

ABSTRACT
Formyl-phloroglucinol meroterpenoids (FPMs) are important types of natural products with various bioactivities. Our antifungal susceptibility assay showed that one of the Eucalyptus robusta-derived FPMs, eucarobustol E (EE), exerted a strong inhibitory effect against Candida albicans biofilms at a concentration of 16 μg/ml. EE was found to block the yeast-to-hypha transition and reduce the cellular surface hydrophobicity of the biofilm cells. RNA sequencing and real-time reverse transcription-PCR analysis showed that exposure to 16 μg/ml of EE resulted in marked reductions in the levels of expressions of genes involved in hyphal growth (EFG1, CPH1, TEC1, EED1, UME6, and HGC1) and cell surface protein genes (ALS3, HWP1, and SAP5). Interestingly, in response to EE, genes involved in ergosterol biosynthesis were downregulated, while the farnesol-encoding gene (DPP3) was upregulated, and these findings were in agreement with those from the quantification of ergosterol and farnesol. Combined with the obvious elevation of negative regulator genes (TUP1, NRG1), we speculated that EE's inhibition of carbon flow to ergosterol triggered the mechanisms of the negative regulation of hyphal growth and eventually led to biofilm inhibition.
KEYWORDS: Candida albicans, eucarobustol E, antibiofilm, formyl-phloroglucinol meroterpenoids, negative regulation, natural antimicrobial products
INTRODUCTION
Candidiasis is the most frequently encountered fungal disease, with manifestations ranging from mild mucosal infections to serious candidemia and disseminated candidiasis (1, 2), and it is notoriously the second most common cause of death from nosocomial infections (3, 4). Candida albicans, the main pathogenic agent responsible for candidiasis, is able to transform from budding yeasts to cells with filamentous growth and, eventually, cells that form biofilms (5, 6). C. albicans biofilm formation exacerbates clinical infections by forming a reservoir for producing recalcitrant pathogenic cells, which can act as seeds to disseminate the organism to the bloodstream, leading to invasive systemic infections of tissues and organs (7). In addition, biofilm cells display phenotypic traits that are dramatically different from those of their planktonic counterparts, resulting in enhanced resistance to antifungal drugs (7, 8). Studies have shown that azoles are ineffective against C. albicans biofilms due to the upregulation of efflux pumps in biofilms (9), while conventional amphotericin B failed to reach biofilm cells because of the enhanced extracellular matrix or beta-glucan synthesis during biofilm growth (10). Clinically, only liposomal amphotericin B and candins are effective against C. albicans biofilms (11–13). Therefore, drugs that specifically and efficiently inhibit C. albicans biofilms are urgently needed.
Formyl-phloroglucinol meroterpenoids (FPMs) are an important class of secondary metabolites that are widely available from the genera Eucalyptus and Psidium (14–16). The past decades have seen increasing reports on structurally diverse FPMs with various bioactivities, such as anti-HIV, antimicrobial, and antitumor effects (17–21). It is to be noted that these explorations of the antifungal activities of FPMs have uncovered that FPMs are effective against a wide spectrum of pathomycetes, including C. albicans, C. glabrata, Trichophyton mentagrophytes, and T. rubrum (22–24), exhibiting great potential and broad prospects to be used as novel antifungal compounds. Nevertheless, rare investigations have been performed to unveil the underlying molecular antifungal mechanisms of FPMs.
During the search for bioactive natural products from the leaves of Eucalyptus robusta (15, 16), we found that eucarobustol E (EE) (Fig. 1), a recently reported FPM, showed potent inhibitory effects against both C. albicans yeast cells and biofilms, while it maintained no toxicity toward human cells. Our in-depth study through RNA sequencing (RNA-seq) and real-time reverse transcription (RT)-PCR revealed that the antibiofilm effect of EE was related to its ability to inhibit the expressions of hypha-specific genes (HSGs), which was probably achieved through reducing ergosterol biosynthesis, stimulating farnesol production, and, consequently, activating negative regulatory mechanisms.
FIG 1.
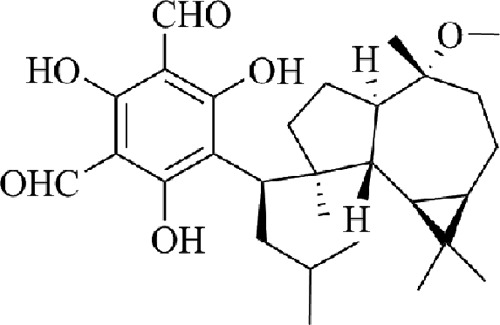
Chemical structure of EE.
RESULTS
Antifungal effect of EE.
Results showed that EE had a broad-spectrum antifungal ability, exerting a potent to moderate killing effect, especially against clinical isolates (see Table S2 in the supplemental material). The MIC50 was between 4 and 16 μg/ml against fluconazole-susceptible strains and was between 32 and 128 μg/ml against fluconazole-resistant isolates. A time-kill curve assay showed that EE inhibited the growth of C. albicans SC5314 cells in a dose-dependent manner (Fig. 2). Cell growth under treatment with EE at a concentration of less than 8 μg/ml was hardly retarded compared to that of the control group, but cell numbers were moderately reduced under treatment with EE at a concentration of less than 16 μg/ml. A further test with 32 μg/ml of EE resulted in a more than a 100-fold reduction in the numbers of viable cells compared to the numbers in the control group.
FIG 2.
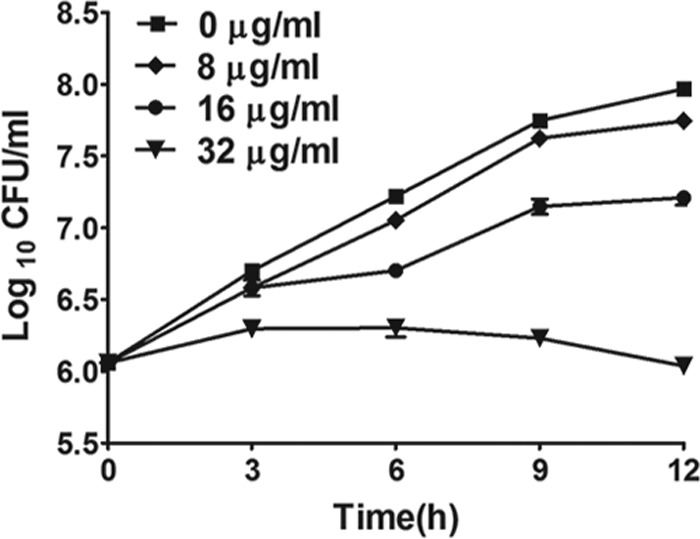
Time-kill curves of EE against C. albicans strain SC5314. Different working concentrations of EE were added to 1 × 106 cells/ml of C. albicans suspensions in RPMI 1640 medium. The cell suspension with 0.5% DMSO was used as a control. Cell numbers are means ± SDs calculated from three independent experiments.
EE inhibits C. albicans biofilms in vitro.
Both a 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT) reduction assay and the crystal violet (CV) method revealed that EE eradicated mature biofilms and inhibited biofilm formation (Fig. 3). The XTT assay found, first, that EE effectively eliminated preformed mature biofilms that had been allowed to grow for 24 h. The effect was dose dependent (Fig. 3a), and almost all mature biofilms (92%) were removed when they were treated with 128 μg/ml EE. Second, a biofilm formation test further revealed that the antibiofilm effect started from the biofilm initiation stage. Levels of suppression of biofilms of nearly 60% and 73% were obtained at EE concentrations of 16 and 32 μg/ml, respectively (P < 0.001). When cells were treated with higher concentrations of EE, no biofilms were formed (Fig. 3b). This effect was further seen in a CV staining experiment (Fig. 3c). The mean absorbance at 570 nm of a biofilm destaining solution in the control group was 1.30, and the values were dose dependently reduced to 0.25, 0.16, and 0.11 at EE concentrations of 32, 64, and 128 μg/ml, respectively, indicating that biofilm growth was disrupted upon EE treatment.
FIG 3.

EE inhibits C. albicans biofilms in vitro. (a and b) An XTT reduction assay was performed to determine EE's effect on mature biofilms (a) and biofilm formation (b). The mature biofilms were left to grow for 24 h, and a biofilm formation assay was conducted after cell adhesion for 1.5 h. (c) The crystal violet method was conducted to quantify biofilm formation. Bars represent means ± SDs from three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Confocal laser scanning microscopy (CLSM) and scanning electron microscopy (SEM) analysis further confirmed the antibiofilm effect of EE. C. albicans biofilms in the control group developed into a crisscrossing network with long filaments expanding robustly, as shown in Fig. 4a and 5a to c. In the EE treatment groups, biofilm formation was disrupted in a dose-dependent manner (Fig. 4b to d and 5d to l). Upon treatment with EE at 16 μg/ml, the viability of the hyphae was reduced, as visualized by fluorescein diacetate (FDA) labeling, and the proportion of dead hyphae increased, as analyzed by CLSM (Fig. 4c); SEM analysis showed that the density of biofilm cells was reduced and large amounts of cells were restricted at the budding yeast stage (Fig. 5k and l, white arrows). At 32 μg/ml EE, no long hyphae were seen, indicating that EE inhibited biofilm formation by blocking hyphal elongation and filamentation.
