

ABSTRACT
Malaria-related mortality has slowly decreased over the past decade; however, eradication of malaria requires the development of new antimalarial chemotherapies that target liver stages of the parasite and combat the emergence of drug resistance. The diminishing arsenal of anti-liver-stage compounds sparked our interest in reviving the old and previously abandoned compound menoctone. In support of these studies, we developed a new convergent synthesis method that was facile, required fewer steps, produced better yields, and utilized less expensive reagents than the previously published method. Menoctone proved to be highly potent against liver stages of Plasmodium berghei (50 percent inhibitory concentration [IC50] = 0.41 nM) and erythrocytic stages of Plasmodium falciparum (113 nM). We selected for resistance to menoctone and found M133I mutations in cytochrome b of both P. falciparum and P. berghei. The same mutation has been observed previously in atovaquone resistance, and we confirmed cross-resistance between menoctone and atovaquone in vitro (for P. falciparum) and in vivo (for P. berghei). Finally, we assessed the transmission potential of menoctone-resistant P. berghei and found that the M133I mutant parasites were readily transmitted from mouse to mosquitoes and back to mice. In each step, the M133I mutation in cytochrome b, inducing menoctone resistance, was confirmed. In summary, this study is the first to show the mechanism of resistance to menoctone and that menoctone and atovaquone resistance is transmissible through mosquitoes.
KEYWORDS: antimalarial agents, drug resistance mechanisms, menoctone
INTRODUCTION
Despite the decline in deaths caused by malaria in the last 10 years, the disease threats from infections with Plasmodium spp. still pose a significant global public health problem. In 2015 alone, the World Health Organization estimates that malaria caused 214 million clinical cases and 438,000 deaths (1). The increased prevalence of drug-resistant parasites and insecticide-resistant vectors continues to drive the need for new treatments for this disease. New drugs are urgently needed, and in some cases old compounds with proven antimalarial activity can serve as the basis for lead optimization of new potent antimalarial drugs (2). Menoctone, also known as 2-hydroxy-3-(8-cyclohexyloctyl)-1,4-naphthoquinone, was first synthesized by Fieser et al. in 1948 (3). Initial studies with menoctone and analogs thereof found that the drug reduced blood stage parasitemia but failed to prevent recrudescence at safe, nontoxic levels of drug administration (3). In 1965, Howland used rat liver mitochondrial preparations to model how menoctone and various naphthoquinones inhibited the respiratory chain between cytochromes b and c1 (35). Intriguingly, Skelton et al. showed that menoctone, when tested in vitro at concentrations of 10 to 25 nM, inhibited the NAD (NADH) peroxidase of Plasmodium berghei by 90% (5). Specific morphological changes, such as swelling and thickening of the mitochondria as well as reduplication of mitochondrial membranes, were observed in intraerythrocytic trophozoites of P. berghei-infected mice exposed to menoctone (6, 7). Peters et al. later reported the 50% effective dose (ED50) and ED90 against strain N of P. berghei as 0.7 and 1.5 mg/kg of body weight, respectively, and showed an effective causal prophylactic dose of 1 to 3 mg/kg against Plasmodium yoelii nigeriensis (4). Later studies with menoctone treatment of the preerythrocytic stages in rodent malaria models demonstrated efficacy 2.5 times greater than that of primaquine (8). All the previous studies led to menoctone progressing to clinical trials, but disappointingly it proved less effective against P. falciparum than antimalarials under development at the time. Thereafter, research on menoctone was discontinued due to the low curative activity and the potential for development of resistance reported in P. berghei. When administered daily at doses ranging from 6.25 to 25 mg/kg for five consecutive days, the cure rate for menoctone was low, as only 4 of 72 mice remained parasite free for 28 days (8). Since resistant parasites were readily selected for, Peters created a highly resistant line of P. berghei against menoctone through a serial technique (4). The menoctone-resistant line was not responsive to challenge by primaquine and cycloguanil; however, it remained sensitive to mefloquine, chloroquine, pyrimethamine, and sulfadoxine. When tested for transmission-blocking activity, menoctone showed no sporontocidal activity, as demonstrated by successful oocyst and sporozoite development (9).
Although menoctone is not currently used for malaria prevention or treatment, it is an effective treatment at low intravenous and intramuscular doses against Theileria parva, a parasitic protozoan that causes East Coast fever in cattle (10). Intramuscular doses were preferred over intravenous administration, and oral administration proved to be ineffective (11). The efficacy of menoctone against T. parva is quite remarkable, but the complexity of compound synthesis and the high manufacturing costs rendered menoctone unmarketable as theileriosis treatment (12).
Previous studies with menoctone did not reveal a mechanism of action, although the chemical structure of menoctone reveals a core scaffold similar to that of atovaquone (Fig. 1). Atovaquone-proguanil (AP) is a fixed-dose combination antimalarial that is currently used for prophylaxis as well as treatment of Plasmodium falciparum infection. Atovaquone affects the malaria parasite by binding to the quinol oxidation (Qo) site of cytochrome b (encoded by cytb) in the mitochondrial respiratory chain. Atovaquone-resistant isolates following atovaquone or AP treatment have been described (13–17). In vitro studies of atovaquone with P. falciparum have selected for point mutations at multiple sites in P. falciparum cytochrome b (encoded by pfcytb); however, the predominant clinical resistance mutation seen in patient isolates is Y268S. In contrast, in vivo studies with P. berghei have selected for mutations in pbcytb, specifically M133I, a mutation commonly seen with P. falciparum in vitro resistance selections (18, 19).
FIG 1.

Synthesis of menoctone. RT, room temperature.
Since menoctone is structurally similar to atovaquone and displays activity against liver stages of P. berghei (8), we hypothesize that menoctone has a target similar to that of atovaquone and may generate similar mutations in pfcytb that confer cross-resistance to atovaquone. In this study, we generated menoctone-resistant parasites in both P. berghei and P. falciparum and sequenced cytb to better understand the mechanism(s) of resistance to menoctone and potential for this compound to serve as a lead for antimalarial drug development.
RESULTS
Synthesis of menoctone.
