

ABSTRACT
Despite continuous efforts to control cariogenic dental biofilms, very few effective antimicrobial treatments exist. In this study, we characterized the activity of the novel synthetic cyclic lipopeptide 4 (CLP-4), derived from fusaricidin, against the cariogenic pathogen Streptococcus mutans UA159. We determined CLP-4's MIC, minimum bactericidal concentration (MBC), and spontaneous resistance frequency, and we performed time-kill assays. Additionally, we assessed CLP-4's potential to inhibit biofilm formation and eradicate preformed biofilms. Our results demonstrate that CLP-4 has strong antibacterial activity in vitro and is a potent bactericidal agent with low spontaneous resistance frequency. At a low concentration of 5 μg/ml, CLP-4 completely inhibited S. mutans UA159 biofilm formation, and at 50 μg/ml, it reduced the viability of established biofilms by >99.99%. We also assessed CLP-4's cytotoxicity and stability against proteolytic digestion. CLP-4 withstood trypsin or chymotrypsin digestion even after treatment for 24 h, and our toxicity studies showed that CLP-4 effective concentrations had negligible effects on hemolysis and the viability of human oral fibroblasts. In summary, our findings showed that CLP-4 is a potent antibacterial and antibiofilm agent with remarkable stability and low nonspecific cytotoxicity. Hence, CLP-4 is a promising novel antimicrobial peptide with potential for clinical application in the prevention and treatment of dental caries.
KEYWORDS: Streptococcus mutans, antimicrobial peptide, cyclic lipopeptide, dental biofilms, dental plaque
INTRODUCTION
Dental caries is the most prevalent bacterial infection in humans. It affects up to 90% of school children and nearly 100% of adults worldwide (1). The main etiological factor of dental caries is the colonization and biofilm formation of pathogenic microorganisms on tooth surfaces (2). When the host's diet is rich in sugars, the microbial composition of dental biofilms is enriched in acidogenic and aciduric bacteria, such as the cariogenic oral pathogen Streptococcus mutans (3, 4). This Gram-positive bacterium is a persistent colonizer of tooth surfaces and interacts with other species of oral bacteria to form complex biofilm communities known as dental plaques (2, 5). S. mutans ferments a variety of different carbohydrates (6). Tooth decay occurs as a result of acid production, the by-product of carbohydrate metabolism, which leads to the demineralization of tooth enamel (3).
Dental plaques are polymicrobial biofilms that are difficult to eradicate. Phylogenetic studies estimate that more than 600 different phylotypes comprise this complex microbial community, although fewer than 100 species are typically found per individual (7, 8). These biofilms are encased in a matrix of extracellular polymers that confer increased recalcitrance to mechanical abrasions and antimicrobial treatments. Poor drug penetration, lowered growth rate, activation of general stress responses, and multidrug resistance and/or tolerance are just a few of the myriad features that protect dental biofilms (9–12). Regular dental hygiene and low-sugar diets decrease dental plaque build-up, thus preventing caries. However, once caries progresses, very few antimicrobial treatments exist. Currently available treatments include commonly used antibiotics (13), prophylactic formulations of chlorhexidine (14), triclosan (15), and cetylpyridinium chloride (16). Clinical studies have shown that these agents are capable of reducing and suppressing the population of cariogenic bacteria found in plaques, as well as oral mucosa, until they recolonize a few weeks after treatment (17, 18). However, these antimicrobial agents have several disadvantages, which include the development of bacterial resistance (19), pronounced cytotoxicity in vitro (20–22), and reduced effectiveness due to poor solubility (23). Other side effects include loss of taste and tooth staining (24). Several antibiotics have been evaluated for the prevention and disruption of dental biofilms, but few have proven to be effective (25, 26).
In the wake of widespread bacterial resistance to common antibiotics, antimicrobial peptides (AMPs) have emerged as promising lead compounds for the development of novel antibiotics. They are short peptides composed of 3 to 50 amino acids and occur naturally in all domains of life as defense mechanisms (27, 28). AMPs are effective not only against planktonic bacteria but also against bacterial biofilms (25, 29, 30). This is especially relevant to the recent paradigm shift in modern medicine, which recognizes the importance of biofilms in chronic bacterial infections and their resistance and/or tolerance to commonly used antibiotics. Although the specific mechanism of action of AMPs varies based on amino acid composition and physicochemical properties, most contain positively charged residues that allow them to interact with the negatively charged bacterial membranes (31–33). This can lead to a fatal membrane depolarization, membrane damage, formation of pores that cause leakage of cell contents, peptide internalization, and damage to intracellular targets, or a combination of these effects (31, 32).
Fusaricidins are AMPs isolated from Paenibacillus polymyxa (34–37). They are cyclic lipodepsipeptides characterized by a common macrocyclic ring consisting of six amino acids, three of which (Thr1, d-aThr4, and d-Ala5) are conserved throughout the family. A 15-guanidino-3-hydroxypentadecanoic acid tail is attached to the N terminus of Thr1 by an amide bond (38). Although their mechanism of action remains to be elucidated, these antibiotics have potent in vitro antimicrobial activities against a variety of Gram-positive bacteria (35, 37, 39). In previous studies, Bionda et al. synthesized and tested several cyclic lipopeptides (29, 34, 38) derived from the naturally occurring fusaricidin A/LI-F04a. These synthetic analogues showed significant advantages over naturally occurring fusaricidins, such as improved stability and lower nonspecific cytotoxicity against human cells (38). In this study, we have tested the stability and activity of cyclic lipopeptide 4 (CLP-4) in vitro against planktonic cultures and biofilms of S. mutans, as well as the safety of CLP-4 for human cells.