FIG 4.
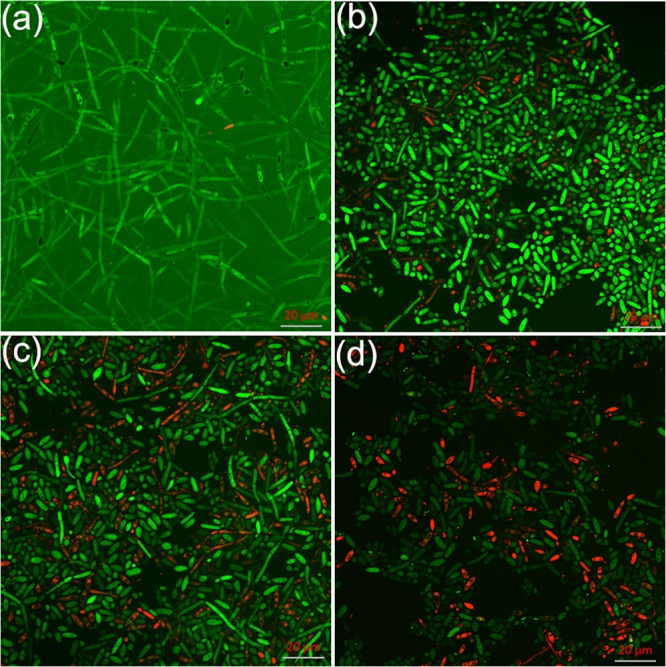
Effects of EE on C. albicans biofilm formation shown in CLSM images. (a) Control group; (b) group treated with 8 μg/ml of EE; (c) group treated with 16 μg/ml of EE; (d) group treated with 32 μg/ml of EE. FDA and PI were used to distinguish viable and dead cells.
FIG 5.

SEM images show C. albicans biofilm inhibition under treatment with different concentrations of EE. Each treatment group was visualized under magnifications of 500×, 2,000×, and 5,000×. The boxes with dashed lines in the first and second columns are the areas enlarged in the second and third columns, respectively.
EE reduces CSH of C. albicans.
C. albicans goes through two important stages during biofilm formation: adhesion and morphological transition (7). The first stage is positively correlated with the cell surface hydrophobicity (CSH) of C. albicans (25). We found the CSH of control group to be 0.68, and the CSH underwent a reduction in a dose-dependent manner in response to EE (Fig. 6). Specifically, with treatment with EE at 4 μg/ml, the CSH was 0.45, which was reduced to 0.17, 0.15, 0.06, and 0.03 with treatment with EE at 8, 16, 32, and 64 μg/ml, respectively. Collectively, the CSH of biofilm cells was reduced upon EE treatment.
FIG 6.
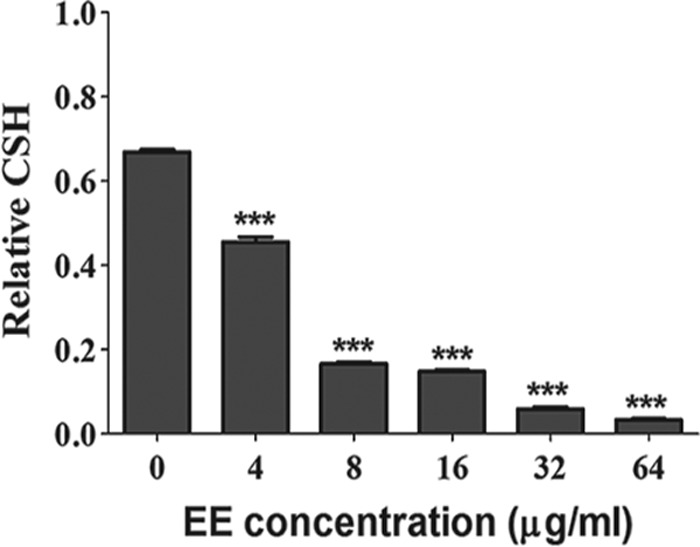
Effects of different concentrations of EE on CSH of C. albicans SC5314. CSH was evaluated using a water-hydrocarbon two-phase assay. Standard deviations based on three independent experiments are depicted. ***, P < 0.001.
EE inhibits the C. albicans yeast-to-hypha transition.
The activity of EE on the C. albicans yeast-to-hypha transition was evaluated using different liquid and solid hypha-inducing media (26–29). In the present study, C. albicans formed long and crisscrossing hyphae in the control group in all liquid media tested (Fig. 7a), including RPMI 1640 medium, yeast extract-peptone-dextrose (YPD) with 10% fetal bovine serum (FBS), and Lee's and Spider media. A morphological study under a microscope showed radial colonies of untreated C. albicans cells with feathery edges on solid Spider medium (Fig. 7b), indicative of true hyphal growth (30, 31). At the lowest concentration of 8 μg/ml, EE blocked hyphal formation; however, pseudohyphae with constrictions at sites of septation were observed (5, 9). At higher EE concentrations, cell densities were significantly reduced and filamentation was greatly inhibited as more yeast cells were observed. Accordingly, cells grown on solid Spider medium demonstrated smooth-edged colonies under treatment with 32 μg/ml of EE, suggesting only yeast growth inside the colony (32). Therefore, EE inhibited the C. albicans yeast-to-hypha morphological transition in a dose-dependent manner in both liquid and solid hypha-inducing media.
FIG 7.

EE inhibits the yeast-to-hypha transition in a dose-dependent manner. (a) Effects of different concentrations of EE on hyphal formation in liquid media. Bars, 50 μm. (b) Effects of different concentrations of EE on colony morphology on solid Spider medium.
Exposure to EE alters gene expression in C. albicans.
Using RNA-seq, approximately 44,000,000 raw reads were obtained from each sample, and after quality filtration, about 93% clean reads were mapped, as shown in Table S3. By using the threshold of significance as a relative fold change of >1.1 and a false discovery rate (FDR) of <0.05, a total of 2,204 differentially expressed genes (DEGs) were obtained (Table S5), including 1,065 upregulated genes and 1,139 downregulated genes under treatment with 16 μg/ml of EE (Fig. S1 and S2). EE treatment significantly changed the expressions of important genes involved in filamentation, encoding cell surface proteins, and involved in ergosterol biosynthesis, and these genes are listed in Tables 1 and 2. Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) database assignments were used to classify the C. albicans genes affected in response to EE. Through GO term analysis, a high percentage of genes were indicated to be involved in metabolic processes, binding, transporter activity, and cell composition (Fig. S3). Ontology assignments obtained through mapping with the KEGG database identified 171 genes in 30 pathways which were significantly enriched (P < 0.05) (Fig. S4). These pathways in which the genes were significantly enriched were mainly associated with carbon metabolism, glycolysis/gluconeogenesis, and amino sugar and nucleotide sugar metabolism (Table S6).
TABLE 1.