Although first identified as an antimalarial by Fieser and coworkers in collaboration with the Sterling-Winthrop Research Institute in 1948 (20), it was not until 1969 that Lorenz patented a synthetic route for menoctone starting from 5-phenylvaleric acid (21). McHardy et al. reported that this synthesis had many shortcomings requiring costly raw materials, harsh reaction conditions, and some low-yielding synthetic steps (10). In an effort to design a facile synthetic route for menoctone, we developed a novel convergent approach involving modern and classical transformations (Fig. 1). Isochroman-1,4-dione (compound 1) was synthesized from 2-acetylbenzoic acid, which was first brominated and then cyclized. The copper-mediated alkane synthesis of 1-bromo-7-chloroheptane and allylmagnesium bromide afforded chloroalkene (compound 2) (22). Replacement of the chloride with iodide via a Finkelstein reaction afforded iodoalkene (compound 3), which was then treated with cyclohexylmagnesium bromide in another copper-mediated alkane synthesis to afford the allylcyclohexane (compound 4). Lemieux-Johnson oxidation of allyl compound 4 yielded aldehyde (compound 5). Condensation of aldehyde (compound 5) with isochroman-1,4-dione (compound 1) afforded 3-substituted isochroman-1,4-dione (compound 6), which was rearranged to menoctone using sodium methoxide in methanol (Fig. 1). This new synthetic route was facile, required fewer steps, produced better yields, and utilized less expensive reagents than the previously published method.
Efficacy of menoctone against exoerythrocytic and blood stage parasites.
Previous studies demonstrated that menoctone is efficacious against asexual blood stage as well as liver stage parasites, yet these studies were done in vivo before the advent of quantitative in vitro susceptibility assays. Therefore, we first profiled the activity of menoctone against asexual blood stages of P. falciparum in vitro. The 50 percent inhibitory concentration (IC50) of menoctone was 113 nM against P. falciparum W2, a clone that is resistant to chloroquine, pyrimethamine, and sulfadoxine. Next, we assessed the activity of menoctone against P. berghei liver stages infecting HepG2 hepatoma cells in vitro. In these assays, menoctone proved to be extremely efficacious (IC50 = 0.41 nM) and was approximately 3-fold more potent than atovaquone (Fig. 2).
FIG 2.
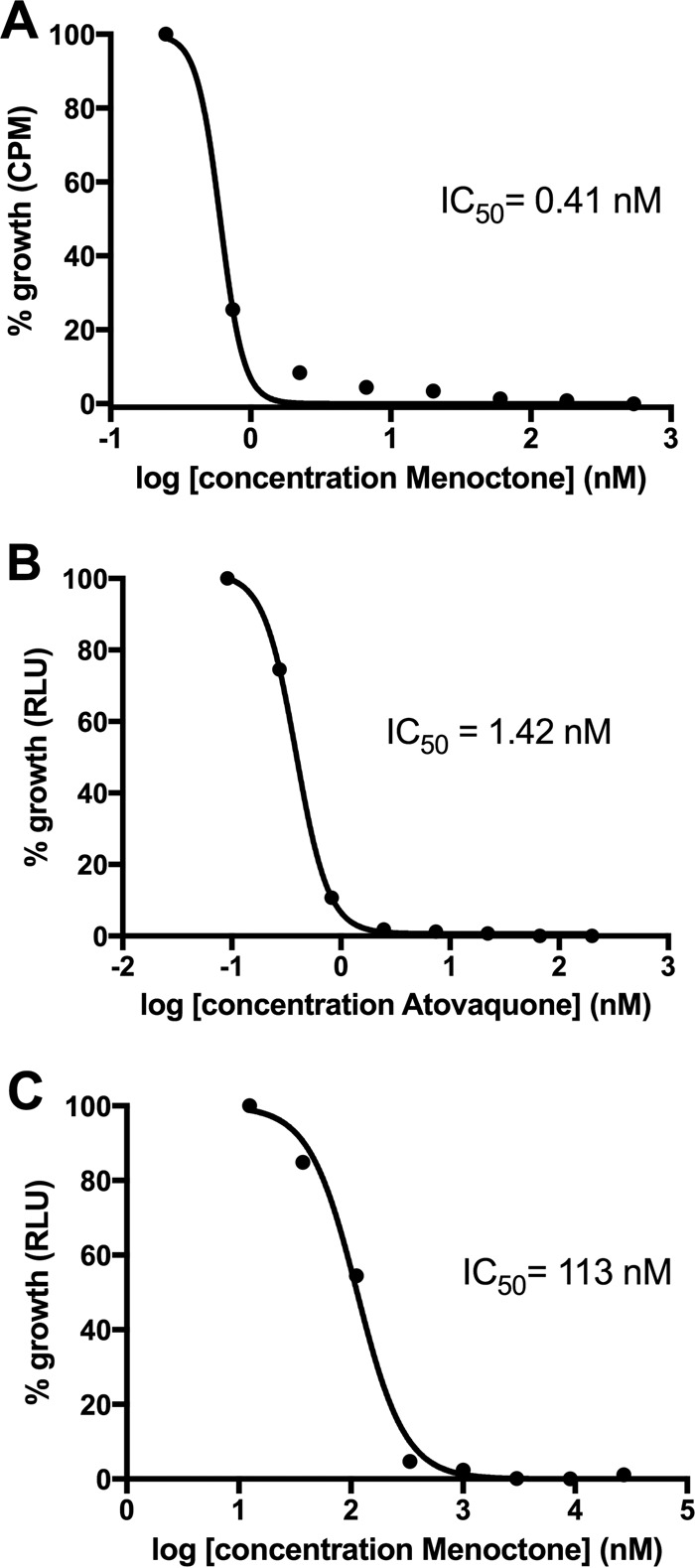
Menoctone and atovaquone efficacy against liver stages of P. berghei luc ANKA (A, B) and blood stages of P. falciparum in vitro (C). RLU, relative light units; CPM, corrected counts per minute.
Menoctone resistance selection in P. falciparum.
Based upon the structural similarity between menoctone and known mitochondrial electron chain inhibitors, we hypothesized that cytochrome bc1 is the site of action of menoctone. A common way to identify mechanisms of action is the selection of resistance to an inhibitor followed by sequencing of the putative target or whole genome. We used standard methods to select for resistance to menoctone in P. falciparum (W2). We grew 108 asexual erythrocytic stage parasites and exposed them to constant menoctone drug pressure (1.5 μM). Initial exposure of W2 to menoctone resulted in pyknotic forms and parasites outside erythrocytes after 48 h, followed by a gradual reduction of parasites observed in thin smears. However, on day 23 of menoctone exposure, parasites recovered to healthy morphology and reached 2% parasitemia, effects that were expanded further under continued menoctone drug pressure. We sequenced the parasites to identify mutations in cytochrome b and found that the menoctone-resistant progeny had a nonsynonymous single nucleotide polymorphism (SNP; G to C at nucleotide 399) that resulted in an M133I mutation (Table 1).
TABLE 1.
