 is downregulated by DACi, which affects the interaction of MM cells with the ECM. ICAM, intracellular adhesion molecule; LFA4, leukocyte function-associated antigen 4; STAT3, signal transducers and activators of transcription 3; VCAM, vascular cell adhesion molecule; VLA4, very late antigen 4.
SUMMARY AND FUTURE DIRECTIONS
The synergy between proteasome inhibitors and DACi is most likely dependent on a number of mechanisms targeting MM cell biology. MM cell proliferation, survival, and progression of disease are dependent on the activation of key pathways within the cell, as well as the interaction with elements in the tumor microenvironment. One of the most compelling mechanisms underlying the synergy remains the disruption of protein degradation by inhibition of the proteasome and aggresome. Because MM cells produce abundant amounts of immunoglobulin that must be properly folded or degraded, they are more dependent on efficient processing of proteins (41–43). This mechanism clearly contributes to synergy observed between the 2 agents; however, it is unlikely to be solely responsible for the synergy observed. Of note, a recent report demonstrated that romidepsin, a DACi with limited HDAC6 inhibitory activity, enhanced the in vitro and in vivo activity of bortezomib in HDAC1-overexpressing MM cells, thus suggesting a role for the interaction of these agents independent of the effects on protein degradation (45). In addition, both proteasome inhibitors and DACi decrease cytokine production and expression of adhesion molecules, key factors supporting the growth and survival of MM cells (39, 46). It is therefore most likely that a number of factors contribute to the synergy between proteasome inhibitors and DACi.
Although bortezomib clearly has proven activity as a single agent and in combination therapy in patients with MM, initial trials with single-agent DACi have not led to significant clinical activity (4, 37, 38, 47). Based on preclinical data, combining DACi with proteasome inhibitors such as bortezomib represents an attractive strategy for the treatment of patients with MM. Preliminary data from phase I studies evaluating either the pan-DACi panobinostat or vorinostat in combination with bortezomib have demonstrated responses in patients who received bortezomib before study enrollment, including patients who failed to previously respond to bortezomib(48, 49). These preliminary observations are now being evaluated further in phase II and III clinical trials. The 2 phase III trials will evaluate the role of DACi as a strategy to increase treatment efficacy in patients with relapsed MM. The VANTAGE 088 trial (NCT00773747) is evaluating the combination of vorinostat plus bortezomib and the PANORAMA 1 trial (NCT01023308) is evaluating the combination of panobinostat plus bortezomib and dexamethasone (50, 51). Both trials are comparing the combination with bortezomib plus placebo. The results of these trials will help to determine if DACi can enhance the efficacy of bortezomib in patients with relapsed MM. Two single-arm phase II studies, VANTAGE 095 (NCT00773838) and PANORAMA 2 (NCT01083602), are evaluating the combination of DACi, bortezomib, and dexamethasone in patients with relapsed and bortezomib-refractory disease. The results from these trials will determine if DACi can sensitize patients with bortezomib-resistant MM(50, 52). The results of these trials, along with further research on other novel DACi and proteasome inhibitors in development, will help guide the ideal setting and combination partners for the treatment of patients with MM.
Acknowledgments
Financial support for editorial assistance was provided by Novartis. We thank William Fazzone for editorial assistance.
This study is supported by National Institutes of Health (NIH) SPORE IP50 CA10070, PO-1 78378, and RO-1 CA 50947 Grants; the LeBow Family Fund to Cure Myeloma (KCA); American Cancer Society Clinical Reward Professorship (KCA).
Footnotes
Disclosure of Potential Conflict of Interest disclosure
T.H. is a consultant for Acetylon Pharmaceuticals. P.G.R. is a consultant and on advisory boards for Millennium and Celgene. K.C.A. is a consultant and on advisory board for Millennium, Celgene, and Novartis.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman J, Gleason C, Lonial S. Treatment of relapsed and refractory myeloma. Curr Hematol Malig Rep. 2009;4:99–107. doi: 10.1007/s11899-009-0014-5. [DOI] [PubMed] [Google Scholar]
- 3.Rajkumar SV, Rosinol L, Hussein M, Catalano J, Jedrzejczak W, Lucy L, et al. Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J Clin Oncol. 2008;26:2171–7. doi: 10.1200/JCO.2007.14.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–17. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 5.Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–32. doi: 10.1056/NEJMoa070594. [DOI] [PubMed] [Google Scholar]
- 6.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–20. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar S, Blade J, Crowley J, Goldschmidt H, Hoering A, Jagannath S, et al. Natural history of multiple myeloma relapsing after therapy with IMiDs and bortezomib: A multicenter international myeloma working group study. Blood. 2009;114:2878. [Google Scholar]
- 8.Teoh G, Anderson KC. Interaction of tumor and host cells with adhesion and extracellular matrix molecules in the development of multiple myeloma. Hematol Oncol Clin North Am. 1997;11:27–42. doi: 10.1016/s0889-8588(05)70413-5. [DOI] [PubMed] [Google Scholar]
- 9.Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, et al. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:3064–73. [PubMed] [Google Scholar]
- 10.Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Fonseca R, et al. Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003;17:2025–31. doi: 10.1038/sj.leu.2403084. [DOI] [PubMed] [Google Scholar]
- 11.Dankbar B, Padro T, Leo R, Feldmann B, Kropff M, Mesters RM, et al. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood. 2000;95:2630–6. [PubMed] [Google Scholar]