Important genes downregulated in EE-treated C. albicans cells compared to their expression in untreated C. albicans cells identified by RNA-seq
| Function and standard or systematic name in CGDa | Log2 fold change in expressionb | Descriptiona |
|---|---|---|
| Filamentation | ||
| ECE1 | −4.83 | Candidalysin; cytolytic peptide toxin essential for mucosal infection; hypha-specific protein |
| UME6 | −3.86 | Zn(II)2Cys6 transcription factor; role in hyphal extension |
| HGC1 | −4.41 | Hypha-specific G1 cyclin-related protein involved in regulation of morphogenesis and biofilm formation |
| TEC1 | −1.54 | TEA/ATTS transcription factor; involved in white cell pheromone response and hyphal gene regulation |
| EFG1 | −0.49 | Transcription factor; required for white-phase cell type and hyphal growth |
| CPH1 | −1.77 | Transcription factor; required for mating, filamentation on solid medium, and pheromone-stimulated biofilms |
| EED1 | −0.80 | RNA polymerase II regulator; Efg1/hypha regulated; role in adhesion and hyphal growth on solid medium |
| RBF1 | −1.16 | HMG domain transcriptional repressor of filamentous growth and hyphal genes |
| CaO19.6705 | −1.91 | Putative guanyl nucleotide exchange factor with Sec7 domain; required for normal filamentous growth |
| CDC11 | −1.81 | Septin; involved in cell and hyphal morphology, agar-invasive growth, and hypha- and cell cycle-regulated phosphorylation |
| CDC10 | −1.01 | Septin; role in wild-type cell, hyphal, or chlamydospore morphology |
| CDC24 | −0.91 | GDP-GTP exchange factor for Cdc42p; phosphorylated; required for maintenance of hyphal growth |
| CDC42 | −0.50 | Rho-type GTPase; required for budding and maintenance of hyphal growth |
| Cell surface proteins | ||
| ALS3 | −4.77 | Cell wall adhesin; involved in epithelial adhesion and endothelial invasion |
| PGA45 | −3.19 | Putative GPI-anchored cell wall protein; repressed in core caspofungin response; Hog1 induced |
| PRA1 | −4.38 | Cell surface protein that sequesters zinc from host tissue; enriched at hyphal tips |
| RBT1 | −4.33 | Cell wall protein with similarity to Hwp1; required for virulence; predicted role in glycosylation |
| CHT2 | −4.18 | GPI-linked chitinase; required for normal filamentous growth; repressed in core caspofungin response |
| HYR1 | −3.91 | GPI-anchored hyphal cell wall protein; macrophage induced |
| HWP1 | −3.64 | Hyphal cell wall protein; host transglutaminase substrate |
| PGA37 | −3.57 | Putative GPI-anchored protein; Hap43 repressed; induced by biofilms grown on Spider medium |
| SCW4 | −3.27 | Putative cell wall protein; substrate for Kex2p processing in vitro; expression regulated by white-opaque switch |
| PRY1 | −3.15 | Pry family pathogenesis-related protein; extracellular |
| OPT4 | −3.05 | Oligopeptide transporter; detected at germ tube plasma membrane |
| SAP5 | −3.03 | Biofilm-specific aspartyl protease; virulence role effected by URA3 |
| Ergosterol biosynthesis | ||
| ERG6 | −2.81 | Acetyl coenzyme A acetyltransferase; role in ergosterol biosynthesis; soluble in hyphae |
| ERG13 | −0.93 | 3-Hydroxy-3-methylglutaryl coenzyme A synthase; role in ergosterol biosynthesis |
| ERG252 | −0.92 | C-4 sterol methyl oxidase; role in ergosterol biosynthesis |
| ERG11 | −0.67 | Lanosterol 14-α-demethylase; cytochrome P450 family; role in ergosterol biosynthesis |
| ERG10 | −0.66 | Acetyl coenzyme A acetyltransferase; role in ergosterol biosynthesis; soluble in hyphae |
| ERG7 | −0.39 | 2,3-Epoxysqualene-lanosterol cyclase (lanosterol synthase); involved in conversion of 2,3-oxidosqualene to lanosterol in sterol biosynthesis |
As reported in the CGD database (http://www.candidagenome.org/).
The log2 fold change in expression was derived from RNA-seq results with an FDR of <0.05.
TABLE 2.
Important genes upregulated in EE-treated C. albicans cells compared to their expression in untreated C. albicans cells identified by RNA-seq
| Function and standard or systematic name in CGDa | Log2 fold change in expressionb | Descriptiona |
|---|---|---|
| Heat shock protein | ||
| HSP30 | 2.78 | Putative heat shock protein |
| HSP21 | 2.17 | Small heat shock protein; role in stress response and virulence |
| HSP70 | 1.75 | Putative Hsp70 chaperone; role in entry into host cells |
| HSP104 | 1.28 | Heat shock protein; roles in biofilm and virulence |
| HSP78 | 1.19 | Heat shock protein; regulated by macrophage response, Nrg1, Mig1, Gcn2, Gcn4, and Mnl1p |
| HSP60 | 0.94 | Heat shock protein; soluble in hyphae; regulated by Nrg1 and by iron |
| HSP90 | 0.64 | Essential chaperone; regulates several signal transduction pathways and temperature-induced morphogenesis |
| Filamentation | ||
| CZF1 | 1.41 | Transcription factor; regulates white-opaque switch; hyphal growth regulator |
| FGR41 | 2.54 | Putative GPI-anchored adhesin-like protein; transposon mutation affects filamentous growth |
| CNT3 | 2.50 | Peroxisomal carnitine acetyltransferase; role in hyphal growth |
| DLD1 | 2.44 | Putative d-lactate dehydrogenase; white cell-specific transcript |
| C1_12570C_A | 2.43 | Putative elongator complex subunit |
| NRG1 | 1.44 | Transcription factor/repressor; regulates formation/hyphal |
| RIM8 | 0.63 | Beta-arrestin-like protein; required for pathogenesis, activation of Rim101, and alkaline pH-induced hyphal growth |
| Cell surface proteins | ||
| CDR4 | 2.72 | Putative ABC transporter superfamily |
| RTA3 | 2.16 | Putative lipid translocase localized in plasma membrane |
| RBE1 | 2.15 | Pry family cell wall protein |
| GCA1 | 1.98 | Extracellular/plasma membrane-associated glucoamylase |
| ALS4 | 1.96 | ALS family protein; role in adhesion, biofilm formation, and germ tube induction |
| YWP1 | 1.48 | Secreted yeast wall protein; mutation increases adhesion and biofilm formation |
As reported in the CGD database (http://www.candidagenome.org/).
The log2 fold change in expression derived from RNA-seq results with an FDR of <0.05.
RNA-seq revealed that the levels of expression of genes involved in C. albicans biofilm formation were significantly changed under EE treatment. To validate the RNA-seq results, 13 of the most differentially expressed genes were selected for real-time RT-PCR, including 8 genes involved in C. albicans filamentation and regulation (EFG1, CPH1, TEC1, EED1, UME6, HGC1, ECE1, and NRG1), 3 genes encoding cell surface proteins (ALS3, HWP1, and SAP5), and 2 genes involved in ergosterol biosynthesis (ERG6 and ERG11). RT-PCR results (Fig. 8) were consistent with those from RNA-seq (Table S4). EFG1, CPH1, and TEC1, three important hyphal initiation regulators (33–35), were downregulated after EE treatment by 1.58-, 4.35-, and 5.00-fold, respectively. EED1, UME6, HGC1, and ECE1, four genes required for the long-term maintenance of hyphal growth and cell elongation (12, 35, 36), were downregulated significantly by 5.26-, 6.25-, 10.00-, and 12.50-fold, respectively. On the other hand, NRG1, the gene encoding a negative regulator of hyphal growth (37), was found to be upregulated by 2.19-fold after EE treatment. The downregulation of the important genes that are positive regulators of hyphal growth and the upregulation of the hyphal growth repressor may collectively be responsible for the inhibitory effect of EE on filamentation. HWP1, ALS3, and SAP5, the three most highly expressed genes in C. albicans biofilm cells contributing to the adhesion ability (38, 39), were downregulated by 5.89-, 9.09-, and 12.82-fold, respectively. ERG6 and ERG11, two of the genes involved in ergosterol biosynthesis pathways (40), were also downregulated by 3.70- and 1.85-fold, respectively, indicating that the synthesis of ergosterol was affected by treatment with EE.
FIG 8.
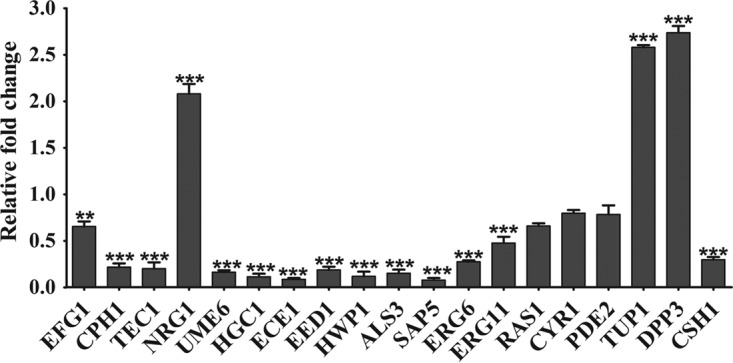
Gene expression levels after EE exposure. RT-PCR was us ed to validate the differential expression of genes found by RNA sequencing (EFG1, CPH1, TEC1, NRG1, UME6, HGC1, ECE1, EED1, HWP1, ALS3, SAP5, ERG6, and ERG11). Other important genes (RAS1, CYR1, PDE2, TUP1, DPP3, and CSH1) related to C. albicans biofilm formation were also quantified by RT-PCR. GSP1 was used to normalize the levels of expression. Data are means ± SDs from three experiments. **, P < 0.01; ***, P < 0.001.
To obtain a detailed understanding of the antibiofilm mechanism of EE, we further tested other biofilm-related genes that were not identified by RNA-seq, including RAS1, CYR1, PDE2, TUP1, DPP3, and CSH1 (Fig. 9). No obvious changes in the levels of expression of RAS1 (the RAS signal transduction GTPase gene), CYR1 (a gene encoding adenylyl cyclase in C. albicans), and PDE2 (a phosphodiesterase-encoding gene) were found. However, the levels of expression of TUP1 (a general transcriptional corepressor that negatively regulates hypha-specific gene expression) and DPP3 (a gene involved in the conversion of farnesyl pyrophosphate into farnesol synthesis) were increased by 2.58- and 2.74-fold, respectively, and the level of expression of CSH1 (cell surface hydrophobicity protein 1) was decreased by 3.36-fold.
FIG 9.