Single-dose treatments not sufficient for mutation selectiona
| P. berghei strain (stage)/treatment | Nucleotide | Mutation | Relevant genotype |
|---|---|---|---|
| luc ANKA (blood stages)/single-dose atovaquone or menoctone | 399 | None | WT |
| MEN-R (blood stages)/single-dose atovaquone or menoctone | 399 | G→T | M133I |
| luc ANKA (sporozoites) | 399 | None | WT |
| MEN-R (sporozoites) | 399 | G→T | M133I |
| MEN (MRA-414) (blood stages) | 397 | A→T | M133L |
| W2-MEN-R (blood stages) | 399 | G→C | M133I |
Sensitive and resistant parasites were challenged with either menoctone or atovaquone at different concentrations to observe whether single-dose treatment was favored. Single rounds of drug are not sufficient to select for M133I point mutation and resulted in WT genotypes at cytb, identical to that of the parent strain, P. berghei luc ANKA. All menoctone-resistant parasites demonstrated the M133I point mutation. Remarkably, when genotyped, sporozoites produced from menoctone-resistant blood stage parasites also carried the M133I point mutation, while sporozoites harvested from the sensitive parasite line displayed WT cytb.
Menoctone resistance selection in P. berghei.
In previous studies, Peters found that resistance to menoctone was selected rapidly in P. berghei in vivo (4); however, these parasites have not been fully characterized or genotyped. Therefore, we aimed to select for resistance in P. berghei luc ANKA by using a stepwise selection method. First, we infected mice with asexual erythrocytic stages and treated them with a single dose (3 mg/kg) of menoctone, and no decrease in parasitemia was observed posttreatment (Fig. 3A). Blood from these mice was then inoculated into two malaria-naive mice, and once patent infections ensued, we treated them with a single dose of menoctone (30 mg/kg). In these mice, parasitemia decreased to <1% 3 days posttreatment but recrudesced to approximately 30% 6 days later. Once again, blood from the recrudescent infections (day 6) was inoculated into malaria-naive mice, and the treatment dose was escalated to 300 mg/kg. As illustrated in Fig. 3C, parasitemia reduced rapidly after treatment yet recrudesced a few days later. Parasitemia in these mice was low, so we immediately subinoculated the infected blood into more malaria-naive mice and repeated the single high dose of menoctone (300 mg/kg). In these mice, parasitemia continued to increase at a high rate despite treatment, thus indicating selection of parasites highly resistant to menoctone. The menoctone-resistant P. berghei luc ANKA parasite is here referred to as P. berghei strain MEN-R (Fig. 3).
FIG 3.

Generation of menoctone resistance in P. berghei luc ANKA. Blood from recrudescent infections was serially passaged in malaria-naive mice following treatment with the drug at 3 (A), 30 (B), and 300 (C, D) mg/kg. Mice were treated on day 0 of the study, and 3 mice were used per group; data shown are the mean parasitemia for each group.
Confirmation of menoctone resistance in MEN-R-infected mice.
Next, we aimed to assess the efficacy of menoctone and atovaquone in the standard Thompson efficacy assay. In these studies, mice were inoculated with parasites, and after parasitemia reached ∼1%, mice were treated once a day for 3 days with drug. Efficacy was assessed by reduction of parasitemia on day 9 (1 day following drug treatment) as well as cure rate (survival to day 30 of the study). The doses used were 1, 3, and 10 mg/kg for atovaquone and 3, 30, and 300 mg/kg for menoctone. In these studies, we confirmed that atovaquone was more efficacious than menoctone against asexual blood stages of P. berghei luc ANKA as well as MEN-R. Interestingly, there was evidence of atovaquone cross-resistance in MEN-R, as evidenced by higher survival rates in MEN-R than in P. berghei luc ANKA following treatment with atovaquone (Fig. 4). These studies confirmed that MEN-R is highly resistant to menoctone in comparison to the response of the parental line P. berghei luc ANKA (Fig. 4).
FIG 4.

Survival curves of menoctone-sensitive (A, C) versus menoctone-resistant (B, D) P. berghei following treatment with menoctone or atovaquone. Menoctone offers some protection to P. berghei luc ANKA-infected mice (A) but demonstrates a marked decrease in protection against menoctone-resistant parasite P. berghei MEN-R (B). Atovaquone provided significantly more efficacy than menoctone (C); however, even at the highest dose of 10 mg/kg, atovaquone was not effective at preventing recrudescence of P. berghei MEN-R (D).
Menoctone resistance genotype in P. berghei.
Next we compared the cytochrome b genotypes of the parent P. berghei luc ANKA line and the menoctone-resistant MEN-R progeny. Sanger sequencing revealed the same M133I mutation in MEN-R as the one we observed in menoctone-resistant P. falciparum (W2 MEN-R) (see Table S1 in the supplemental material). The SNP identified (G to T, nucleotide [nt] 399) is located outside the quinol oxidation site (Table S1) and is the same SNP as the one observed in previous studies in which atovaquone was used to select for resistance (18). Although we did not have access to the parental line, we obtained the P. berghei MEN (MRA-414) line selected for resistance to menoctone by Peters (4) and sequenced the cytochrome b gene. Interestingly, in P. berghei MEN (MRA-414), we observed a A-to-T mutation at nucleotide position 397 that confers an M133L mutation (Table 1). These data suggest that menoctone resistance is conferred by similar mutations in cytochrome b in both P. berghei and P. falciparum and that these mutations confer cross-resistance with atovaquone.
A previous study suggested rapid selection of menoctone resistance in P. berghei; therefore, we next aimed to evaluate if M133I is readily selected in a single passage. In this study, we challenged parasites both sensitive and resistant to menoctone with single-dose exposures of different doses of menoctone or atovaquone. After one round of atovaquone or menoctone treatment, we found wild-type (WT) cytb in all the menoctone-sensitive parasites. All the P. berghei MEN-R parasites, regardless of the treatment, maintained the M133I mutation (Table 1).
Assessment of atovaquone cross-resistance on P. berghei MEN resistant parasites.
Upon initial treatment with 3 mg/kg of atovaquone, parasitemia decreased to <1% in P. berghei MEN-infected mice; however, parasites never completely cleared. Parasites recrudesced by day 6 posttreatment, and a 10% parasitemia was reestablished by day 9. Blood was passaged into naive BALB/c mice, and a higher dose of 30-mg/kg atovaquone decreased the parasitemia to <1%. Resistant parasite populations recrudesced faster, 4 days posttreatment, than after the first drug exposure, 9 days. Again, a higher dose of atovaquone (100 mg/kg) brought the parasitemia down to <1%, and parasites recrudesced by day 7 posttreatment. Successive treatments with a high dose of atovaquone (100 mg/kg) failed to clear parasites from peripheral blood (Fig. 5).
FIG 5.

Assessment of atovaquone cross-resistance with menoctone with P. berghei MEN (MRA-414). P. berghei MEN-infected mice were treated with atovaquone at 3 (A), 30 (B), and 100 (C, D) mg/kg on day 0 (n = 2 per group). Parasitemia decreased to <1% following atovaquone treatment after each treatment; however, atovaquone failed to completely clear all parasites after each round and low concentrations of parasites were observed. Blood from recrudescent-parasitemia mice treated with 100 mg/kg atovaquone (C) was inoculated into malaria-naive mice, which were treated with 100 mg/kg of the drug (D); these infections also confirmed resistance against atovaquone.
