

ABSTRACT
Clostridium bolteae, which belongs to the Clostridium clostridioforme complex, is a member of the human gut microbiota. Recent analysis of seven genomes of C. bolteae revealed the presence of an arr-like gene. Among these strains, only 90A7 was found to be resistant to rifampin in the absence of alteration of RpoB. Cloning of arr-cb from 90A7 in Escherichia coli combined with directed mutagenesis demonstrated that Arr-cb was functional but that a Q127→R variant present in 90A9 and 90B3 was inactive. Quantitative reverse transcription-PCR analysis indicated that arr-cb was silent in the four remaining strains because of defective transcription. Thus, two independent mechanisms can make the probably intrinsic arr-cb gene of C. bolteae cryptic.
KEYWORDS: rifampin ADP-ribosyltransferase, Clostridium bolteae, antibiotic resistance
INTRODUCTION
Rifampin belongs to the first-line drug treatment of tuberculosis, leprosy, and brucellosis. This antibiotic has been recommended as an alternative treatment for Borrelia burgdorferi infections and Legionnaires' disease. Rifampin use has expanded in combination therapy for the treatment of various infections, such as those due to Staphylococcus aureus, multiresistant Acinetobacter baumannii, and recurrent Clostridium difficile colitis (1, 2). It has also been used to prevent meningococcal disease in people exposed to meningococci (3). Rifampin binds in a pocket of the RNA polymerase β subunit deep within the DNA-RNA channel, blocking the exit tunnel from the growing RNA chain and transcription initiation (4). The most common mechanism of rifampin resistance is a nucleotide mutation within the rpoB gene, which encodes the β subunit of RNA polymerase (5). Decreased permeability of the membrane barriers or overexpression of efflux pumps represents a second mechanism (6). In addition, three enzymatic inactivation mechanisms attenuating the affinity of rifampin for the β subunit of RNA polymerase have been reported. Rifampin glycosylation and phosphorylation have been found initially in actinomyces (7–9), while ADP ribosylation is widely distributed on the basis of the presence of genes encoding predicted Arr enzymes in various pathogenic and nonpathogenic bacteria (10). Three proteins have been shown to be able to catalyze the ADP ribosylation of rifampin. The first, Arr-2 encoded by an integron cassette is the most famous representative and is widespread among Gram-negative pathogenic bacteria. The second, Arr-ms, confers intrinsic resistance to rifampin on the opportunistic pathogen Mycobacterium smegmatis (11), and the third, Arr-sc, is from environmental Streptomyces coelicolor (10).
Recent genomic comparison inside the Clostridium clostridioforme group has revealed the presence of an arr-like determinant in Clostridium bolteae (12). C. bolteae was previously reported as a member of the C. clostridioforme complex including C. clostridioforme (formally), Clostridium aldenense, Clostridium citroniae, and Clostridium hathewayi. A divergence of 3% in the 16S rRNA separates C. bolteae and C. clostridioforme, which are distinguishable by phenotypic tests (13). These bacteria are members of normal human gut microbiota but can cause intra-abdominal infections when the natural barrier is altered. Anaerobes of the gut microbiota represent a reservoir for antibiotic resistance determinants (14). The aim of this study was to evaluate the capacity of arr-cb of C. bolteae to confer rifampin resistance.
RESULTS AND DISCUSSION
Susceptibility of C. bolteae to rifampin.
C. bolteae 90A5, 90A9, 90B3, 90B7, 90B8, and ATCC BAA613 were very susceptible to rifampin (MICs, ≤0.006 μg/ml); meanwhile, 90A7 was resistant (MIC, >32 μg/ml) (Table 1).
TABLE 1.
Rifampin susceptibilities of C. bolteae strains and E. coli with and without arr-cb90A7
| Strain(s) | Rifampin MIC (μg/ml) |
|---|---|
| C. bolteae | |
| 90A7 | >32 |
| 90A5, 90A9, 90B3, 90B7, 90B8, BAA613 | ≤0.006 |
| E. coli | |
| TG1(pUC18) | 4 |
| TG1(pUC18Ωarr-cb90A7) | >32 |
| TG1(pUC18Ωarr-cb90A7/Q127→R) | 4 |
In silico analysis of rifampin resistance in C. bolteae.