RESULTS
Peptide synthesis.
CLP-4 was synthesized using the standard 9-fluorenylmethoxy carbonyl (Fmoc) methodology and purified by preparative reversed-phase high-performance liquid chromatography (RP-HPLC). The final purity of synthesized CLP-4 was confirmed by analytical RP-HPLC and matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) and was ≥95%. CLP-4 was used in this study against the cariogenic pathogen S. mutans UA159. The chemical structure of CLP-4 is shown in Fig. 1. CLP-4 has a molecular mass of 1,023.28 Da and a lipid tail consisting of 12-guanidinododecanoic acid instead of the naturally occurring 15-guanidino-3-hydroxypenta-decanoic acid, and its peptide sequence is Dap1-d-Leu2-Trp3-d-Lys4-d-Asn5-d-Arg6.
FIG 1.
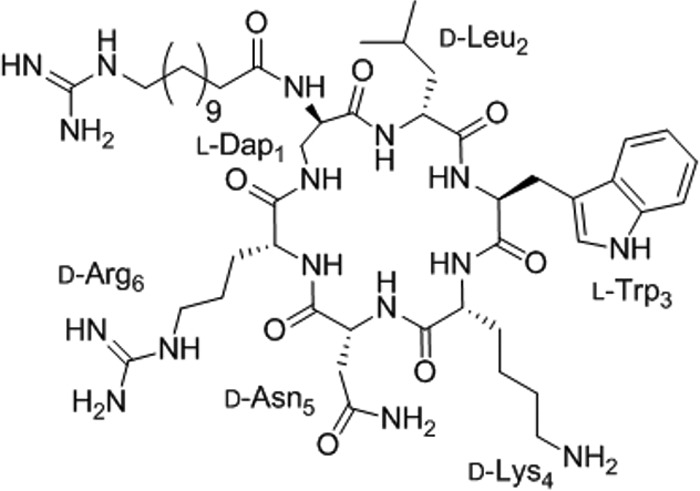
Chemical structure of CLP-4.
CLP-4 is stable against proteolytic degradation.
In the oral cavity, AMPs are exposed to a number of proteases originating from saliva, white blood cells, and bacteria from dental plaques (40, 41). Therefore, peptide stability represents one of the key characteristics required for its therapeutic potential. To investigate the stability of CLP-4, we incubated intact CLP-4 with either trypsin or chymotrypsin and monitored its proteolysis. Samples were taken at 0, 1, 2, 4, 18, and 24 h and analyzed using analytical RP-HPLC. Approximately 82% and 94% of CLP-4 remained intact after incubation with trypsin and chymotrypsin, respectively (Fig. 2), indicating significant resistance toward proteolytic degradation.
FIG 2.

Stability of CLP-4 against proteolytic degradation. One milligram of CLP-4 was digested with TPCK trypsin or chymotrypsin for 1, 2, 4, 18, and 24 h and compared to untreated CLP-4 (control). Fractions were subjected to analytical RP-HPLC to measure the percent recovery of intact CLP-4. The average of the results from three independent experiments is shown. Error bars represent standard deviations. *, P < 0.05 compared to control.
CLP-4 is effective against S. mutans.
In order to evaluate the antibacterial properties of CLP-4, we first examined the MIC on S. mutans UA159 planktonic cultures. Two different concentrations of starting inoculum were used in these experiments: 6 × 105 CFU/ml, according to the recommendations of the Clinical and Laboratory Standards Institute (CLSI), or 2 × 107 CFU/ml, which was the optimal starting inoculum used in our biofilm experiments. The MIC of CLP-4 was determined as the lowest concentration at which UA159 growth was completely inhibited after treatment for 24 h. Commonly used antibiotics and antiseptics, such as erythromycin and chlorhexidine, were included for comparison purposes. The results are presented in Table 1 and showed that CLP-4 effectively inhibited the growth of planktonic UA159 culture. The MIC for the culture with the starting inoculum of 6 × 105 CFU/ml was 2.8 μg/ml, while the MIC for the culture with the starting inoculum of 2 × 107 CFU/ml was 5 μg/ml. The MIC of CLP-4 was comparable to that of chlorhexidine, which was 1.25 μg/ml for both inocula. Erythromycin displayed a stronger potency, with an MIC of 16 ng/ml for the lower inoculum concentration and 62 ng/ml for the higher inoculum concentration. Additionally, we evaluated the susceptibilities of different serotypes and clinical isolates of S. mutans to CLP-4. The results are presented in Table 2 and demonstrate similar MICs (2 to 3 μg/ml). These results showed that CLP-4 is a promising agent that can effectively inhibit planktonic growth of S. mutans.
TABLE 1.