Quantification of ergosterol and farnesol content under EE treatment. (a) EE decreased the level of ergosterol biosynthesis, as measured by spectrophotometric assay; (b) EE treatment elevated the farnesol content, as measured by HPLC. Each experiment was performed in triplicate. ***, P < 0.001.
EE inhibits ergosterol production and enhances farnesol production.
The production of ergosterol in C. albicans was quantified through a spectrophotometric method. Extracted sterols demonstrated a characteristic four-peak spectral absorption pattern at between 240 and 300 nm (41–43). In our experiment, EE reduced the level of ergosterol production in C. albicans. With treatment with 8 and 16 μg/ml of EE, the ergosterol content was reduced by 9.2% and 65.3%, respectively, compared to that in the control group (Fig. 9a).
The amount of farnesol secreted by C. albicans cells with or without EE treatment was examined by using high-performance liquid chromatography (HPLC). Farnesol showed a retention time of 16.97 min and a pseudo molecular ion peak at m/z 245.1875 [M + Na]+, consistent with the formula C15H26O (Fig. S5). The farnesol content of each sample was calculated on the basis of the HPLC integral area compared with that on the standard curve (Fig. S6). Samples treated with EE showed a larger amount of extracellular farnesol. Specifically, by treatment with EE at 8 and 16 μg/ml, the level of farnesol production showed 3.95- and 5.43-fold increases, respectively (Fig. 9b).
Assessment of EE toxicity for LO2 cells.
Finally, we evaluated the safety profile of EE using cells of the human normal liver cell line LO2. We found that EE at concentrations up to 128 μg/ml did not influence the viability of LO2 cells (data not shown), suggesting that EE is safe at the concentrations tested and may not have adverse effects on human cells.
DISCUSSION
In its biofilm form, C. albicans is less responsive to commonly used antifungal drugs, such as azoles and conventional amphotericin B, than planktonic cells (9, 10). Given the severity of biofilm-related diseases, it is important to find alternative antifungal drugs that can efficiently control biofilms. Interestingly, some phytochemicals have the ability to inhibit the yeast-to-hypha conversion and, hence, block C. albicans biofilm formation (44, 45). The expanding repertoire of FPMs with antifungal activity suggests that FPMs are potentially valuable natural sources of antifungal agents (22–24). In our screening of antifungal FPMs, EE showed a strong in vitro inhibitory effect toward multiple C. albicans strains, including fluconazole-susceptible and -resistant clinical isolates, while it was not toxic against LO2 human normal liver cells. More importantly, EE suppressed 73% of biofilm formation at 32 μg/ml, destroyed nearly all mature biofilms (92%) at 128 μg/ml, and significantly reduced the stained biofilm absorbance by nearly 80% at 32 μg/ml. This effect was further confirmed through morphological studies using SEM, CLSM, and a filamentation assay. Because biofilm infections are challenging to treat clinically, we were inspired to elucidate the underlying molecular mechanisms of naturally derived FPMs, and to the best of our knowledge, this is the first report on the antibiofilm effect of such compounds.
Relative hydrophobicity is considered an important pathogenic attribute of Candida spp. pertaining to adhesion (46). Our data showed that 16 μg/ml EE significantly reduced the CSH of C. albicans to 0.15 (the CSH was 0.68 for the control group). Thus, it could be inferred that the CSH-reducing ability played an important role in EE's biofilm inhibition.
The highly structured biofilms were constructed on crisscrossing hyphae; thus, interruption of the yeast-to-hypha transition would prevent biofilm development (7, 8). The filamentation assay showed that EE obviously inhibited the morphological transition (Fig. 7). Starting at 16 μg/ml of EE, large numbers of cells were locked in the yeast form in all test media, and at 32 μg/ml, only smooth-edged colonies were found on solid Spider medium. The ability of EE to inhibit C. albicans transformation was one of the main contributors to its antibiofilm effect.
RNA-seq and RT-PCR were further performed to explore the antibiofilm mechanisms of EE at the genetic level (47). First, both of the independent tests mentioned above showed that EE inhibited the expression of hypha-specific genes (HSGs) at the hyphal initiation (EFG1, CPH1, and TEC1) and long-term maintenance (EED1, UME6, HGC1, and ECE1) stages (40, 44, 45). Transcription factors like Efg1, Cph1, Tec1, Czf1, and Flo8 play important roles in initializing hyphal growth (5, 34, 35). For example, Efg1 and Cph1, the first identified regulators of hyphal development, synergistically regulate virulence genes (35), while Tec1 has been shown to regulate hyphal development and virulence in C. albicans (48, 49). On the other hand, C. albicans filamentation requires cell polarized growth as well as the simultaneous suppression of cell separation, during which EED1, UME6, HGC1, etc., are activated (12). Therefore, the downregulation of these hyphal growth genes upon EE treatment contributed to the antibiofilm effect of EE. Second, the reduction of CSH upon EE treatment could possibly be due to the reduced expression of those genes involved in adhesion and the cell wall component. In our test, the downregulation of HWP1, ALS3, and SAP5 was evidenced to reduce the cell adhesion and virulence ability, which could further cause the detachment of biofilms from an abiotic substrate (38, 39). In addition, RNA-seq showed that many genes encoding glycosylphosphatidylinositol (GPI) proteins (PGA45, CHT2, PRA1, RBT1, HYR1, etc.) (Table 1) were also downregulated under EE treatment. Since GPI proteins undertake various roles in enzymatic activities that are related to adhesion and morphogenesis (50, 51), the downregulation of genes for these proteins could directly lead to changes in CSH. Third, and most importantly, the inhibition of ergosterol biosynthesis by EE might mainly account for its antibiofilm effect. As we found, six ergosterol biosynthetic genes (ERG6, ERG13, ERG252, ERG11, ERG10, and ERG7) were collectively downregulated after EE treatment, which directly resulted in the reduction of ergosterol biosynthesis by half (Fig. 9a). The depletion of ergosterol directly alters cell membrane functions, which eventually leads to cell death (52). That explained the increased numbers of dead hyphae and cells in the CLSM images (Fig. 4). On the other hand, it has been reported that the inhibition of carbon flow to ergosterol elevates the level of farnesol production (53, 54). Farnesol is an important quorum-sensing molecule (55) which also plays a major role in biofilm formation (56). In fact, natural products, like bisbibenzyls, have been reported to inhibit the switch in morphogenesis and biofilm formation directly through upregulating DPP3 (45). In accordance with the findings presented in previous reports, the key gene involved in farnesol synthesis (DPP3) was upregulated 3-fold in our tests, and the level of farnesol production showed a 5-fold increase after EE treatment (Fig. 9b). TUP1 and NRG1 are both upregulated in response to the overexpressed farnesol (5, 37). Consequently, the overflowing Tup1-Nrg1 complex launched a series of negative regulators, resulting in the above-mentioned inhibition of hypha-specific genes. In addition, the molecular response to farnesol in C. albicans can be multifaceted. Besides the above-mentioned Tup1-Nrg1 complex genes, many others, like heat shock protein genes, drug resistance genes, and adhesion genes (Table 1 and 2), are regulated by farnesol (57, 58), thus affecting the cell or biofilm in many aspects. Collectively, we postulate that the antibiofilm effect of EE is attributed to its inhibition of ergosterol biosynthesis, which in turn elevates the production of farnesol and further activates the Tup1-Nrg1 complex to negatively regulate biofilm formation.
Since RNA-seq also showed the downregulation of EFG1, we further verified the changes to the main genes in the Ras/cyclic AMP (cAMP) pathway by RT-PCR, including RAS1, CYR1, PDE2, and EFG1 (5, 59). The results showed that, except for EFG1, all these genes remained unchanged. In addition, exogenous addition of 10 mM cAMP did not reverse the filamentation inhibition effect of EE (data not shown). Therefore, EE treatment exerted a minor influence on the Ras/cAMP pathway.
It is to be noted that biofilms are resistant to most clinically used antifungal agents targeting sterol synthesis, including both the azoles, which target sterol-14-α-demethylase, and the allylamines, which inhibit squalene epoxidase (60). Biofilms treated with fluconazole show the upregulated expression of genes involved in ergosterol biosynthesis (ERG1, ERG3, ERG11, ERG25, etc.) (61, 62). Therefore, it is interesting to figure out why EE exerts antibiofilm activity through reducing sterol production in C. albicans biofilms. Since ERG6 was found to be the most reduced, we presumed that EE targets the expression of ERG6 and inhibits the sterol C24-methyltransferase (Erg6p), a catalyst not found in humans (63). Blocking ergosterol biosynthesis could be effective in both yeasts and biofilms. In addition, EE's antibiofilm activity might also be achieved through a multitarget action, as the expression of many genes encoding important cell wall proteins (PGA45, RBT1, CHT2, etc.) was significantly affected after EE treatment, and this altered expression would greatly influence structural integrity and the mediation of adherence (64). Although further explorations are needed to verify our hypothesis, EE and its potential bioactive FPM analogs differ from clinical antifungal agents in their antibiofilm mechanisms and are worth consideration for further development as antifungal drugs.