Mosquito transmission of menoctone resistance.
Recent studies have suggested that the resistance to atovaquone that is mediated by cytochrome b mutations is not transmissible through mosquitoes (23). We aimed to test that hypothesis with the P. berghei MEN-R parasites generated in this study. First, we inoculated mice intraperitoneally (i.p.) with P. berghei MEN-R- or P. berghei ANKA-infected erythrocytes, collected blood for cytochrome b genotyping, and then fed adult female Anopheles stephensi mosquitoes on the infected animals. On day 10, midgut dissections revealed that 32.6% of mosquito midguts were infected with oocysts of P. berghei MEN-R (Table 2). Mercurochrome staining of midguts showed healthy, round oocysts with definitive borders. Infected midguts contained approximately 130 oocysts and produced a qualitative score of +++ (see Materials and Methods). On day 21, salivary glands were dissected and yielded approximately 200,000 sporozoites of P. berghei MEN-R. In contrast, infections with P. berghei luc ANKA produced higher rates of oocyst development and sporozoites per mosquito than did P. berghei MEN-R. Some sporozoites were used to extract genomic DNA (gDNA), and the rest were used to inoculate intravenously (i.v.) five malaria-naive BALB/c mice. Three of the mice infected with P. berghei MEN-R were not treated with drug and produced patent blood stage infections 12 days postinfection. The two P. berghei MEN-R-infected mice that were treated with menoctone (300 mg/kg) demonstrated menoctone resistance as evidenced by continued growth of the parasite despite treatment with the highest dose of drug.
TABLE 2.
Menoctone resistance point mutation is capable of mosquito developmental stage transmission and produces next-generation blood stage infectiona
| P. berghei strain | Pre-mosquito feed cytochrome b genotype | Post-mosquito feed |
Mice after sporozoite inoculation |
||||
|---|---|---|---|---|---|---|---|
| No. of infected midguts/total no. (%) | Midgut infection scoreb | Sporozoites per mosquito | Cyt b genotype | No. of mice with patent infections/total no. (%) | Cytochrome b genotype | ||
| luc ANKA | WT | 37/50 (74) | ++ | 21,071 | WT | 3/3 (100) | WT |
| MEN-R | M133I | 19/54 (35.2) | +++ | 1,593 | M133I | 5/5 (100) | M133I |
A menoctone-resistant point mutation was identified in pre-mosquito-feed blood stage parasites. The blood meal produced positive oocysts and sporozoite infections in mosquitoes. Sporozoites produced next-generation blood stage infection. The M133I point mutation was fully maintained throughout the transmission in menoctone-resistant parasites, while the sensitive parasites demonstrated WT cytb.
Symbols: ++, 51 to 100 oocysts; +++, 101 to 150 oocysts.
As noted above, at each stage of the study we collected gDNA and then sequenced parasites to elucidate the cytochrome b genotype (Table 2). Genotype analysis confirmed that P. berghei MEN-R parasites used for mosquito infections had the M133I mutation, as did sporozoites dissected from the salivary glands of the mosquitoes day 21 postfeed. Furthermore, the M133I mutation was maintained with subsequent sporozoite challenge, and menoctone-resistant erythrocytic stages of P. berghei MEN-R maintained the resistance phenotype and M133I genotype. In simultaneous studies with the parental line P. berghei luc ANKA, we found the WT cytochrome b sequence throughout the transmission study. These studies demonstrate that menoctone and atovaquone resistance associated with M133I mutations in cytochrome b are transmissible through mosquitoes. The only differences observed were somewhat lower oocyst and sporozoite yields with P. berghei MEN-R than with P. berghei luc ANKA (Table 2). Mosquito transmission studies with menoctone-resistant P. falciparum were not possible since neither W2 nor W2 MEN-R produces viable gametocytes for mosquito infections.
DISCUSSION
In previous studies, we have shown that old, potent drug scaffolds can be optimized into clinical candidates (2). Menoctone represents an interesting case in which there was evidence of efficacy against multiple stages, although much was not known since the drug was abandoned shortly after its discovery. Early studies were limited mostly to in vivo evaluation in rodent models and a few biochemical analyses. In addition, the limited reports date back almost 70 years, laying down the initial groundwork for determining efficacy, yet the mechanism of action of menoctone against malaria was still not fully understood. Therefore, the major aim of this study was to better profile the parasitological efficacy of menoctone and to generate resistance among the parasites in order to better understand the mode of action. Limited availability of the compound prompted us to develop a new synthesis of menoctone using contemporary synthetic approaches. In comparison to the original menoctone synthesis (20, 21), our new synthetic route is convergent and requires fewer synthetic steps. At the synthetic scale at which we prepared menoctone, the overall yield of our synthetic approach also surpassed the overall yield of the original synthesis. Availability of menoctone then enabled us to generate encouraging efficacy data, especially the fact that menoctone is much more active against liver stages of P. berghei than the approved drug atovaquone. We also were successful at generating menoctone-resistant malaria parasites, which has allowed us to evaluate the possible mechanism(s) of action exhibited by menoctone on Plasmodium species.
In this study, we demonstrated the high potency of menoctone against liver and blood stage parasites. We tested menoctone in a quantitative dose-response assay for P. berghei liver stages and found that menoctone was approximately 3-fold more potent than atovaquone. Further investigation with human blood stage parasites demonstrated nanomolar activity against P. falciparum (W2). These results for a putative cytochrome b inhibitor are different from those of most other scaffolds that we have studied. Usually, the potency of those other drugs against blood and liver stages is more similar (in fold differences) to that of atovaquone. In contrast, menoctone is much more potent against liver stages than blood stages. Further studies to better understand the <1 nM potency of menoctone for liver stages are warranted.
In the course of generating the menoctone-resistant parasite line P. berghei MEN-R, we demonstrated that WT P. berghei under menoctone pressure generates a mutation at M133I, which is identical to results from previous reports of P. berghei under atovaquone pressure (18). Since the mutation is located directly outside the Qo site, the known binding site for atovaquone, we believe that menoctone shares the same mechanism of action as atovaquone, which consists of collapsing the mitochondrial membrane potential and disrupting pyrimidine biosynthesis, leading to parasite death (24). The stability of resistance in P. berghei MEN-R following passage in untreated mice has not been verified beyond three passages, but resistance remains stable following cryopreservation, which is similar to observations of P. berghei MEN after cryopreservation (4). P. berghei MEN demonstrated cross-resistance with atovaquone and when sequenced, selected for a point mutation allowing for parasite survival. Interestingly, P. berghei MEN selected for an adenine-to-thymine nucleotide change at nucleotide position 397. This confers an amino acid change from methionine to leucine. Although P. berghei MEN and P. berghei MEN-R differ in nucleotide mutations, it is interesting that the resultant amino acids in resistant parasites are isomers of one another. The data suggest that the M133I or M133L mutation inhibits the activity of menoctone or atovaquone to the binding site. Our results show, for the first time, the menoctone resistance genotype and suggest a potential mechanism of action of menoctone.