In vitro susceptibilities of planktonic S. mutans UA159
| Antimicrobial agent | MIC and MBC (μg/ml) by inoculum density of: |
|||
|---|---|---|---|---|
| 6 × 105 CFU/ml |
2 × 107 CFU/ml |
|||
| MIC | MBC | MIC | MBC | |
| CLP-4 | 2.8 | 6 | 5 | 20 |
| Erythromycin | 0.016 | 0.6 | 0.062 | 1 |
| Chlorhexidine dihydrochloride | 1.25 | 3.5 | 1.25 | 5 |
TABLE 2.
S. mutans strains used in this study and their in vitro susceptibilities to CLP-4
CLP-4 possesses potent bactericidal properties against actively dividing cells.
We also examined the bactericidal properties of CLP-4 by measuring the minimum bactericidal concentrations (MBCs) of UA159 planktonic cultures. Cell suspensions were adjusted to a final density of 6 × 105 CFU/ml or 2 × 107 CFU/ml and treated with CLP-4 at concentrations of 1 to 20 μg/ml. Cell viability was measured by determining the CFU per milliliter after treatment for 24 h. The MBC was defined as the minimum concentration required to kill at least 99.99% of bacteria in the culture. The results are presented in Table 1 and show that CLP-4 exhibited strong bactericidal activities, with MBCs of 6 μg/ml (for culture density of 6 × 105 CFU/ml) and 20 μg/ml (for culture density of 2 × 107 CFU/ml). The ratios of MBC and MIC were 2.1-fold and 4-fold, respectively. Similar results were obtained when the different isolates of S. mutans were tested, as shown in Table 2. At a final cell density of 6 × 105 CFU/ml, the MBCs of different S. mutans strains ranged between 5 μg/ml and 9 μg/ml. The MBC/MIC ratios were between 1.67- and 3-fold, suggesting that CLP-4 was bactericidal.
Additionally, we examined the killing kinetics of CLP-4 under two conditions: (i) active growth and (ii) carbon starvation. We performed this experiment by monitoring the cell viability (by colony counting) of actively growing UA159 cultures that had a starting cell density of 6 × 105 CFU/ml. These cultures were treated with 5, 10, or 25 μg/ml CLP-4 for 1, 3, 6, 9, 24, and 30 h. The results are presented in Fig. 3A. When actively growing bacteria were treated with CLP-4 at the concentration of 5 μg/ml, there was an overall 3.6-log10 reduction in the number of viable cells, corresponding to 99.97% killing after 24 h (Fig. 3A, closed circles). A strong dosage-dependent bactericidal activity was observed when bacteria were treated with increasing concentrations of CLP-4. Specifically, at 10 μg/ml, a 2.7-log10 reduction in cell viability (99.80% killing) was achieved after 6 h of treatment, up to a 5.9-log10 reduction (>99.99% killing) by 24 h, and a complete killing by 30 h (Fig. 3A, closed triangle). At 25 μg/ml, the bactericidal effect of CLP-4 was even more pronounced, with a 2.8-log10 reduction in cell viability (99.84% killing) after 3 h of treatment and complete killing by 9 h (Fig. 3A, open squares).
FIG 3.

Comparative killing kinetics of CLP-4. S. mutans UA159 cultures at a cell density of 6 × 105 CFU/ml were challenged with 5, 10, and 25 μg/ml CLP-4 under conditions of active growth in CDM supplemented with 0.5% (wt/vol) glucose (A) and against growth-arrested cells in CDM lacking any carbon source (B). Samples at time zero were enumerated prior to peptide treatment. Data shown are the means and standard deviations of three biological replicates from three independent experiments.
To determine if the CLP-4 bactericidal activity extended to growth-arrested S. mutans cultures, we examined the killing kinetics in media lacking any carbon source. Under this condition, S. mutans is not able to grow. Bacterial cultures at a density of 6 × 105 CFU/ml were treated similarly as in the previous experiment. The results are presented in Fig. 3B and show that CLP-4 did not exhibit any bactericidal activity. A slow but steady decline in cell viability was observed for all samples, regardless of whether CLP-4 was present. An initial decrease in cell viability was observed in all cultures after 3 h of incubation, and cell viability steadily decreased up to 30 h (overall 2.1-log10 reduction corresponding to 98.74% death due to starvation). Together, these results indicate that CLP-4 possessed potent bactericidal properties only against actively growing bacteria.
Spontaneous resistance frequency of S. mutans UA159 to CLP-4.
A common concern for novel antimicrobial therapeutics is the development of bacterial resistance. In order to determine if S. mutans UA159 can develop spontaneous resistance to CLP-4, we plated 109 CFU of actively dividing S. mutans UA159 on agar plates containing 5× or 10× the MBCs. This experiment was independently repeated three times. After 72 h of incubation at 37°C, we did not detect resistant colonies on plates with 5× and 10× the MBCs of CLP-4. Altogether, these results yielded a calculated spontaneous resistance frequency of less than 10−9.
CLP-4 is a potent inhibitor of S. mutans biofilm formation.