To rule out the possibility that the rifampin resistance of 90A7 resulted from a mutation of the target, the RpoB sequence was compared to that of the susceptible C. bolteae strains included in this study. A ClustalW alignment of the deduced RpoB sequences showed that the β subunit of RNA polymerase of the seven C. bolteae strains differed, at most, by three amino acid residues located outside the four clusters involved in rifampin resistance (6). A single amino acid change (E359→K) distinguished 90A7 from 90A9 and 90B3. Involvement of this mutation in the rifampin resistance of 90A7 was unlikely, since this position has never been reported to be involved in resistance.
NCBI GenBank analysis including other C. bolteae genomes, such as that of CAG:59 strain WAL-14578, indicated an arr gene as part of the core genome (12), whereas it was not found in other species. The arr-cb gene encoded a 137-amino-acid protein that displayed similarity to Arr-2, Arr-ms, and Arr-sc (51, 57, and 55% identity, respectively) (Fig. 1).
FIG 1.
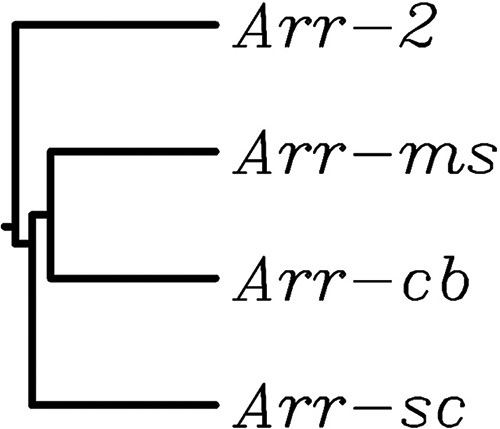
Phylogeny of Arr enzymes. The tree shown was constructed from sequences available in the Protein Data Bank by the neighbor-joining method. At each branch, the Arr protein is indicated. Accession numbers of proteins: Arr-2, WP_020442390; Arr-cb90A7, WP_002589901.1; Arr-ms, WP_063842202; Arr-sc, WP_011028626.
Four Arr-cb variants differing by a single amino acid change were detected among the seven C. bolteae strains (Fig. 2A). Two features were found in the regulatory region of the arr-cb genes. The common region was disrupted 36 bp upstream of the initiation codon by a 107-bp sequence in 90A7, 90A9, and 90B3 versus a 283-bp sequence in 90A5, 90B7, 90B8, and ATCC BAA613 (Fig. 2B).
FIG 2.

Differences in Arr-cb proteins and in the regulatory regions of arr-cb in C. bolteae. (A) Deduced amino acid sequences encoded by arr-cb90A7 and its variants. Accession numbers of proteins: Arr-cb90A7, WP_002589901.1; Arr-cb90A5 and Arr-cbBAA613, WP_002569919; Arr-cb90B8, WP_002578201; Arr-cb90A9 and Arr-cb90B3, WP_002578201. The ClustalW program was used to recognize the identical amino acids encoded by arr-cb, arr-2, arr-ms, and arr-sc, which are indicated by asterisks, the highly isofunctional amino acids are indicated by two dots, and those that are isofunctional are indicated by single dots. (B) The putative regulatory region of arr-cb. The conserved sequence in C. bolteae strains is represented by the bold line. The +1 transcription initiation site identified by the 5′ RACE technique is indicated. The initiation codon of arr-cb is in bold type. The ribosome binding site is underlined. The −35 and −10 motifs, separated by 17 bp, are in gray boxes; they are conserved compared to the canonical promoter of the σ70 factor of the RNA polymerase of E. coli (TGGACA and TATAAT). In 90A5, 90B7, 90B8, and BAA613, a change from 107 to 283 bp led to a greater distance between the +1 transcription site and the initiation codon (393 to 559 bp, respectively) and acquisition of a GC-rich stem-loop (located at the beginning of the 283-bp fragment), two factors that can contribute to transcription attenuation.