Furthermore, we used a modified Thompson model to demonstrate the in vivo activity of menoctone against sensitive and resistant erythrocytic stages of rodent malaria (25). A summary of the in vivo results is presented in Table S1 in the supplemental material. In the menoctone drug-administered groups, all mice inoculated with sensitive parasites exhibited low survival outcomes, and menoctone administration did not provide protection for mice inoculated with P. berghei MEN-R. Alternatively, atovaquone at 10 mg/kg, 3 mg/kg, and 1 mg/kg offered almost complete protection against sensitive P. berghei throughout the 30-day study. It is interesting that even though 12 of the 15 mice survived P. berghei luc ANKA infection after atovaquone administration, some of the surviving mice did present with low levels of parasitemia on day 30 of the study. Overall, atovaquone imparted better protection against both P. berghei luc ANKA and P. berghei MEN-R than did menoctone, and only high dosages of menoctone, 30 mg/kg and 300 mg/kg, respectively, offered low levels of protection against sensitive P. berghei infection (Fig. 4). These data bolster the initial findings by Fieser et al. that menoctone fails to prevent recrudescence of blood stage parasites (3). In addition, our study confirms that menoctone resistance confers cross-resistance to atovaquone, which is characterized by the healthy and uninterrupted growth of P. berghei MEN-R in the presence of atovaquone in vivo. When comparing menoctone and atovaquone administration against resistant P. berghei MEN-R, there was a slight delay in parasite recrudescence for atovaquone-treated mice, indicating perhaps that atovaquone clears more parasites than menoctone, thus requiring more time for the parasites remaining in peripheral circulation to recrudesce. This finding was expected, since the parasites were resistant to menoctone at high levels. Our results demonstrate that cross-resistance is clearly present between the two drugs and the mechanisms by which atovaquone and menoctone act on the parasite affect very similar targets, if not the same target.
Previously, Coleman et al. demonstrated menoctone failure to block transmission and successfully produced both oocysts and sporozoites in P. berghei and P. falciparum at a dose of 100 mg base drug/kg mouse body weight (9). More recently, a study has shown that atovaquone resistance cannot be transmitted via mosquitoes (23). This study reported that atovaquone drug pressure selects for the M133I cytb mutation in P. berghei, which is consistent with our data on menoctone drug pressure. Furthermore, they found that the P. berghei strains with M133I or other cytb mutations could not be transmitted through mosquitoes. In contrast, we found that P. berghei MEN-R was easily transmitted through mosquitoes. We confirmed that the M133I mutation existed in the prefeed P. berghei MEN-R-infected blood, which after being fed to Anopheles stephensi produced healthy oocysts and infective sporozoites in the mosquito. Genotyping of the resulting sporozoites uncovered the same M133I point mutation, which we then compared to its sensitive parent parasites displaying WT cytb. These sporozoites carrying the M133I mutation then successfully produced a blood stage infection in the next generation of mice. We confirmed that the mutation is fully transmittable from sporozoites to mice, since all sporozoite-infected mice produced a blood stage infection and carried the M133I mutation. To further demonstrate that the resistance phenotype was maintained, we administered high doses of menoctone to two of the sporozoite-inoculated mice to observe parasite growth; despite treatment with a high dose of menoctone (300 mg/kg), parasitemia continued to rise and healthy parasites propagated. Parasites from these mice also maintained the M133I mutation. We conclude that menoctone- and atovaquone-resistant parasites containing the M133I mutation in cytb are indeed transmittable by the mosquito vector. At present, we do not understand why P. berghei with M133I mutations were transmitted in our study but not in the recent study by Goodman et al. (23). The difference could be due to inherent transmissibility of the different P. berghei strains or mosquitoes used in the respective studies. Another possibility is that yet-to-be-identified mutations in the nuclear genome may contribute to resistance and transmission potential. The transmission potential of parasites resistant to mitochondrial electron chain inhibitors warrants additional studies.
The diminishing arsenal of anti-liver-stage compounds sparked our interest in reviving the old and previously abandoned compound menoctone. In summary, menoctone is a potent liver stage antimalarial with corresponding erythrocytic stage efficacy. The compound readily selects for cytochrome b M133I mutation in P. falciparum and P. berghei; this is the mutation often selected for under atovaquone drug pressure. This leads us to hypothesize that menoctone most likely targets the mitochondrial respiration chain of Plasmodium. In these studies, we have demonstrated for the first time that cytb mutations are transmittable from mouse to mosquito to mouse and have provided insight into a potential mechanism of action of menoctone. In addition, we developed a new synthesis method for menoctone that could be used in medicinal chemistry optimization of the scaffold. Further exploratory investigations would provide support toward whether menoctone may serve as a lead for novel antimalarial drug development.
MATERIALS AND METHODS
Parasites and animals.
We used P. falciparum (W2) that was cloned from an Indochina isolate by Oduola et al. in 1988 (26). Human O+ red blood cells and plasma for culturing P. falciparum were obtained from Interstate Blood Bank (Memphis, TN). All in vitro-cultured parasites were maintained according to previously published methods (27).
Menoctone-resistant P. berghei MEN (MRA-414) was obtained from the Malaria Research and Reference Reagent Resource Center (MR4); the line was originally generated by Wallace Peters et al. (28, 29). P. berghei 1052 Cl1 was obtained from C. J. Janse at Leiden University and was used for menoctone resistance selections. This line was generated from the reference ANKA clone c1115cyl and expresses both green fluorescent protein (GFP) and firefly luciferase (30). P. berghei 1052 Cl1 has two copies of the GFP-luc genes incorporated into its genome. One copy is regulated by a constitutively expressed eef1aα promoter, and the other is an ama1 promoter (30, 31).
All mice used in these experiments were female BALB/c mice (average weight, approximately 20 g) obtained from Harlan/EnVigo (Indianapolis, IN) and from Charles River (Wilmington, MA). Rodent malaria parasites were monitored via modified Giemsa-stained blood from tail vein smears, and parasitemia was determined via light microscopy. Parasites were passaged by removing blood via cardiac venipuncture and inoculating the intraperitoneal cavity of a naive mouse with parasite-infected blood. Mice were humanely euthanized when parasitemia reached 40% or the animal showed severe symptoms of malaria. This study was conducted in compliance with the Guide for the Care and Use of Laboratory Animals of the National Research Council for the National Academies, and the University of South Florida Institutional Animal Care and Use Committee approved the animal use protocols.