MATERIALS AND METHODS
Strains, culture, and agents.
Candida albicans standard strains SC5314 and ATCC 24433 were purchased from ATCC, USA. Isolates of 10 fluconazole-resistant strains (strains 24D, 28I, CA102, CA901, CA112869, CA311, CA331, CA23C, CA652, and CA20051) and 8 fluconazole-susceptible strains (strains CA13, CA14, CA19, CA21, CA163, CA381, CA422, and CA592) were also used in our study. All strains were routinely grown in yeast extract-peptone-dextrose (YPD; 1% yeast extract, 2% peptone, and 2% dextrose) (26) liquid medium at 30°C in a shaking incubator. EE was isolated from the leaves of E. robusta, and its purity (>99.80%) was analyzed by high-performance liquid chromatography (HPLC). For in vitro experiments, 10 mg/ml of EE in dimethyl sulfoxide (DMSO) was used as a stock solution and was further diluted in RPMI 1640 medium before use. Other media used included YPD plus 10% (vol/vol) fetal calf serum (26), Spider medium (26), Lee's medium (pH 4.8) (27), synthetic low-ammonium dextrose (SLAD) medium (28), and RPMI 1640 medium (29).
Antifungal assay.
The antifungal activity of EE was determined in 96-well plates in accordance with the broth microdilution protocol of the Clinical and Laboratory Standards Institute (described in document M27-A3 [65]) with minor modifications (66). Briefly, the initial fungal suspension used for inoculation was adjusted to 103 CFU/ml in RPMI 1640 medium, and the final concentrations of chemicals were adjusted from 0.063 to 64 μg/ml for fluconazole and 1 to 128 μg/ml for EE. The plates were incubated at 37°C for 24 h. Growth inhibition was determined by a spectrophotometer. Optical densities at 540 nm (OD540s) (67) were measured, and the MIC50 was defined as the concentration of drugs that inhibited 50% of cell growth. Each strain was tested in triplicate.
Time-kill curve assay.
For the time-kill curve assay, C. albicans cells in exponential growth phase were washed with phosphate-buffered saline (PBS), resuspended in RPMI 1640 medium to 1 × 106 cells/ml (59, 68), and divided into 4 bottles. Different concentrations of EE were added to the suspensions. The samples were cultured at 30°C under constant shaking (200 rpm), and small aliquots were withdrawn at predetermined time points (0, 3, 6, 9, and 12 h). After incubation for 48 h at 30°C, serially diluted cell suspensions were plated on YPD agar to obtain colony counts. The experiments were performed in triplicate (59).
In vitro antibiofilm assay.
The antibiofilm effect of EE was measured through a 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT) reduction assay as described by Ramage and colleagues (69) with minor modifications. Briefly, 96-well plates were seeded with 100-μl aliquots of cell suspensions of 1.0 × 106 cells/ml in RPMI 1640 medium and incubated statically at 37°C for 1.5 h for initial adhesion. The supernatant was aspirated to remove nonadherent cells. Fresh RPMI 1640 medium with or without different concentrations of EE was added, followed by another 24 h of incubation. For detection of EE on mature biofilms, biofilms pregrown for 24 h were treated with different concentrations of EE and were incubated for 24 h at 37°C. After incubation, the supernatant was discarded and XTT (Sigma, USA) was added to the prewashed biofilms (69). The plate was incubated in the dark for 2 to 3 h at 37°C. An aliquot of 70 to 80 μl of the resulting colored supernatant from each well was transferred to the wells of a new 96-well plate. Finally, the plate was read in a plate reader at 490 nm for determination of the antibiofilm effect of EE.
Biofilm quantification.
Biofilm biomass was indirectly measured using the crystal violet (CV) staining method (70). Following incubation, biofilms were washed three times with sterile PBS to remove nonadherent cells. The plate was dried, and 200 μl of 99% methanol was added to the biofilms for 15 min to allow cell fixation. The methanol was then removed, and following drying, 200 μl of CV stain (1%, vol/vol) was added into each well. After 5 min, the excess CV was removed and the plate was washed with sterile distilled water three times and dried at room temperature. Afterwards, 200 μl of acetic acid (33%, vol/vol) was added to each well to dissolve the CV stain from the biofilms. Then, 100 μl of the destaining solution was transferred to a new plate, and the absorbance at 570 nm was determined using a microplate reader (71). The experiment was performed in triplicate, and the mean absorbance of each well was plotted against the EE concentration.
CLSM.
Confocal laser scanning microscopy (CLSM) was performed as previously described (32, 72) by using six-well plates containing plastic disks. Biofilms were incubated following the method described above and were costained with 10 μg/ml of fluorescein diacetate (FDA) and 5 μg/ml of propidium iodide (PI) for 30 min (72, 73). Images were taken with a confocal laser scanning microscope (LSM700; Carl Zeiss). Laser beams with 488- and 555-nm excitation wavelengths were used for FDA and PI imaging, respectively. FDA was hydrolyzed by viable cells, resulting in the accumulation of green fluorescence, whereas PI stained the dead cells (30).
SEM.
Scanning electron microscopy (SEM) was used to investigate the ultrastructure of C. albicans biofilms with and without EE treatment. Sterile glass disks coated with poly-l-lysine hydrobromide (Sigma) were used to develop the biofilms. For the EE treatment and the control groups, biofilms were prewashed using PBS and placed in a fixative consisting of 2% (vol/vol) glutaraldehyde in 0.15 M sodium cacodylate buffer (pH 7.2) for 2 h. Then, samples were rinsed twice in cacodylate buffer, garnished with 1% osmic acid for 2 h, and dehydrated in an ascending ethanol series. After treatment with hexamethyldisilazane (Polysciences Europe GmbH, Eppelheim, Germany), the samples were dried overnight. The specimens were coated with gold and observed through a Carl Zeiss SEM (EVO LS10) in high-vacuum mode (27, 74).
CSH assay.
Cellular surface hydrophobicity (CSH) was measured by using a water-hydrocarbon two-phase assay as described earlier (28, 74). In brief, the preformed C. albicans biofilms were removed from the flask surface to obtain a cell suspension (OD600, 1.0 in YPD medium). Then, 1.2 ml of the suspension was pipetted into a clean glass tube and overlaid with 0.3 ml of octane. The mixture was vortexed for 3 min and then allowed to stand at room temperature for another 3 min for phase separation. Then, the OD600 of the aqueous phase was determined. The OD600 of the group without the octane overlay was used as a control. Each experiment was performed in triplicate. Relative hydrophobicity was calculated as follows: relative CSH = [(OD600 of control − OD600 after octane overlay)/OD600 of control].
Filamentation assay.
A filamentation assay was carried out in 12-well plates. In brief, cells of C. albicans SC5314 from an overnight growth in YPD medium were centrifuged and washed in PBS. Suspensions of 1 × 106 cells/ml were prepared in RPMI 1640 medium, YPD with 10% FBS, Spider medium, or Lee's liquid medium, which are known to induce hyphal growth (26–28). Different concentrations of the compound EE were added, followed by incubation at 37°C for 4 h. Images of cells were obtained using a microscope (IX53; Olympus). The experiments were performed in triplicate.
The C. albicans filamentation assay was also carried out on solid Spider agar plates with or without various concentrations of EE. C. albicans cells pregrown overnight in YPD medium were diluted with Spider liquid medium to a final concentration of 1,000 cells/ml (67). Then, 50 μl of the suspension was spread onto the plates. The plates were incubated at 37°C for 3 days. Images of the colony edges were obtained using a microscope (IX53; Olympus). Each experiment was performed in triplicate.
RNA extraction and sequencing.
Exponentially growing C. albicans cells were washed and suspended in RPMI 1640 medium to a density of 106 cells/ml. Biofilms were cultured as mentioned above. Fresh RPMI 1640 medium containing 16 μg/ml of EE or DMSO was added. After incubation for 8 h, the biofilm samples were collected and washed with PBS. Total RNA was extracted using the TRIzol reagent (Invitrogen) and was quantified by use of an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) prior to RNA-seq library construction using a NEBNext Ultra RNA library preparation kit for Illumina (NEB, USA) according to the manufacturer's protocol (75, 76). Briefly, the poly(A) mRNA was purified from the total RNA using poly(A) selection to remove rRNA and contamination with small RNAs. The mRNA was then chemically fragmented and converted into single-stranded cDNA using ProtoScript II reverse transcriptase, and a second cDNA strand was synthesized using a second-strand synthesis enzyme. Adapters containing the full complement of the sequencing primer hybridization sites for multiplexed reads were added to the DNA to generate the RNA-seq library. RNA-seq was performed by the Genewiz Company (South Plainfield, NJ) on an Illumina HiSeq platform. Differential gene expression analysis was performed by using the DESeq Bioconductor package, in which the false discovery rate (FDR) was <0.05 and the threshold of significance was a relative fold change of >1.1. Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichments were analyzed to detect the functions and pathways of differentially expressed genes (DEGs) (77, 78). Three technical replicates were conducted for each sample.