Dental plaque formation by cariogenic S. mutans is an important etiological factor for dental caries. To evaluate the effectiveness of CLP-4 as an antibiofilm agent, we determined the minimum biofilm inhibitory concentration (MBIC) required to prevent the formation of UA159 biofilms. Bacterial cultures (2 × 107 CFU/ml) were seeded in 48-well plates, and biofilms were formed in the presence of CLP-4, erythromycin, or chlorhexidine at concentrations of 0.6×, 0.8×, 1×, and 2× their respective MICs. The biofilm biomass was quantified after 24 h by staining with crystal violet, and the results are presented in Fig. 4A. Biofilm formation of UA159 was completely inhibited, and no biofilm biomass could be detected in CLP-4 concentrations corresponding to 1× the MIC (5 μg/ml). Below this concentration, UA159 was able to form biofilms. While the CLP-4 MBIC coincided with the MIC of the starting culture (Fig. 4B and Table 1), chlorhexidine exhibited increased potency because it inhibited biofilm formation at 0.8× the MIC (1 μg/ml). On the other hand, erythromycin was less effective at inhibiting biofilm formation and required 2× the MIC (125 ng/ml) to achieve complete inhibition.
FIG 4.

CLP-4 prevents S. mutans biofilm formation. (A) Biofilms inoculated with 2 × 107 CFU/ml were grown for 24 h in the presence of CLP-4, chlorhexidine, or erythromycin at concentrations ranging between 0.6× and 2× their respective MICs. Biofilm formation was quantified using crystal violet staining and expressed in percentage relative to untreated control. Shown are the means and standard deviations of three biological replicates from three independent experiments. *, P < 0.05; ***, P < 0.001 compared to untreated control. (B) Corresponding growth curve kinetics showing the MIC of CLP-4 on S. mutans UA159.
CLP-4 is effective against established biofilms.
We determined if CLP-4 can also eradicate preformed biofilms. S. mutans UA159 cultures were seeded in 48-well plates at a starting cell density of 2 × 107 CFU/ml, and biofilms were established for 24 h. The biofilms were then treated with CLP-4, erythromycin, or chlorhexidine for 24 h at concentrations of 1×, 2×, 5×, and 10× their respective MICs. The cell viability of treated biofilms was quantified by colony enumeration, and the results are presented in Fig. 5A. A dosage-dependent reduction in cell viability was observed. Treatment with 1× the MIC (5 μg/ml) of CLP-4 was not effective and yielded a reduction in viability of only 0.64 log10, which corresponded to a killing of 77.09%. Treatment with 2× the MIC (10 μg/ml) resulted in 2.21-log10 reduction (99.38% killing), and treatment with 5× the MIC (25 μg/ml) resulted in 3.4-log10 reduction (99.96% killing). The minimum biofilm eradication concentration (MBEC) was achieved at 10× the MIC (50 μg/ml), with an overall 4.5-log10 reduction in the number of viable cells compared to the untreated control. This viability reduction corresponded to 99.997% killing. In comparison, no significant change in cell viability reduction was observed for chlorhexidine until biofilms were treated with 5× the MIC (6.25 μg/ml) (Fig. 5A), which resulted in 0.73-log10 viability decrease (81.34% killing). At 10× the MIC (12.5 μg/ml), chlorhexidine exhibited a 3.05-log10 viability reduction, corresponding to 99.91% killing. In contrast, erythromycin was not effective at killing established biofilms (Fig. 5A). The maximum cell viability reduction observed for erythromycin was 1.91 log10, corresponding to 98.59% killing.
FIG 5.

Effects of CLP-4 on preformed biofilms. S. mutans UA159 biofilms were established for 24 h and then treated with increasing concentrations (1× to 10× the MIC) of CLP-4, chlorhexidine, or erythromycin. (A) Antibiofilm activities were assessed by quantifying the cell viability of treated biofilms by colony enumeration on agar plates. The means and standard deviations of three biological replicates from three independent experiments are shown. **, P < 0.01; ***, P < 0.001 compared to untreated control. (B) Biofilms treated with 10× the MICs for each antimicrobial were fluorescently labeled using the LIVE/DEAD BacLight viability stain and visualized by confocal laser scanning microscopy. Shown are the top-down three-dimensional (3D) volume rendering of biofilms at a total magnification of ×400. Bottom images represent optical planes in the xz, and vertical thin images represent yz dimensions. Membrane-compromised bacteria are stained red with propidium iodide, while intact bacteria are stained green with SYTO 9. Areas highlighted by dashed lines indicate regions of interest (ROIs) viewed at a higher magnification. Dimensions shown are 387.5 μm by 387.5 μm by 16 μm. (C) ROIs are presented at ×2,300 magnification. Dimensions shown are 68.1 μm by 68.1 μm by 16 μm.
In addition to quantitative analysis, biofilms treated with 10× the MICs were also visualized using LIVE/DEAD staining and confocal microscopy. This differential staining discriminates membrane-compromised bacteria from those that have intact membranes, because the red fluorescent propidium iodide dye can only penetrate damaged bacterial membranes. Microscopy images showed that almost all biofilm bacteria exposed to CLP-4 were killed, confirming the colony enumeration results (Fig. 5B). The biofilms visualized under the confocal microscope had an average height of 17 μm. All biofilm cells located 5 μm above the glass surface were killed by CLP-4, whereas the vast majority of cells below were killed, except for a few patches that did not respond to treatment (Fig. 5C). Chlorhexidine treatment resulted in a similar response, but the antibiofilm activities observed were heterogeneous, as intact bacteria could be found interspersed throughout the biofilms. Erythromycin treatment, on the other hand, did not appear to have any killing effect on preestablished biofilms. Overall, the different patterns of LIVE/DEAD staining observed indicated that CLP-4 and chlorhexidine acted upon bacterial membranes, while erythromycin did not (Fig. 5B and C).