No DNA sequence related to genes involved in the mobilization or transfer was detected in the environment of arr-cb in the seven C. bolteae genomes analyzed, except an ISL3 family insertion sequence integrated just downstream from arr-cb in strain 90A7. This was a feature that differed from arr-2 and numerous arr-like genes (10).
Cloning of the arr gene of C. bolteae.
The arr-cb90A7 gene cloned into pUC18 conferred rifampin resistance on E. coli (Table 1). Deduced protein sequences from available C. bolteae genomes indicated the presence of four variants of Arr-cb differing by a single amino acid change (Fig. 2A). The lack of resistance in the hosts could be due to either an amino acid substitution leading to a nonfunctional protein or failure of transcription. The Q127→R change was likely the most significant since K17→R and D123→N consisted of substitutions between isofunctional residues. pUCΩarr-cb90A7 was mutagenized to introduce the Q127→R modification. The mutated gene was unable to confer rifampin resistance on E. coli (Table 1), confirming that position 127 is crucial for that activity. The structure of Arr-ms in complex with rifampin has been determined and compared with that of other ADP-ribosyltransferases, leading to identification of the residues contacting both NAD+ and rifampin (10). A three-dimensional model analysis based on the structure of Arr-ms suggested that residue 127 of Arr-cb is similar to that of other Arr proteins outside the active site (data not shown).
Transcriptional analysis of arr-cb.
The site of arr-cb transcription initiation was identified by the 5′ rapid amplification of cDNA ends (RACE) technique in 90A7 (Fig. 2B), showing a −35 motif (TTGACA) and a −10 motif (TAGAGT) separated by 17 bp according to consensus sequences recognized by the σ70 factor and binding of RNA polymerase (15). These promoters were separated from the start codon by 393 bp in 90A7, 90A9, and 90B3 versus 559 bp in 90A5, 90B7, 90B8, and ATCC BAA613 (Fig. 1). The 559-bp spacing sequence, which contained a GC-rich stem-loop sequence (TATCCGCCTTTGGCGGATA) may be responsible for transcription attenuation.
To quantify the influence of the two arr-cb regulatory regions, specific mRNA for arr-cb was measured by quantitative reverse transcription (qRT)-PCR in 90A7, 90B3, and 90A5. The expression of arr-cb in 90A7 and 90B3 was found to be similar and ca. 114-fold higher than that in 90A5. This result was in agreement with changes found in the regulatory region of the arr-cb gene (Fig. 2B).
Conclusions.
Arr-cb is a new member of the ADP-ribosyltransferase family modifying rifampin. This determinant, borne by the core genome of C. bolteae, was able to confer resistance; however, it was also found to be cryptic in certain members of the species because of either a gene mutation or a polymorphism in the regulatory region leading to transcription attenuation. The presence of an IS element near arr-cb in 90A7 raises the question of mobilization leading to horizontal transfer. Searching for arr-cb by PCR assay combined with bacterial identification by matrix-assisted laser desorption ionization–time of flight analysis would allow the detection of such an event.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. bolteae 90A5, 90A7, 90A9, 90B3, 90B7, and 90B8 were isolated in France from intra-abdominal infections, and their genomes were previously analyzed (12). Strain 90A7 was first identified as C. clostridioforme on the basis of ambiguity at the 16S rRNA level, but analysis of its complete genome enabled a formal correction of its identification. C. bolteae ATCC BAA613 was also included in this study. C. bolteae strains were cultured in Muller-Hinton (MH) agar supplemented with 5% defibrinated horse blood for antibiotic susceptibility tests or brain heart infusion (BHI) agar or BHI broth (Oxoid) for DNA or RNA extraction and incubated in an anaerobic chamber (atmosphere of 5% CO2, 5% H2, and 90% N2) at 37°C. E. coli TG1 was cultured under aerobic conditions at 37°C in MH agar or broth (Oxoid) supplemented with 100 μg/ml ampicillin for cloning experiments.