Quantification of gene expression by real-time RT-PCR.
Real time RT-PCR was performed for validation of the results derived from RNA-seq (31). Total RNAs were isolated from the preformed biofilms using the hot phenol method. Approximately 1 μg of total RNA was used to synthesize cDNA using a ReverTra Ace quantitative PCR RT kit (Toyobo Ltd.), and cDNA was diluted (1:5), followed by amplification with SYBR green real-time PCR master mix (Toyobo Ltd., Osaka, Japan) in a final volume of 20 μl. Quantitative RT-PCR was carried out using a LightCycler 480 instrument (Roche Molecular Biochemicals, Mannheim, Germany). The relative differences in mRNA abundance obtained with the LightCycler 480 instrument were calculated by determination of the changes in the cycle threshold (CT) values. The primers used in this study are listed in Table S1 in the supplemental material. The data were normalized to the level of expression of GSP1, as its expression level remains unchanged in both yeast cells and biofilms. The relative fold change in gene expression levels was calculated using the formula 2−ΔΔCT.
Sterol quantification.
Total cellular ergosterol from the cell cultures was quantified as previously reported (41–43). Briefly, suspensions of 106 cells/ml with or without EE were incubated at 35°C for 12 h under constant shaking at 150 rpm. Cells were then harvested by centrifugation at 2,000 × g for 5 min, followed by washing with sterile PBS. The wet weight of the cell pellet was determined. The alcoholic potassium hydroxide solution used for saponification of the cells was prepared by adding 25 g KOH to 36 ml of sterile water, which was then brought to 100 ml with 100% ethanol. To each pellet, 3 ml of the aforementioned solution was added and the mixture was vortexed for 1 min, followed by incubation at 85°C in a water bath for 1 h. The suspensions were allowed to cool to room temperature, and then 1 ml sterile water and 3 ml petroleum ether were added and the mixture was vortexed for 3 min and left still for extraction of sterols. The upper layer was transferred to a clean tube, and the tube was stored at −20°C for spectrophotometric analysis. The presence of ergosterol and the late sterol intermediate 24(28)-dihydroergosterol [24(28)-DHE] in the extracted sample resulted in a characteristic four-peak curve at between 200 and 300 nm. The ergosterol content was calculated as a percentage of the wet weight of the cells using the following equations: percent ergosterol + percent 24(28)-DHE = [(A281.5/290) × F]/pellet weight, percent 24(28)-DHE = [(A230/518) × F]/pellet weight, and percent ergosterol = [percent ergosterol + percent 24(28)-DHE] − percent 24(28)-DHE, where F is the factor for dilution in ethanol, and 290 and 518 are the E values (in percent per centimeter) determined for crystalline ergosterol and 24(28)-DHE, respectively (79).
HPLC quantification of farnesol.
Suspensions of 105 yeast cells/ml were divided into three groups, with each group containing 12 ml, and each group was treated with EE at a final concentration of 0, 8, or 16 μg/ml. Cell samples were incubated at 37°C for 8 h. The cell pellets were dried and weighed with an electronic balance. Farnesol was extracted using ethyl acetate-hexanes (1:4) and analyzed by HPLC as previously reported (55, 80). Chromatographic separation was performed on a Shim-pack VP-ODS column (250 by 4.6 mm; Agilent Technologies, Palo Alto, CA, USA) with a solvent flow rate of 1 ml/min at a column temperature of 30°C. Samples were eluted by methanol-H2O (4:1) and were detected at 210 nm. The existence of farnesol was verified by HPLC-quadrupole time of flight mass spectroscopy (HPLC-QTOF-MS; Agilent Technologies, USA). Eight different concentrations of farnesol standard samples were used to create a standard curve of integral areas eluting at its retention time. Each experiment was performed in triplicate, and the farnesol content of each sample was calculated on the basis of the HPLC integral area compared with that on the standard curve.
Assessment of EE cytotoxicity against LO2 cells.
Finally, we investigated the safety profile of the compound EE against LO2 human normal liver cells using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method (81).
In brief, a 100-μl inoculum of LO2 cells was seeded in a 96-well plate at a density of 104 cells/ml. After overnight growth, the cells were treated with various concentrations of EE for further incubation at 37°C for 48 h. The supernatant was carefully removed, and 20 μl MTT solution (5 mg/ml) was added to each well for further culture for 4 h. Following removal of the supernatant, 150 μl DMSO was added to dissolve the resulting formazan. The absorbance at 570 nm was measured by using a microplate reader (Molecular Devices, CA, USA).
Accession number(s).
All of the RNA sequencing data were deposited in the NCBI database under BioProject accession number PRJNA356183.
Supplementary Material
ACKNOWLEDGMENTS
This research work was funded by the National Natural Science Foundation of China (81503218), the Program for Changjiang Scholars and the Innovative Research Team in University (IRT_15R63), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and Fundamental Research Funds for the Central Universities (2016ZZD010).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02707-16.
REFERENCES
- 1.Odds FC. 1988. Candida and candidosis: a review and bibliography. Bailliere Tindall, London, United Kingdom. [Google Scholar]
- 2.Eggimann P, Garbino J, Pittet D. 2003. Management of Candida species infections in critically ill patients. Lancet Infect Dis 3:772–785. doi: 10.1016/S1473-3099(03)00831-4. [DOI] [PubMed] [Google Scholar]
- 3.Kibbler CC, Seaton S, Barnes RA, Gransden WR, Holliman RE, Johnson EM, Perry JD, Sullivan DJ, Wilson JA. 2003. Management and outcome of bloodstream infections due to Candida species in England and Wales. J Hosp Infect 54:18–24. doi: 10.1016/S0195-6701(03)00085-9. [DOI] [PubMed] [Google Scholar]
- 4.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 5.Sudbery PE. 2011. Growth of Candida albicans hyphae. Nat Rev Microbiol 9:737–748. doi: 10.1038/nrmicro2636. [DOI] [PubMed] [Google Scholar]
- 6.Chandra J, Kuhn DM, Mukherjee PK, Hoyer LL, Mccormick T, Ghannoum MA. 2001. Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J Bacteriol 183:5385–5394. doi: 10.1128/JB.183.18.5385-5394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gulati M, Nobile CJ. 2016. Candida albicans biofilms: development, regulation, and molecular mechanisms. Microbes Infect 18:310–321. doi: 10.1016/j.micinf.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Y, Su C, Liu HP. 2014. Candida albicans hyphal initiation and elongation. Trends Microbiol 22:707–714. doi: 10.1016/j.tim.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker LA, Gow NAR, Munro CA. 2010. Fungal echinocandin resistance. Fungal Genet Biol 47:117–126. doi: 10.1016/j.fgb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiori B, Posteraro B, Torelli R, Tumbarello M, Perlin DS, Fadda G, Sanguinetti M. 2011. In vitro activities of anidulafungin and other antifungal agents against biofilms formed by clinical isolates of different Candida and Aspergillus species. Antimicrob Agents Chemother 55:3031–3035. doi: 10.1128/AAC.01569-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katragkou Α, Chatzimoschou A, Simitsopoulou M, Dalakiouridou M, Dizamataftsi E, Tsantali C, Roilides E. 2008. Differential activities of newer antifungal agents against Candida albicans and Candida parapsilosis biofilms. Antimicrob Agents Chemother 52:357–360. doi: 10.1128/AAC.00856-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn DM, George T, Chandra J, Mukherjee PK, Ghannoum MA. 2002. Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob Agents Chemother 46:1773–1780. doi: 10.1128/AAC.46.6.1773-1780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramage G, Jose A, Sherry L, Lappin DF, Jones B, Williams C. 2013. Liposomal amphotericin B displays rapid dose-dependent activity against Candida albicans biofilms. Antimicrob Agents Chemother 57:2369–2371. doi: 10.1128/AAC.02344-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pal Singh I, Bharate SB. 2006. Phloroglucinol compounds of natural origin. Nat Prod Rep 23:558–591. doi: 10.1039/b600518g. [DOI] [PubMed] [Google Scholar]