CLP-4 has minimal toxicity on human cells.
Having established the in vitro stability and potent antimicrobial activities of CLP-4 against S. mutans, we also evaluated its toxicity in human cells. First, hemolytic activity was determined against human red blood cells (hRBCs) by treating with CLP-4 for 1 h at concentrations of 0 to 1,000 μg/ml. Phosphate-buffered saline (PBS) and 1% Triton X-100 were used as controls for 0 and 100% hemolysis, respectively. As shown in Fig. 6, CLP-4 showed low hemolytic activity of approximately 9% at the highest tested concentration of 1,000 μg/ml. Importantly, at the MIC, MBC, MBIC, and MBEC of CLP-4, no appreciable hemolytic activity was observed.
FIG 6.

Hemolytic activity of CLP-4. Human erythrocytes (hRBCs) were treated with CLP-4 at concentrations ranging between 8 μg/ml and 1,000 μg/ml for 1 h at 37°C. Hemoglobin release was quantified as a percentage relative to the hemolysis caused by 0.5% Triton X-100. The average of three biological replicates and standard deviations from three independent experiments are shown. *, P < 0.05 compared to untreated control.
Next, we examined the cytotoxicity of CLP-4 in human oral fibroblasts (human embryonic palatal mesenchyme [HEPM] cells). This experiment was performed by treating fibroblasts with CLP-4 for 24 h, 48 h, and 72 h at final concentrations of 0, 5, 51, 154, and 256 μg/ml. The known cytotoxic drug adriamycin (Doxorubicin; Sigma-Aldrich, St. Louis, MO) was used as a positive control in these assays. The toxicity of CLP-4 for HEPM cells is shown in Fig. 7. At the concentration of 51 μg/ml, no statistically significant change in cell viability was observed after treatment for 24 h and 48 h. A moderate cytotoxic effect was observed after 72 h (20.4% reduction in cell viability). In this study, the effective CLP-4 concentrations (MIC, MBC, MBIC, and MBEC) corresponded only to a negligible reduction in cell viability.
FIG 7.

Cytotoxicity of CLP-4. The cell viability of oral fibroblasts (HEPM) treated with CLP-4 for 24, 48, and 72 h at concentrations of 0 to 256 μg/ml was determined using the CellTiter-Glo luminescent cell viability assay. Doxorubicin (10 μM) was included as a positive control. The results shown are the average of three biological replicates and standard deviations from three independent experiments. *, P < 0.05; ***, P < 0.001 compared to untreated control.
DISCUSSION
Four types of naturally occurring fusaricidins (A, B, C, and D) produced by Paenibacillus polymyxa have been well characterized. Among them, fusaricidin A/LI-F04a was shown to have the most potent in vitro antimicrobial activity against a variety of clinically relevant fungi and Gram-positive bacteria (35, 36). In previous studies, Bionda et al. (29, 34) synthesized analogues of this fusaricidin with a modified lipidic tail, enhanced stability, and reduced cytotoxicity toward human cells (38). In this study, we synthetized a novel analogue of fusaricidin called cyclic lipopeptide 4 (CLP-4) and showed that it was effective against several clinical isolates and serotypes of the Gram-positive cariogenic pathogen S. mutans (Tables 1 and 2). We found that the MIC of CLP-4 was 2.8 μg/ml, and the MBC was 6 μg/ml for S. mutans UA159 when a starting inoculum of 6 × 105 CFU/ml was used, as recommended by the CLSI (42) (Tables 1 and 2). Similar results were obtained when different S. mutans clinical isolates and serotypes were tested (Table 2), further supporting the efficacy of CLP-4. Because MICs and MBCs are known to vary depending on the density of the starting inoculum, we also determined the MIC when the inoculum size was adjusted to 2 × 107 CFU/ml. This cell density was the inoculum used in our biofilm experiments and allowed us to compare the MIC and the minimum biofilm inhibitory concentrations (MBICs). At high cell density, the MIC of CLP-4 was 5 μg/ml, while the MBC was 20 μg/ml (Table 1). The ratio of MBC to MIC is used to determine if an antimicrobial agent is bacteriostatic or bactericidal. An antimicrobial agent is considered bactericidal if this ratio is ≤4-fold (43). Our results showed that these ratios were approximately 2-fold for the lower-density inoculum and 4-fold for the higher-density inoculum, which indicated that CLP-4 is bactericidal. The MIC and MBC values for CLP-4 were in agreement with the range of concentrations reported for other antimicrobial cyclic lipopeptides (44, 45). For example, MICs for daptomycin were reported to be ≥0.2 to 8 μg/ml and ≥1 to 8 μg/ml for ramoplanin against clinically relevant Gram-positive bacteria (44, 45). CLP-4 exhibited antibacterial activity comparable to that of the antiseptic chlorhexidine, which is commonly used to treat oral infections, such as periodontitis (Table 1).