Antimicrobial susceptibility assays.
Testing for rifampin susceptibility was carried out with the Etest (bioMérieux). For E. coli harboring arr-cb, 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added to the growth medium to induce the expression of the cloned gene.
Cloning of the arrA7 gene.
BLAST software was used for gene and protein comparisons. The arr gene preceded by its ribosome binding site was amplified with primers Arr_F (5′ CCCCCGAATTCGAGGAGGTTTGACATGAGCAATTCAG) and Arr_R (5′ CCCCCAAGCTTATTCGACAGCTTTAATTCCCATC) and genomic DNA of C. bolteae strain A7 as a template. The resulting 447-bp fragment was cloned into vector pUC18 by placing the resistance gene under the control of the Plac promoter. The ligation product was introduced into E. coli strain TG1 by electrotransformation.
Site-directed mutagenesis of arrA7.
Site-directed mutagenesis was performed to demonstrate in E. coli that the unique substitution change between Arr-cb from 90A7 and that from 90B3 and 90A9 is crucial for resistance activity. Overlap extension PCR was performed as described by Zheng et al. (16) to introduce the replacement of glutamine with arginine at position 127 in Arr-cb90A7. Briefly, plasmid pUC18 containing arrA7 was used as a template for amplification by PCR with primers ArrQ127R_F (5′GGTATGCGCGATCATTTGAAACGGTTAGATGAGATGGGAATTAAA) and ArrQ127R_R (5′TTTAATTCCCATCTCATCTAACCGTTTCAAATGATCGCGCATACC). The purified PCR product was treated with the DpnI enzyme and used to transform E. coli TG1. One transformant examined by sequencing for the presence of the mutation was selected for further experiments.
Gene expression analysis.
The transcriptional expression of the arr gene from C. bolteae strains 90A7, 90B3, and 90A5 was determined by qRT-PCR. Overnight cultures of bacterial strains were cultured in BHI. Three-milliliter volumes of these cultures were centrifuged, and RNA extraction was performed with the RNeasy kit (Qiagen) in accordance with the manufacturer's instructions for Gram-positive bacteria. To remove genomic DNA contamination, the samples were treated with DNase I (Invitrogen) for 30 min at 37°C and then heat inactivated at 65°C for 5 min. A 1-μg sample of total RNA was used to synthesize cDNA with the SuperScript II Reverse Transcriptase kit (Invitrogen). For each qPCR, reactions were set up with 500 nM primers and 4 μl of the cDNA template (diluted 1:100). The primers used were arr90A7_qRT for (5′AGCAGCCATTTTTGTTGTG) and arr90A7_qRT rev (5′AAATGATCGCGCATACCTTG). All reactions were carried out in triplicate with at least two biological replicates and SsoAdvanced SYBR green Supermix (Bio-Rad). Relative expression was determined by the cycle threshold method on the Bio-Rad C1000 CFX96 real-time system (Bio-Rad). Target gene expression was measured by determining expression relative to that of the rpoB reference gene.
The transcriptional start site of arr-cb was determined by 5′ RACE with a 5′/3′ RACE kit (Roche). First-strand cDNA synthesis was performed with 1 μg of DNase-treated RNA and arr-cb-specific primer 2620 (5′ATACTTCCGGTGAATGTCCTTTCC). The dA-tailed cDNA was amplified by PCR with the oligo(dT) anchor primer provided by the manufacturer and arr-cb-specific primer 2621 (5′TCCGTTACATTTGGGTCGTCTTC). A nested PCR assay was performed with the PCR anchor primer and arr-cb-specific primer 2623 (5′TCTCCCGTTGGCTCCACAAC). We then directly sequenced the PCR products with primer 2626 (5′ACGCTTAAGTCTGCTTTGGTTC).