- 15.Shang ZC, Yang MH, Jian KL, Wang XB, Kong LY. 2016. 1H NMR-guided isolation of formyl-phloroglucinol meroterpenoids from the leaves of Eucalyptus robusta. Chemistry 22:11778–11784. doi: 10.1002/chem.201601732. [DOI] [PubMed] [Google Scholar]
- 16.Yu Y, Gan LS, Yang SP, Sheng L, Liu QF, Chen SN, Li J, Yue JM. 2016. Eucarobustols A-I, conjugates of sesquiterpenoids and acylphloroglucinols from Eucalyptus robusta. J Nat Prod 79:1365–1372. doi: 10.1021/acs.jnatprod.6b00090. [DOI] [PubMed] [Google Scholar]
- 17.Yang SP, Zhang XW, Ai J, Gan LS, Xu JB, Wang Y, Su ZS, Wang L, Ding J, Geng MY. 2012. Potent HGF/c-Met axis inhibitors from Eucalyptus globulus: the coupling of phloroglucinol and sesquiterpenoid is essential for the activity. J Med Chem 55:8183–8187. doi: 10.1021/jm3007454. [DOI] [PubMed] [Google Scholar]
- 18.Tian LW, Xu M, Li XC, Yang CR, Zhu HJ, Zhang YJ. 2014. Eucalmaidials A and B, phloroglucinol-coupled sesquiterpenoids from the juvenile leaves of Eucalyptus maideni. RSC Adv 4:21373–21378. doi: 10.1039/c4ra01078g. [DOI] [Google Scholar]
- 19.Varshney VK, Pandey A, Onial PK, Dayal R. 2012. Antifungal activity of phytochemicals from Eucalyptus hybrid leaves against some plant pathogenic and wood decay fungi. Arch Phytopathol Plant Prot 45:2347–2354. doi: 10.1080/03235408.2012.727073. [DOI] [Google Scholar]
- 20.Li XC, ElSohly HN, Nimrod AC, Clark AM. 1999. Two auronols from Pseudolarix amabilis. J Nat Prod 62:767–769. doi: 10.1021/np980539z. [DOI] [PubMed] [Google Scholar]
- 21.Yin S, Xue JJ, Fan CQ, Miao ZH, Ding J, Yue JM. 2007. Eucalyptals A-C with a new skeleton isolated from Eucalyptus globulus. Org Lett 9:5549–5552. doi: 10.1021/ol7025075. [DOI] [PubMed] [Google Scholar]
- 22.Shou Q, Smith JE, Mon H, Brkljaca Z, Smith AS, Smith DM, Griesser HJ, Wohlmuth H. 2014. Rhodomyrtals A-D, four unusual phloroglucinol-sesquiterpene adducts from Rhodomyrtus psidioides. RSC Adv 4:13514–13517. doi: 10.1039/c4ra00154k. [DOI] [Google Scholar]
- 23.Wong JH, Lau KM, Wu YO, Cheng L, Wong CW, Yew DT, Leung PC, Fung KP, Hui M, Ng TB. 2015. Antifungal mode of action of macrocarpal C extracted from Eucalyptus globulus Labill (Lan An) towards the dermatophyte Trichophyton mentagrophytes. Chin Med 10:34. doi: 10.1186/s13020-015-0068-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bharate SB, Khan SI, Yunus NAM, Chauthe SK, Jacob MR, Tekwani BL, Khan IA, Singh IP. 2007. Antiprotozoal and antimicrobial activities of O-alkylated and formylated acylphloroglucinols. Bioorg Med Chem 15:87–96. doi: 10.1016/j.bmc.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Ellepola ANB, Samaranayake LP. 1998. The effect of limited exposure to antimycotics on the relative cell-surface hydrophobicity and the adhesion of oral Candida albicans to buccal epithelial cells. Arch Oral Biol 43:879–887. doi: 10.1016/S0003-9969(98)00064-8. [DOI] [PubMed] [Google Scholar]
- 26.Maidan MM, De Rop L, Serneels J, Exler S, Rupp S, Tournu H, Thevelein JM, Van Dijck P. 2005. The G protein-coupled receptor Gpr1 and the Galpha protein Gpa2 act through the cAMP-protein kinase A pathway to induce morphogenesis in Candida albicans. Mol Biol Cell 16:1971–1986. doi: 10.1091/mbc.E04-09-0780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee KL, Buckley HR, Campbell CC. 1975. An amino acid liquid synthetic medium for the development of mycelial and yeast forms of Candida albicans. Sabouraudia 13:148–153. doi: 10.1080/00362177585190271. [DOI] [PubMed] [Google Scholar]
- 28.Gimeno C. 1992. Unipolar cell divisions in the yeast S. cerevisiae lead to filamentous growth: regulation by starvation and RAS. Cell 68:1077–1090. doi: 10.1016/0092-8674(92)90079-R. [DOI] [PubMed] [Google Scholar]
- 29.Espinel-Ingroff A, Cuenca-Estrella M, Cantón E. 2013. EUCAST and CLSI: working together towards a harmonized method for antifungal susceptibility testing. Curr Fungal Infect Rep 7:59–67. doi: 10.1007/s12281-012-0125-7. [DOI] [Google Scholar]
- 30.Li Y, Ma Y, Zhang L, Guo F, Ren L, Yang R, Li Y, Lou H. 2012. In vivo inhibitory effect on the biofilm formation of Candida albicans by liverwort derived riccardin D. PLoS One 7:e35543. doi: 10.1371/journal.pone.0035543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun L, Liao K, Wang D. 2015. Effects of magnolol and honokiol on adhesion, yeast-hyphal transition, and formation of biofilm by Candida albicans. PLoS One 10:e0117695. doi: 10.1371/journal.pone.0117695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson DN, Yang L, Wu S, Kennelly EJ, Lipke PN. 2015. Garcinia xanthochymus benzophenones promote hyphal apoptosis and potentiate activity of fluconazole against Candida albicans biofilms. Antimicrob Agents Chemother 59:6032–6038. doi: 10.1128/AAC.00820-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naseem S, Araya E, Konopka JB. 2015. Hyphal growth in Candida albicans does not require induction of hyphal-specific gene expression. Mol Biol Cell 26:1174–1187. doi: 10.1091/mbc.E14-08-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stoldt VR, Sonneborn A, Leuker CE, Ernst JF. 1997. Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J 16:1982–1991. doi: 10.1093/emboj/16.8.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane S, Birse C, Zhou S, Matson R, Liu H. 2001. DNA array studies demonstrate convergent regulation of virulence factors by Cph1, Cph2, and Efg1 in Candida albicans. J Biol Chem 276:48988–48996. doi: 10.1074/jbc.M104484200. [DOI] [PubMed] [Google Scholar]
- 36.Ramage G, Saville SP, Thomas DP, López-Ribot JL. 2005. Candida biofilms: an update. Eukaryot Cell 4:633–638. doi: 10.1128/EC.4.4.633-638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams SM. 2011. Investigating the role of Nrg1p and Tup1p during Candida albicans chlamydospore formation. McNair Scholars J 15:11. [Google Scholar]
- 38.Nobile CJ, Andes DR, Nett JE, Smith FJ Jr, Yue F, Phan Q-T, Edwards JE Jr, Filler SG, Mitchell AP. 2006. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathog 2:e63. doi: 10.1371/journal.ppat.0020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tronchin G, Pihet M, Lopes-Bezerra LM, Bouchara J-P. 2008. Adherence mechanisms in human pathogenic fungi. Med Mycol 46:749–772. doi: 10.1080/13693780802206435. [DOI] [PubMed] [Google Scholar]
- 40.Borecká-Melkusová S, Moran GP, Sullivan DJ, Kucharíková S, Chorvát D Jr, Bujdáková H. 2009. The expression of genes involved in the ergosterol biosynthesis pathway in Candida albicans and Candida dubliniensis biofilms exposed to fluconazole. Mycoses 52:118. doi: 10.1111/j.1439-0507.2008.01550.x. [DOI] [PubMed] [Google Scholar]
- 41.Sun LM, Lv BB, Cheng AX, Wu XZ, Lou HX. 2009. The effect of plagiochin E alone and in combination with fluconazole on the ergosterol biosynthesis of Candida albicans. Biol Pharm Bull 32:36–40. doi: 10.1248/bpb.32.36. [DOI] [PubMed] [Google Scholar]
- 42.Brilhante RS, de Lima RA, Caetano EP, Leite JJ, Castelo-Branco DDS, Ribeiro JF, Bandeira TDJ, Cordeiro RDA, Monteiro AJ, Sidrim JJ, Rocha MF. 2013. Effect of farnesol on growth, ergosterol biosynthesis, and cell permeability in Coccidioides posadasii. Antimicrob Agents Chemother 57:2167–2170. doi: 10.1128/AAC.02457-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun LM, Cheng AX, Wu XZ, Zhang HJ, Lou HX. 2010. Synergistic mechanisms of retigeric acid B and azoles against Candida albicans. J Appl Microbiol 108:341–348. doi: 10.1111/j.1365-2672.2009.04429.x. [DOI] [PubMed] [Google Scholar]
- 44.Wang B, You J, King JB, Cai S, Park E, Powell DR, Cichewicz RH. 2014. Polyketide glycosides from Bionectria ochroleuca inhibit Candida albicans biofilm formation. J Nat Prod 77:2273–2279. doi: 10.1021/np500531j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L, Chang W, Sun B, Groh M, Speicher A, Lou H. 2011. Bisbibenzyls, a new type of antifungal agent, inhibit morphogenesis switch and biofilm formation through upregulation of DPP3 in Candida albicans. PLoS One 6:e28953. doi: 10.1371/journal.pone.0028953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borecká-Melkusová S, Bujdáková H. 2008. Variation of cell surface hydrophobicity and biofilm formation among genotypes of Candida albicans and Candida dubliniensis under antifungal treatment. Can J Microbiol 54:718–724. doi: 10.1139/W08-060. [DOI] [PubMed] [Google Scholar]