The results of time-kill assays performed in our study confirmed that CLP-4 was bactericidal and that it exhibited a dosage-dependent rate of killing (Fig. 3A). At the concentration of 5 μg/ml, the rate of killing was low, requiring at least 24 h to achieve a 3-log10 reduction in cell viability. At twice this concentration (10 μg/ml), a 3-log10 reduction was achieved in less than 7 h, and at 25 μg/ml, a 3-log10 reduction was achieved in just 3 h. Most antimicrobial peptides that lyse bacterial membranes are fast acting, displaying detectable lysis within 15 to 90 min (31, 46). The action of CLP-4 is slow and dose dependent, which might indicate that it has multiple targets and lyses bacterial membranes only at higher concentrations.
Our study also demonstrated that CLP-4 is an effective antibiofilm agent. CLP-4 was able to completely prevent UA159 biofilm formation in vitro, even when biofilms were cultured with a high density of bacteria (2 × 107 CFU/ml). The MBIC and MIC of CLP-4 were the same (5 μg/ml), which was expected because the same inoculum was used in both experiments. This result suggests that the same ratio of CLP-4 molecules to the bacterial cell number is required to inhibit both planktonic and biofilm growth. Furthermore, the MBIC of CLP-4 was much lower than the MBC (20 μg/ml) for the same inoculum, suggesting that arresting UA159 growth and preventing biofilm formation do not require 99.99% killing efficiency. This result led us to speculate that CLP-4 might have separate mechanisms for bacterial growth arrest and killing. At low concentrations, it is possible that CLP-4 disrupts some of the cellular processes required for growth. This hypothesis is further supported by our observations that CLP-4 is effective only against metabolically active bacteria (Fig. 3A and B). However, when present at a higher concentration, CLP-4 might exert killing through bacterial membrane damage, as shown by our confocal microscopy results.
In addition to preventing biofilm formation, we demonstrated that CLP-4 is also effective at killing established biofilms. Estimation of bacterial viability by colony enumeration showed that biofilms grown for 24 h had, on average, 3.6 × 108 CFU/ml live bacteria (Fig. 5A). When these biofilms were treated with CLP-4, a dosage-dependent reduction in bacterial viability was observed. Specifically, 2 log10 of established biofilms were killed by treatment with 2× the MIC of CLP-4. However, when we treated established biofilms with CLP-4 at 10× the MIC, there was a 4-log10 reduction in bacterial viability. In contrast, chlorhexidine exhibited a less efficient response (Fig. 5A). At least 5× the MIC of chlorhexidine was required for minor bacterial killing, and at 10× the MIC, there was a 3-log10 reduction in viability. Erythromycin was inefficient, exhibiting only a 1.91-log10 reduction in viability at tested concentrations.
Our confocal microscopy results showed that at high concentrations, CLP-4 damaged bacterial membranes. The LIVE/DEAD staining discriminates damaged bacterial membranes from those that are intact by allowing the red fluorescent propidium iodide dye to penetrate cells. Almost all biofilms exposed to CLP-4 at 10× the MIC were killed (Fig. 5B), except for a few small sections on the bottom of the biofilms that were not affected (Fig. 5B and C). These sections might be dormant (metabolically inactive and/or nondividing) cells that usually do not respond or are tolerant to antimicrobial treatment (47–49). Chlorhexidine also showed a membrane-damaging response; however, unlike CLP-4, intact cells were found interspersed throughout the biofilms. The staining pattern observed for erythromycin, on the other hand, indicated that no membrane damage occurred even at a high concentration (10× the MIC). Taken together, these results suggest that CLP-4 is more potent against biofilms than chlorhexidine or erythromycin.
To this day, the mechanism of action for the fusaricidin family of cyclic lipopeptides and their synthetic analogues is still poorly understood. A previous study using a global transcriptomic analysis approach in Bacillus subtilis implicated fusaricidin C in membrane damage (50). After 5 min of exposure, fusaricidin C caused a rapid induction of the σw regulon involved in envelope stress and detoxification. Additionally, after 3 h, the genes involved in cell wall modification, general stress responses, fatty acid and amino acid catabolism, and cation transport were found to be upregulated, while glucose and gluconeogenesis genes were repressed. The same group recently demonstrated that the bactericidal activity of fusaricidin C coincided with an increased production of free hydroxyl radicals (51). Although a greater emphasis has been placed on membrane damage, it is also possible that fusaricidins inhibit intracellular targets. Our study showed that at low concentrations, CLP-4 does not exhibit rapid killing, suggesting that membrane damage might not be its primary mechanism of action. Furthermore, CLP-4 did not kill S. mutans growth-arrested cultures (Fig. 3A and B), and this result indicated that CLP-4 targets might be those required for cell growth. We are in the process of investigating CLP-4's mechanism(s) of action.
In the wake of widespread bacterial resistance to commonly used antibiotics, it is prudent to develop novel antibacterial therapeutics that exhibit low resistance frequencies. Our results showed that S. mutans did not develop spontaneous resistance to CLP-4 under tested conditions. This result indicates that CLP-4 is a promising agent for future use against the cariogenic pathogen S. mutans.
CLP-4 is a cyclic lipopeptide consisting of not only l-amino acids, but also d-amino acids, and is thus predicted to be more resistant against proteases than linear AMPs (41, 52). Our results confirmed this hypothesis. CLP-4 displayed remarkable resistance to proteolysis in vitro when treated with trypsin or chymotrypsin (Fig. 2). This stability is especially important in the oral cavity where proteases are abundant in saliva, dental biofilms, and oral mucosa (40).