ACKNOWLEDGMENT
We sincerely thank Claudine Deloménie (Region Ile de France, IFR141 IPSIT) for technical assistance with qRT-PCR analysis.
REFERENCES
- 1.Forrest GN, Tamura K. 2010. Rifampin combination therapy for nonmycobacterial infections. Clin Microbiol Rev 23:14–34. doi: 10.1128/CMR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Connor JR, Galang MA, Sambol SP, Hecht DW, Vedantam G, Gerding DN, Johnson S. 2008. Rifampin and rifaximin resistance in clinical isolates of Clostridium difficile. Antimicrob Agents Chemother 52:2813–2817. doi: 10.1128/AAC.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zalmanovici Trestioreanu A, Fraser A, Gafter-Gvili A, Paul M, Leibovici L. 2011. Antibiotics for preventing meningococcal infections. Cochrane Database Syst Rev 8:CD004785. doi: 10.1002/14651858.CD004785.pub4. [DOI] [PubMed] [Google Scholar]
- 4.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912. doi: 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein BP. 2014. Resistance to rifampicin: a review. J Antibiot (Tokyo) 67:625–630. doi: 10.1038/ja.2014.107. [DOI] [PubMed] [Google Scholar]
- 6.Tupin A, Gualtieri M, Roquet-Banères F, Morichaud Z, Brodolin K, Leonetti J-P. 2010. Resistance to rifampicin: at the crossroads between ecological, genomic and medical concerns. Int J Antimicrob Agents 35:519–523. doi: 10.1016/j.ijantimicag.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 7.Yazawa K, Mikami Y, Maeda A, Akao M, Morisaki N, Iwasaki S. 1993. Inactivation of rifampin by Nocardia brasiliensis. Antimicrob Agents Chemother 37:1313–1317. doi: 10.1128/AAC.37.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spanogiannopoulos P, Thaker M, Koteva K, Waglechner N, Wright GD. 2012. Characterization of a rifampin-inactivating glycosyltransferase from a screen of environmental actinomycetes. Antimicrob Agents Chemother 56:5061–5069. doi: 10.1128/AAC.01166-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spanogiannopoulos P, Waglechner N, Koteva K, Wright GD. 2014. A rifamycin inactivating phosphotransferase family shared by environmental and pathogenic bacteria. Proc Natl Acad Sci U S A 111:7102–7107. doi: 10.1073/pnas.1402358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baysarowich J, Koteva K, Hughes DW, Ejim L, Griffiths E, Zhang K, Junop M, Wright GD. 2008. Rifamycin antibiotic resistance by ADP ribosylation: structure and diversity of Arr. Proc Natl Acad Sci U S A 105:4886–4891. doi: 10.1073/pnas.0711939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dabbs ER, Yazawa K, Mikami Y, Miyaji M, Morisaki N, Iwasaki S, Furihata K. 1995. Ribosylation by mycobacterial strains as a new mechanism of rifampin inactivation. Antimicrob Agents Chemother 39:1007–1009. doi: 10.1128/AAC.39.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dehoux P, Marvaud JC, Abouelleil A, Earl AM, Lambert T, Dauga C. 2016. Comparative genomics of Clostridium bolteae and Clostridium clostridioforme reveals species-specific genomic properties and numerous putative antibiotic resistance determinants. BMC Genomics 17:819. doi: 10.1186/s12864-016-3152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, Cai J, Hippe H, Farrow JA. 1994. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol 44:812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- 14.van Schaik W. 2015. The human gut resistome. Philos Trans R Soc Lond B Biol Sci. 370:20140087. doi: 10.1098/rstb.2014.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulligan ME, Hawley DK, Entriken R, McClure WR. 1984. Escherichia coli promoter sequences predict in vitro RNA polymerase selectivity. Nucleic Acids Res 12:789–800. doi: 10.1093/nar/12.1Part2.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng L, Baumann U, Reymond J-L. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]