- 12.Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001;98:428–35. doi: 10.1182/blood.v98.2.428. [DOI] [PubMed] [Google Scholar]
- 13.Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: Therapeutic applications. Oncogene. 2001;20:4519–27. doi: 10.1038/sj.onc.1204623. [DOI] [PubMed] [Google Scholar]
- 14.Mitsiades CS, Mitsiades N, Poulaki V, Schlossman R, Akiyama M, Chauhan D, et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: Therapeutic implications. Oncogene. 2002;21:5673–83. doi: 10.1038/sj.onc.1205664. [DOI] [PubMed] [Google Scholar]
- 15.Adams J. The proteasome: A suitable antineoplastic target. Nat Rev Cancer. 2004;4:349–60. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 16.An B, Goldfarb RH, Siman R, Dou QP. Novel dipeptidyl proteasome inhibitors overcome bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 1998;5:1062–75. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- 17.Orlowski RZ, Eswara JR, Lafond-Walker A, Grever MR, Orlowski M, Dang CV. Tumor growth inhibition induced in a murine model of human burkitt’s lymphoma by a proteasome inhibitor. Cancer Res. 1998;58:4342–8. [PubMed] [Google Scholar]
- 18.Masdehors P, Merle-Beral H, Maloum K, Omura S, Magdelenat H, Delic J. Deregulation of the ubiquitin system and p53 proteolysis modify the apoptotic response in B-CLL lymphocytes. Blood. 2000;96:269–74. [PubMed] [Google Scholar]
- 19.LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 20.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–6. [PubMed] [Google Scholar]
- 21.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 22.Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996;87:1104–12. [PubMed] [Google Scholar]
- 23.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–47. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 24.Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66:184–91. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- 25.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100:9946–51. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–16. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 28.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 29.StatBite. FDA oncology drug product approvals in 2009. J Natl Cancer Inst. 2010(102):219. doi: 10.1093/jnci/djq030. [DOI] [PubMed] [Google Scholar]
- 30.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 31.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009;280:233–41. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 32.Campbell RA, Sanchez E, Steinberg J, Shalitin D, Li ZW, Chen H, et al. Vorinostat enhances the antimyeloma effects of melphalan and bortezomib. Eur J Haematol. 2010;84:201–11. doi: 10.1111/j.1600-0609.2009.01384.x. [DOI] [PubMed] [Google Scholar]
- 33.Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–22. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez E, Shen J, Steinberg J, Li M, Wang C, Bonavida B, et al. The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk Res. 2011;35:373–9. doi: 10.1016/j.leukres.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 35.Khan SB, Maududi T, Barton K, Ayers J, Alkan S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br J Haematol. 2004;125:156–61. doi: 10.1111/j.1365-2141.2004.04882.x. [DOI] [PubMed] [Google Scholar]
- 36.Ocio EM, Vilanova D, Atadja P, Maiso P, Crusoe E, Fernandez-Lazaro D, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95:794–803. doi: 10.3324/haematol.2009.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma. 2008;49:502–7. doi: 10.1080/10428190701817258. [DOI] [PubMed] [Google Scholar]
- 38.Niesvizky R, Ely S, Mark T, Aggarwal S, Gabrilove JL, Wright JJ, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011;117:336–42. doi: 10.1002/cncr.25584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–62. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 40.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: Biological and clinical implications. Proc Natl Acad Sci U S A. 2004;101:540–5. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006;108:3441–9. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simms-Waldrip T, Rodriguez-Gonzalez A, Lin T, Ikeda AK, Fu C, Sakamoto KM. The aggresome pathway as a target for therapy in hematologic malignancies. Mol Genet Metab. 2008;94:283–6. doi: 10.1016/j.ymgme.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102:8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 45.Kikuchi J, Wada T, Shimizu R, Izumi T, Akutsu M, Mitsunaga K, et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116:406–17. doi: 10.1182/blood-2009-07-235663. [DOI] [PubMed] [Google Scholar]
- 46.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A. 2002;99:14374–9. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–17. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 48.San-Miguel JF, Richardson PGG, Sezer O, Guenther A, Siegel DSD, Blade J, et al. A phase lb study of oral panobinostat and IV bortezomib in relapsed or relapsed and refractory multiple myeloma. J Clin Oncol. 2011;29:8075. doi: 10.1200/JCO.2012.46.7068. [DOI] [PubMed] [Google Scholar]
- 49.Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15:5250–7. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siegel DS, Jagannath S, Hajek R, Dimopoulos MA, Yoon SS, Lonial S, et al. Vorinostat combined with bortezomib in patients with relapsed or relapsed and refractory multiple myeloma: Update on the vantage study program. Blood. 2010;116:1952. [Google Scholar]
- 51.San Miguel JF, Lonial S, Hungria V, Moreau P, Einsele H, Lee JH, et al. PANORAMA1: A randomized, double-blind, placebo controlled phase III study of panobinostat in combination with bortezomib and dexamethasone in patients with relapsed multiple myeloma. J Clin Oncol. 2011;29:TPS227. [Google Scholar]
- 52.Schlossman R, Alsina M, Weber D, Coutre S, Lonial S, Khan M, et al. PANORAMA 2: A phase II study of panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory multiple Myeloma. Haematologica. 2011;96:0900. doi: 10.1182/blood-2013-01-481325. [DOI] [PubMed] [Google Scholar]