Many AMPs cause some degree of toxicity against human cells. In contrast, CLP-4 exhibited negligible levels of hemolysis against human erythrocytes (Fig. 6) and low toxicity against human oral fibroblasts (Fig. 7). In our study, we found that CLP-4 did not induce nonspecific toxicity when used in concentrations equal to the MIC, MBC, MBIC, and MBEC. These results are consistent with our previous finding showing low cytotoxicity for similar cyclic lipopeptides (38).
The findings in this study showed that CLP-4 is a cyclic lipopeptide with excellent stability, low cytotoxicity, and potent bactericidal and antibiofilm activities against S. mutans. Thus, CLP-4 is a novel antimicrobial peptide with potential for further development into an antibiotic for the prevention and treatment of dental caries.
MATERIALS AND METHODS
Bacterial strains, culture media, and growth conditions.
All strains used in this study are listed in Table 2. S. mutans strains were routinely cultured in Todd Hewitt broth supplemented with 0.3% yeast extract (THY) (BD Biosciences San Jose, CA) overnight at 37°C for 16 h. Chemically defined medium (CDM [pH 7.2]) (6, 53) supplemented with 0.5% (wt/vol) glucose was used for MIC, MBC, and killing kinetics assays. Half-strength CDM (0.5× CDM) supplemented with 10 mM sucrose was used for biofilm studies.
Peptide synthesis, purification, and preparation.
CLP-4 was synthesized using standard 9-fluorenylmethyloxy carbonyl (Fmoc) methodology on a PS3 automated peptide synthesizer (Protein Technologies, Inc., Tucson, AZ). Synthesis was carried out on amide TentaGel XV RAM resin obtained from Rapp Polymere, GmbH (Tübingen, Germany) (substitution, 0.21 mmol/g; 0.25 mmol scale). All amino acids and coupling reagents (hydroxybenzotriazole [HOBt], O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate [HBTU], and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate [PyBOP]) were purchased from Chem-Impex, Inc. (Wood Dale, IL). All solvents were purchased from Sigma-Aldrich (St. Louis, MO) and were analytical reagent grade or better. Synthesized peptides were cleaved from the resin using trifluoroacetic acid-triisopropylsilane-water (TFA-TIS-H2O) (95:2.5:2.5 [vol/vol/vol]) for 3 h, precipitated with cold methyl tert-butyl ether, and purified using preparative RP-HPLC on an Agilent 1260 Infinity LC system (Agilent Technologies, Santa Clara, CA). Elution was carried out in a C18 monomeric column (250 by 22 mm, 10 mm, 120 Å, Vydac; Grace) eluted with a linear gradient of 2 to 98% with 0.1% TFA in H2O and 0.08% TFA in CH3CN at a flow rate of 19 ml/min over 30 min. HPLC fractions were analyzed for purity, combined, and lyophilized to give a white powder. The final purity was confirmed to be ≥95% using analytical RP-HPLC with a C18 monomeric column (250 by 4.6 mm, 5 mm, 120 Å, Vydac; Grace) at a flow rate of 1 ml/min and MALDI-TOF mass spectrometry on a Bruker microflex LT system (Bruker, Billerica, MA) in reflector mode using α-cyano-4-hydroxycinnamic acid matrix (positive-ion mode). Prior to assessing antimicrobial activity, the peptide was dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mg/ml and stored at −20°C.
Peptide stability test.
Proteolytic digestion of CLP-4 was performed using tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin (Thermo Fisher Scientific, Grand Island, NY) or chymotrypsin (ProteoChem, Hurricane, UT) immobilized on beaded agarose, according to manufacturer's recommendations. Proteases immobilized on beaded agarose helped eliminate enzyme digest contamination and thus simplify sample analysis. Briefly, 100 μl of resin containing 20 TAME (p-toluenesulfonyl-l-arginine methyl ester) units of immobilized TPCK trypsin or chymotrypsin was washed three times with 500 μl of digestion buffer (0.1 M NH4HCO3 [pH 8.0]); the gel was then suspended in 200 μl of digestion buffer, and 1 mg of CLP-4 was added for proteolytic digestion. After incubation at 37°C for 1, 2, 4, 18, and 24 h, 20 μl of treated samples was collected and analyzed by analytical RP-HPLC.
Bacterial susceptibility assays.
MICs and minimum bactericidal concentrations (MBCs) were measured by the standard broth microdilution method, according to the recommendations of the Clinical and Laboratory Standards Institute (CLSI) (42). Briefly, planktonic cultures of S. mutans UA159 were grown overnight in THY, washed, suspended at a dilution of 1:10 in CDM supplemented with 0.5% glucose, and incubated at 37°C until cultures reached the mid-logarithmic phase of growth to an optical density at 600 nm (OD600) of 0.65 (corresponding to 6 × 108 CFU/ml). Cultures were adjusted to a final density of 6 × 105 CFU/ml or 2 × 107 CFU/ml and treated with CLP-4, erythromycin (USP grade; Amresco, Solon, OH), or chlorhexidine dihydrochloride (catalog no. C8527; Sigma-Aldrich, St. Louis, MO). The final concentrations of CLP-4 were 1 to 20 μg/ml, and the final concentrations of erythromycin and chlorhexidine were 0.08 to 5 μg/ml. The experiments were performed in 100-well Honeycomb plates (Oy Growth Curves AB Ltd., Finland), with a final volume of 300 μl per well. Growth curves were generated using a Bioscreen C analyzer, version 2.4 (Oy Growth Curves AB Ltd.) by incubating the plates at 37°C for 24 h. Optical densities were measured using the wideband 420- to 580-nm filter. To measure the MBC, 20-μl aliquots of treated bacterial cell suspensions were plated onto THY agar and enumerated after incubation at 37°C for 2 days. All experiments included three biological replicates and were independently repeated three times.