Drugs and chemicals.
Atovaquone was obtained from Sigma-Aldrich (St. Louis, MO). Menoctone was not commercially available at reasonable costs, and the published synthesis methods required improvement; therefore, a new production method was devised and is described here.
Susceptibility assessment of P. berghei liver stages in vitro. (i) Mosquito infections and sporozoite isolation.
Mice were infected using previously described methods above. When parasites reached >4% parasitemia, mice were anesthetized using ketamine (100 mg/kg) and xylazene (10 mg/kg) and placed on cartons containing 200 naive female 3- to 4-day-old Anopheles stephensi. Mosquitoes fed for 25 min and were maintained on 10% sucrose ad libitum inside environmental chambers at 22°C. On day 10 postexposure (PE), 50 mosquitoes were checked for infection through the visualization of oocysts on the midgut. On day 21 PE, infected salivary glands were dissected and sporozoites were isolated and counted.
(ii) Liver stage drug susceptibility assay.
HepG2 cells (17,500 cells/well) were seeded in black 384-well collagen-coated plates with optically clear bottoms (Becton Dickinson, Franklin Lakes, NJ). Cells were seeded using the Tecan Freedom Evo robotic system (Tecan Group Ltd., Männedorf, Switzerland) at 50 μl per well and were maintained at 37°C in 5% mixed-gas humidified incubators. Complete medium consisted of Eagle's minimum essential medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% l-glutamine. Mosquito dissections were harvested as described above, and 4,000 sporozoites were added to each well. Plates were incubated at 37°C for 3 h; then, medium was removed and serially diluted drugs that were previously prepared were added to the respective wells (32). After a 44-hour incubation period, we lysed the cells with 5 μl of Promega cell lysis reagent buffer and added 20 μl Promega luciferase assay reagent (Promega, Madison, WI). Luciferase activity in parasite lysates was quantified by using the PerkinElmer EnVision (PerkinElmer, Waltham, MA). An IC50 dose-response curve was generated from these data in GraphPad Prism (GraphPad Software, Inc., La Jolla, CA) to determine the efficacy of drugs against liver stage parasites.
P. falciparum asexual blood stage susceptibility.
Parasitemia was cultured and allowed to grow to approximately 5%, at which point it was synchronized at ring stages by using sorbitol as previously described (33). Drug dilutions were prepared in 96-well plates and were tested in duplicate using a 1:3 dilution series with a starting concentration of 10 μg/ml for menoctone or 62.5 ng/ml for atovaquone. Drugs were allowed to act on parasite development for 48 h. [3H]hypoxanthine was diluted 1:20 and added at 10 μl per well to the whole plate. Plates were placed into a specially contained incubator reserved only for hypoxanthine-treated plates for another 24 h. Plates were then removed and placed in a −80°C freezer for an additional 24 h. Cells were harvested using the PerkinElmer MicroBeta FilterMate-96 Harvester (PerkinElmer, Waltham, MA). We used the TopCount Luminometer (PerkinElmer, Waltham, MA) to read the plates and used the data to determine the IC50 (34).
Selection of menoctone resistance in P. falciparum in vitro and mutation detection. (i) W2 menoctone resistance selection.
Parasite line W2 was grown to 10% parasitemia, and then 108 parasites were seeded in fresh parasite culture medium containing ∼10× IC50 of menoctone (1.5 μM). Medium was changed with fresh menoctone, and blood smears were made twice per week, with fresh erythrocytes added as needed. Cultures were split 1:3 every 10 days and cultured until parasite recovery was observed (2% parasitemia) in the presence of menoctone.
(ii) DNA extraction.
Parasite genomic DNA extraction was performed with an initial lysis step in 1× phosphate-buffered saline (PBS) plus 0.01% saponin, followed by extraction using the Qiagen MiniBlood Prep kit according to the manufacturer's instructions. gDNA was assessed for concentration and purity on a NanoDrop spectrophotometer.
(iii) PCR amplification and sequencing of P. falciparum cytochrome b.
PCR primers were designed for amplification based on the annotated P. falciparum 3D7 cytochrome b gene sequence (MAL_MITO_3) on Plasmodb.org v27 as follows: Pf-cytb-PCRFOR, 5′-TGCCTAGACGTATTCCTG-3′, and Pf-cytb-PCRREV, 5′-GAAGCATCCATCTACAGC-3′. PCRs were amplified using the Phusion HS II High-Fidelity PCR master mix (ThermoFisher Scientific) with ∼20 ng parasite gDNA template according to the manufacturer's instructions with the following program: 98°C for the 30-s initial denaturation step, followed by 35 cycles of 98°C for 10 s, 54°C for 40 s, and 72°C for 30s and a final extension of 72°C for 7 min. PCR products were confirmed as a single, discrete band of 1,382 bp in length on a 1% agarose gel and then subsequently purified using the Qiagen PCR purification kit according to the manufacturer's instructions. Purified PCR products were prepared for Sanger sequencing service at Genewiz (Genewiz, South Plainfield, NJ) using the following sequencing primers: pf-cytb-SEQFOR1, 5′-GTGGAGGATATACTGTGAGTG-3′; pf-cytb-SEQFOR2, 5′-TACAGCTCCCAAGCAAAC-3′; pf-cytb-SEQREV1, 5′-GACATAACCAACGAAAGCAG-3′; and pf-cytb-SEQREV2, 5′-GTTCCGCTCAATACTCAG-3′. PCR primers Pf-cytb-PCRFOR and Pf-cytb-PCRREV were also used for sequencing purposes.
(iv) Analysis of P. falciparum cytochrome b sequences for mutation detection.
Sequence files were aligned to P. falciparum 3D7 cytochrome b sequence (MAL_MITO_3) using the open source software A Plasmid Editor (ApE v2.0.49). Trace files were checked at all mutant positions to validate any potential mutations found.
Generation of menoctone resistance in P. berghei and mutation detection.
We used methods similar to those that W. Peters used to generate a new menoctone-resistant parasite from the well-characterized P. berghei luc ANKA (4). Infected mice were treated with increasing levels of menoctone at doses of 3 mg/kg, 30 mg/kg, and 300 mg/kg. Recrudescent parasites were inoculated into malaria-naive mice, and subsequent treatment with higher doses of drug led to resistance at a dose of 300 mg/kg. Resistance was confirmed by the lack of response to subsequent inoculation of high doses of menoctone. A maintenance dose of 300 mg/kg was used to maintain a high level of parasite resistance.
DNA extraction.
Parasite genotyping was performed by sequencing cardiac venipuncture blood containing parasites at high parasitemia (>40%) that were for the majority schizonts. Murine leukocytes were removed by treating the blood with Ficoll-Paque Plus (GE Healthcare Life Sciences, Waltham, MA). Parasite genomic DNA extraction was then performed as described earlier.