- 47.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schweizer A, Rupp S, Taylor BN, Röllinghoff M, Schröppel K. 2000. The TEA/ATTS transcription factor CaTec1p regulates hyphal development and virulence in Candida albicans. Mol Microbiol 38:435–445. doi: 10.1046/j.1365-2958.2000.02132.x. [DOI] [PubMed] [Google Scholar]
- 49.García-Sánchez S, Aubert S, Iraqui I, Janbon G, Ghigo JM, D'Enfert C. 2004. Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns. Eukaryot Cell 3:536–545. doi: 10.1128/EC.3.2.536-545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruiz-Herrera J, Elorza MV, Valentín E, Sentandreu R. 2006. Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res 6:14–29. doi: 10.1111/j.1567-1364.2005.00017.x. [DOI] [PubMed] [Google Scholar]
- 51.Hazen B, Hazen K. 1988. Dynamic expression of cell surface hydrophobicity during initial yeast cell growth and before germ tube formation of Candida albicans. Infect Immun 56:2521–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajput SB, Karuppayil SM. 2013. Small molecules inhibit growth, viability and ergosterol biosynthesis in Candida albicans. Springerplus 2:26. doi: 10.1186/2193-1801-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornby JM, Kebaara BW, Nickerson KW. 2003. Farnesol biosynthesis in Candida albicans: cellular response to sterol inhibition by zaragozic acid B. Antimicrob Agents Chemother 47:2366–2369. doi: 10.1128/AAC.47.7.2366-2369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Rao R. 2010. Beyond ergosterol: linking pH to antifungal mechanisms. Virulence 1:551–554. doi: 10.4161/viru.1.6.13802. [DOI] [PubMed] [Google Scholar]
- 55.Hornby JM, Jensen EC, Lisec AD, Tasto JJ, Jahnke B, Shoemaker R, Dussault P, Nickerson KW. 2001. Quorum sensing in the dimorphic fungus Candida albicans is mediated by farnesol. Appl Environ Microbiol 67:2982–2992. doi: 10.1128/AEM.67.7.2982-2992.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kebaara BW, Langford ML, Navarathna DH, Dumitru R, Nickerson KW, Atkin AL. 2008. Candida albicans Tup1 is involved in farnesol-mediated inhibition of filamentous-growth induction. Eukaryot Cell 7:980–987. doi: 10.1128/EC.00357-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao YY, Cao YB, Xu Z, Ying K, Li Y, Xie Y, Zhu ZY, Chen WS, Jiang YY. 2005. cDNA microarray analysis of differential gene expression in Candida albicans biofilm exposed to farnesol. Antimicrob Agents Chemother 49:584–589. doi: 10.1128/AAC.49.2.584-589.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Enjalbert B, Whiteway M. 2005. Release from quorum-sensing molecules triggers hyphal formation during Candida albicans resumption of growth. Eukaryot Cell 4:1203–1210. doi: 10.1128/EC.4.7.1203-1210.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.An MM, Shen H, Cao YB, Zhang JD, Cai Y, Wang R, Jiang YY. 2009. Allicin enhances the oxidative damage effect of amphotericin B against Candida albicans. Int J Antimicrob Agents 33:258–263. doi: 10.1016/j.ijantimicag.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 60.Ramage G, Rajendran R, Sherry L, Williams C. 2012. Fungal biofilm resistance. Int J Microbiol 2012:528521. doi: 10.1155/2012/528521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nailis H, Vandenbosch D, Deforce D, Nelis HJ, Coenye T. 2010. Transcriptional response to fluconazole and amphotericin B in Candida albicans biofilms. Res Microbiol 161:284–292. doi: 10.1016/j.resmic.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 62.Yu L, Wei X, Ma M, Chen X, Xu S. 2012. Possible inhibitory molecular mechanism of farnesol on the development of fluconazole resistance in Candida albicans biofilm. Antimicrob Agents Chemother 56:770–775. doi: 10.1128/AAC.05290-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nes WD, Zhou W, Ganapathy K, Liu J, Vatsyayan R, Chamala S, Hernandez K, Miranda M. 2009. Sterol 24-C-methyltransferase: an enzymatic target for the disruption of ergosterol biosynthesis and homeostasis in Cryptococcus neoformans. Arch Biochem Biophys 481:210–218. doi: 10.1016/j.abb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 64.Chaffin LJ. 2008. Candida albicans cell wall proteins. Microbiol Mol Biol Rev 72:495–544. doi: 10.1128/MMBR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts, vol 28, no. 14. Approved standard, 3rd ed CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 66.Li DD, Zhao LX, Mylonakis E, Hu GH, Zou Y, Huang TK, Yan L, Wang Y, Jiang YY. 2014. In vitro and in vivo activities of pterostilbene against Candida albicans biofilms. Antimicrob Agents Chemother 58:2344–2355. doi: 10.1128/AAC.01583-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodríguez-Tudela JL, Cuencaestrella M, Díazguerra TM, Mellado E. 2001. Standardization of antifungal susceptibility variables for a semiautomated methodology. J Clin Microbiol 39:2513–2517. doi: 10.1128/JCM.39.7.2513-2517.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manavathu EK, Cutright JL, Loebenberg D, Chandrasekar PH. 2000. A comparative study of the in vitro susceptibilities of clinical and laboratory-selected resistant isolates of Aspergillus spp. to amphotericin B, itraconazole, voriconazole and posaconazole (SCH 56592). J Antimicrob Chemother 46:229–234. doi: 10.1093/jac/46.2.229. [DOI] [PubMed] [Google Scholar]
- 69.Pierce CG, Uppuluri P, Tristan AR, Wormley FL, Mowat E, Ramage G, Lopezribot JL. 2008. A simple and reproducible 96 well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing. Nat Protoc 3:1494–1500. doi: 10.1038/nprot.2008.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bruder-Nascimento A, Camargo CH, Mondelli AL, Sugizaki MF, Sadatsune T, Bagagli E. 2014. Candida species biofilm and Candida albicans ALS3 polymorphisms in clinical isolates. Braz J Microbiol 45:1371–1377. doi: 10.1590/S1517-83822014000400030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Monteiro DR, Silva S, Negri M, Gorup LF, de Camargo ER, Oliveira R, Barbosa DB, Henriques M. 2012. Silver nanoparticles: influence of stabilizing agent and diameter on antifungal activity against Candida albicans and Candida glabrata biofilms. Lett Appl Microbiol 54:383. doi: 10.1111/j.1472-765X.2012.03219.x. [DOI] [PubMed] [Google Scholar]
- 72.Mishra NN, Ali S, Shukla PK. 2014. Arachidonic acid affects biofilm formation and PGE2 level in Candida albicans and non-albicans species in presence of subinhibitory concentration of fluconazole and terbinafine. Braz J Infect Dis 18:287–293. doi: 10.1016/j.bjid.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Dhaheri RS, Douglas LJ. 2009. Apoptosis in Candida biofilms exposed to amphotericin B. J Med Microbiol 59:149–157. doi: 10.1099/jmm.0.015784-0. [DOI] [PubMed] [Google Scholar]
- 74.Zhao LX, Li DD, Hu DD, Hu GH, Yan L, Wang Y, Jiang YY. 2013. Effect of tetrandrine against Candida albicans biofilms. PLoS One 8:e79671. doi: 10.1371/journal.pone.0079671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Q, Gao L, Tao M, Chen Z, Yang X, Cao Y. 2016. Transcriptomics analysis of Candida albicans treated with Huanglian Jiedu Decoction using RNA-seq. Evid Based Complement Altern Med 2016:3198249. doi: 10.1155/2016/3198249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dhamgaye S, Bernard M, Lelandais G, Sismeiro O, Lemoine S, Coppée J-Y, Le Crom S, Prasad R, Devaux F. 2012. RNA sequencing revealed novel actors of the acquisition of drug resistance in Candida albicans. BMC Genomics 13:396. doi: 10.1186/1471-2164-13-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH. 2006. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 79.Sun L, Sun S, Cheng A, Wu X, Zhang Y, Lou H. 2009. In vitro activities of retigeric acid B alone and in combination with azole antifungal agents against Candida albicans. Antimicrob Agents Chemother 53:1586–1591. doi: 10.1128/AAC.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hornby JM, Nickerson KW. 2004. Enhanced production of farnesol by Candida albicans treated with four azoles. Antimicrob Agents Chemother 48:2305–2307. doi: 10.1128/AAC.48.6.2305-2307.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y, Qu L, Gong L, Sun L, Gong R, Si J. 2013. Targeting and eradicating hepatic cancer cells with a cancer-specific vector carrying the Buforin II gene. Cancer Biother Radiopharm 28:623. doi: 10.1089/cbr.2012.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.