PCR amplification and sequencing of P. berghei cytochrome b.
PCR primers were designed for amplification based on the annotated P. berghei cytochrome b gene sequence (PBANKA_MITO1900) on Plasmodb.org v27 as follows: Pb-cytb-PCRFOR, 5′-TGCCTAGACGTATTCCTG-3′, and Pb-cytb-PCRREV, 5′-GCTGAGCATGTTAACTCG-3′. PCRs were amplified similarly with the following program: 98°C for 30 s for the initial denaturation step, followed by 35 cycles of 98°C for 5 s, 54°C for 10 s, and 72°C for 39s and a final extension of 72°C for 7 min. PCR products were confirmed as a single, discrete band of 1,307 bp in length on a 1% agarose gel and then subsequently purified and sequenced as described earlier, using the following primers: pb-cytb-SEQFOR1, 5′-GTGGAGGATACACTGTTAGTG-3′; pb-cytb-SEQREV1, 5′-CATAACCTATAAAAGC-3′; and pb-cytb-SEQREV2, 5′-GTTTGCTTGGGAGCTGTA-3′. PCR primers Pb-cytb-PCRFOR and Pb-cytb-PCRREV were also used for sequencing purposes.
Analysis of P. berghei cytochrome b sequences for mutation detection.
Sequence files were aligned to the P. berghei cytochrome b gene sequence (PBANKA_MITO1900) as described previously.
In vivo antimalarial efficacy against blood stages of the P. berghei life cycle (modified Thompson test).
In brief, five mice were randomly assigned to each dosage group. P. berghei luc ANKA and P. berghei MEN-R were inoculated at 2 × 106 parasites per mouse. Menoctone was administered orally at 3 mg/kg, 30 mg/kg, and 300 mg/kg, and atovaquone was administered at 1 mg/kg, 3 mg/kg, and 10 mg/kg. Both drugs were suspended in polyethylene glycol 400 (PEG 400; Sigma-Aldrich, St. Louis, MO) and administered via oral gavage at 100 μl per mouse. Dosing by oral gavage occurred once daily for 3 days on days 6 to 8. Blood films were made on days 3, 5, 6, 9, 12, 15, 18, 21, 24, 27, and 30 postinfection from tail vein blood to monitor parasite recrudescence.
Elucidation of mutation selection by atovaquone or menoctone.
Sensitive and resistant parasites were inoculated into mice and challenged with either atovaquone or menoctone at different concentrations. Atovaquone was administered to mice at dosages of 1 mg/kg, 3 mg/kg, and 10 mg/kg, and menoctone was given at 3 mg/kg, 30 mg/kg, and 300 mg/kg. Parasitized blood was then removed and sequenced for cytb mutations.
P. berghei MEN (MRA-414) assessment of atovaquone cross-resistance.
Mice were infected with P. berghei MEN, and parasitemia was allowed to grow to >4%, at which point the parasites were challenged with atovaquone at 3 mg/kg, 30 mg/kg, and 100 mg/kg. Subsequent administration of atovaquone at 100 mg/kg was used as confirmation of atovaquone resistance.
Transmission of menoctone-resistant P. berghei to mosquitoes.
Resistant parasites were passaged and maintained using the methods described above. Once the parasitemia reached 3 to 5%, the mice were anesthetized with a solution of xylazene (10 mg/kg) and ketamine (100 mg/kg) and then placed on cartons of 200 3- to 4-day-old, naive female Anopheles stephensi mosquitoes. Mosquitoes were allowed to feed for approximately 25 min and maintained on 10% sucrose inside environmental chambers at 22°C. On day 10 PE, 50 mosquitoes were checked for infection through the visualization of oocysts on the midgut wall. Oocysts were scored based on two different characteristics: the percentage of midguts infected with oocysts and the quality of infection. The percentage of midguts infected was calculated by dividing the number of infected midguts by the total number of midguts dissected and then multiplying the result by 100. Each midgut was also graded on the following scale to determine the quality of infection: +, 0 to 50 oocysts; ++, 51 to 100 oocysts; +++, 101 to 150 oocysts; ++++, 151 to 200 oocysts; +++++, greater than 200 oocysts. On day 21 PE, the salivary glands were dissected from mosquitoes to determine the presence of sporozoites.
P. berghei luc menoctone-resistant sporozoite liver and blood stage infection.
On day 21 PE, we harvested sporozoites and infected five female mice with 10,000 sporozoites per mouse via tail vein injection. Once parasites were visualized in peripheral blood smears, two of the five mice were treated with 300 mg/kg of menoctone to confirm parasite resistance, while the remaining three were kept as controls. Cardiac venipuncture was used to collect blood from all five mice, and leukocytes were removed prior to parasite genotyping.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chris Janse for providing the transgenic P. berghei ANKA luc line and Steven Maher for comments on the manuscript.
This work was funded by the National Institutes of Health (R01GM097118 and R21AI109530).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00689-17.
REFERENCES
- 1.WHO. April 2016. Malaria: fact sheet. http://www.who.int/mediacentre/factsheets/fs094/en/ Accessed 8 April 2016.
- 2.Monastyrskyi A, Kyle DE, Manetsch R. 2014. 4(1H)-pyridone and 4(1H)-quinolone derivatives as antimalarials with erythrocytic, exoerythrocytic, and transmission blocking activities. Curr Top Med Chem 14:1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fieser LF, Leffler MT. 1948. Naphthoquinone antimalarials. I. General survey. J Am Chem Soc 70:3151–3155. doi: 10.1021/ja01190a001. [DOI] [PubMed] [Google Scholar]
- 4.Peters W. 1987. Chemotherapy and drug resistance in malaria, vol 1 Academic Press, London, England. [Google Scholar]
- 5.Skelton FS, Pardini RS, Heidker JC, Folkers K. 1968. Inhibition of coenzyme Q systems by chloroquine and other antimalarials. J Am Chem Soc 90:5334–5336. doi: 10.1021/ja01021a084. [DOI] [PubMed] [Google Scholar]
- 6.Howells RE, Peters W, Fullard J. 1970. The chemotherapy of rodent malaria. 13. Fine structural changes observed in the eryhrocytic stages of Plasmodium berghei berghei following exposure to primaquine and menoctone. Ann Trop Med Parasitol 64:203–207. [PubMed] [Google Scholar]
- 7.Raether W, Mehlhorn H. 1984. Action of a new floxacrine derivative (S 82 5455) on asexual stages of Plasmodium berghei: a light and electron microscopical study. Zentralbl Bakteriol Mikrobiol Hyg A 256:335–341. [PubMed] [Google Scholar]
- 8.Berberian DA, Slighter RG, Freele HW. 1968. Causal prophylactic effect of menoctone (a new hydroxynaphthoquinone) against sporozoite-induced Plasmodium berghei infection in mice. J Parasitol 54:1181–1189. doi: 10.2307/3276988. [DOI] [PubMed] [Google Scholar]
- 9.Coleman R, Nath A, Schneider I, Song G, Klein T, Milhous W. 1994. Prevention of sporogony of Plasmodium falciparum and P. berghei in Anopheles stephensi mosquitoes by transmission-blocking antimalarials. Am J Trop Med Hyg 50:646–653. doi: 10.4269/ajtmh.1994.50.646. [DOI] [PubMed] [Google Scholar]
- 10.McHardy N, Haigh AJ, Dolan TT. 1976. Chemotherapy of Theileria parva infection. Nature 261:698–699. doi: 10.1038/261698a0. [DOI] [PubMed] [Google Scholar]
- 11.Kuttler K, Kreier J. 1986. Trypanosomiasis, babeiosis, theileriosis and anaplasmosis. Plenum Press, New York, NY. [Google Scholar]
- 12.Aspock H, Behr C, Combes C, Daugschies J, De Bont G, Dobler G, Dubremetz J, Freeman J, Frenkel J, Gessner A, Gustafsson M, Haas W, Hanel H, Hansen O, Harder A, Julsing M, Mehlhorn H, Pereira Da Silva L, Raether W, Reiter-Owona I, Richter D, Rollinghoff M, Schaub G, Schnieder T, Seitz H, Smulian A, Spielman A, Spindler K, Taraschewski H, Tielens A, Turberg A, Vercruysse J, Walldorf V, Wernsdorfer W. 2008. Theileriacidal drugs, p 1366–1367. In Mehlhorn H. (ed), Encyclopedia of parasitology, 3rd ed, vol 2 Springer, New York, NY. [Google Scholar]
- 13.Musset L, Pradines B, Parzy D, Durand R, Bigot P, Le Bras J. 2006. Apparent absence of atovaquone/proguanil resistance in 477 Plasmodium falciparum isolates from untreated French travellers. J Antimicrob Chemother 57:110–115. doi: 10.1093/jac/dki420. [DOI] [PubMed] [Google Scholar]
- 14.Kuhn S, Gill MJ, Kain KC. 2005. Emergence of atovaquone-proguanil resistance during treatment of Plasmodium falciparum malaria acquired by a non-immune north American traveller to west Africa. Am J Trop Med Hyg 72:407–409. [PubMed] [Google Scholar]
- 15.Fivelman QL, Butcher GA, Adagu IS, Warhurst DC, Pasvol G. 2002. Malarone treatment failure and in vitro confirmation of resistance of Plasmodium falciparum isolate from Lagos, Nigeria. Malar J 1:1. doi: 10.1186/1475-2875-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.David KP, Alifrangis M, Salanti A, Vestergaard LS, Ronn A, Bygbjerg IB. 2003. Atovaquone/proguanil resistance in Africa: a case report. Scand J Infect Dis 35:897–898. doi: 10.1080/00365540310016862. [DOI] [PubMed] [Google Scholar]
- 17.Looareesuwan S, Viravan C, Webster HK, Kyle DE, Hutchinson DB, Canfield CJ. 1996. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am J Trop Med Hyg 54:62–66. doi: 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- 18.Syafruddin D, Siregar JE, Marzuki S. 1999. Mutations in the cytochrome b gene of Plasmodium berghei conferring resistance to atovaquone. Mol Biochem Parasitol 104:185–194. doi: 10.1016/S0166-6851(99)00148-6. [DOI] [PubMed] [Google Scholar]
- 19.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother 44:2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fieser LF, Berliner E, Bondhus FJ, Chang FC, Dauben WG, Ettlinger MG, Fawaz G, Fields M, Heidelberger C, Heymann H, Vaughan WR, Wilson AG, Wilson E, Wu M, Leffler MT, Hamlin KE, Matson EJ, Moore EE, Moore MB, Zaugg HE. 1948. Naphthoquinone antimalarials. IV-XI. Synthesis. J Am Chem Soc 70:3174–3175.18891815 [Google Scholar]
- 21.Lorenz RR. October 1969. Process of producing certain 1,4-naphthoquinones. US patent 3,471,525.
- 22.Kennedy AJ, Mathews TP, Kharel Y, Field SD, Moyer ML, East JE, Houck JD, Lynch KR, Macdonald TL. 2011. Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J Med Chem 54:3524–3548. doi: 10.1021/jm2001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman CD, Siregar JE, Mollard V, Vega-Rodriguez J, Syafruddin D, Matsuoka H, Matsuzaki M, Toyama T, Sturm A, Cozijnsen A, Jacobs-Lorena M, Kita K, Marzuki S, McFadden GI. 2016. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 352:349–353. doi: 10.1126/science.aad9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baggish AL, Hill DR. 2002. Antiparasitic agent atovaquone. Antimicrob Agents Chemother 46:1163–1173. doi: 10.1128/AAC.46.5.1163-1173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson PE, Bayles A, Olszewski B, Waitz J. 1965. Quinine-resistant Plasmodium berghei in mice. Science 148:1240–1241. doi: 10.1126/science.148.3674.1240. [DOI] [PubMed] [Google Scholar]
- 26.Oduola AM, Milhous WK, Weatherly NF, Bowdre JH, Desjardins RE. 1988. Plasmodium falciparum: induction of resistance to mefloquine in cloned strains by continuous drug exposure in vitro. Exp Parasitol 67:354–360. doi: 10.1016/0014-4894(88)90082-3. [DOI] [PubMed] [Google Scholar]
- 27.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 28.Peters W, Li ZL, Robinson BL, Warhurst DC. 1986. The chemotherapy of rodent malaria, XL. The action of artemisinin and related sesquiterpenes. Ann Trop Med Parasitol 80:483–489. [DOI] [PubMed] [Google Scholar]
- 29.Peters W, Robinson B. 1999. Malaria, p 757–774. In Sande MA, Zak O (ed), Handbook of animal models in antimicrobial chemotherapy. Academic Press, San Diego, CA. [Google Scholar]
- 30.Janse CJ, Ramesar J, Waters AP. 2006. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc 1:346–356. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- 31.Franke-Fayard B, Trueman H, Ramesar J, Mendoza J, Van der Keur M, Van der Linden R, Sinden RE, Waters AP, Janse CJ. 2004. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol 137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Derbyshire ER, Prudencio M, Mota MM, Clardy J. 2012. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci U S A 109:8511–8516. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 34.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16:710–718. doi: 10.1128/AAC.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howland JL. 1965. Inhibition of mitochondrial succinate oxidation by alkyl hydroxy napthoquinones. Biochem Biophys Acta 1–5:205–213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.