Time-kill assay.
The killing kinetics of CLP-4 were assessed under conditions of active cell growth and carbon starvation. To study the antimicrobial effects on actively dividing cells, overnight cultures of S. mutans UA159 were washed, suspended at a dilution of 1:10 in CDM supplemented with 0.5% glucose, and incubated at 37°C until cultures reached the mid-logarithmic phase of growth, with an OD600 of 0.65. Cell cultures were adjusted to 6 × 105 CFU/ml in CDM and then treated with CLP-4 at concentrations of 5, 10, or 25 μg/ml. The cell survival was estimated by colony enumeration on THY agar after treatment at 37°C for 0, 1, 3, 6, 9, 24, and 30 h. Killing curves were constructed by plotting the log10 of the CFU per milliliter versus time. To study the antimicrobial effects on starving cells, killing curves were constructed similarly but using overnight cultures of S. mutans UA159 that were washed, adjusted to 6 × 105 CFU/ml, and then treated with CLP-4 in CDM lacking any carbon source. All experiments were independently repeated three times and included three biological replicates each.
Spontaneous resistance assay.
Overnight cultures of S. mutans UA159 were washed and diluted 1:10 in CDM supplemented with 0.5% glucose and incubated at 37°C until cultures reached the mid-logarithmic phase of growth, with an OD600 of 0.65. Cell cultures were adjusted to 1 × 109 CFU/ml in CDM and then challenged with different CLP-4 concentrations (5× or 10× the MBC) on THY agar plates and incubated at 37°C for 72 h. The experiments were repeated independently at least three times.
Biofilm susceptibility assays.
Inhibition of biofilm formation was examined using the crystal violet-based microtiter plate assay originally described by O'Toole and Kolter (54). Overnight cultures of S. mutans UA159 were washed, suspended to a final cell density of 2 × 107 CFU/ml in 0.5× CDM supplemented with 10 mM sucrose, and treated with CLP-4, chlorhexidine, or erythromycin. The concentrations tested were 0.6×, 0.8×, 1×, and 2× their respective MICs. Treated and nontreated cell suspensions were seeded in 48-well plates at a volume of 0.5 ml per well and allowed to form biofilms at 37°C in approximately 5% CO2 for 24 h. After incubation, nonadherent planktonic cells were removed by gently washing the wells twice with 1 ml of PBS, and the adherent biomass was stained with 1 ml of filtered 0.1% (wt/vol) crystal violet at room temperature for 10 min. Any excess staining was removed by washing the wells five times with 1 ml of PBS. To quantify the biomass, crystal violet stains were solubilized in 0.5 ml of 30% (vol/vol) acetic acid for 10 min, and the optical densities at the wavelength of 595 nm were measured using a microplate reader. Biofilm formation was expressed as a ratio of crystal violet-stained biofilms compared to the untreated control.
The antibiofilm activities of CLP-4 were examined against preestablished biofilms as follows. Overnight cultures of S. mutans UA159 were washed, adjusted to a cell density of 2 × 107 CFU/ml in 0.5× CDM supplemented with 10 mM sucrose, and seeded in 48-well plates at a volume of 200 μl per well. Biofilms were established at 37°C in approximately 5% CO2 for 24 h. After incubation, nonadherent planktonic cells were removed by gently washing the wells with 1 ml of PBS. Biofilms were then treated with CLP-4, chlorhexidine, or erythromycin at concentrations of 0, 1×, 2×, 5×, and 10× of their respective MICs in fresh 0.5× CDM supplemented with 10 mM sucrose for 24 h. To quantify the cell viability of treated biofilms, the exopolysaccharides were digested with 5 U of dextranase (catalog no. D8144; Sigma-Aldrich, St. Louis, MO) at 37°C for 30 min, and biofilms were homogenized by rigorous pipetting and then plated on THY agar for colony enumeration. All biofilm experiments were repeated three times, and each experiment included three biological replicates.
Hemolytic activity assay.
Human red blood cells (hRBCs; Innovative Research, Novi, MI) were diluted in PBS to 1% and dispensed in clear flat-bottom 96-well plates at a volume of 50 μl per well. Following the addition of 50 μl of CLP-4 dissolved in PBS to give final concentrations of 8 to 1,000 μg/ml, the plate was incubated for 1 h at 37°C. PBS and 0.5% Triton X-100 were used as negative and positive controls, respectively. After incubation, 100 μl of PBS was added to each well, and 150 μl of the supernatants was collected after centrifugation for 10 min at 1,000 × g. To measure the release of hemoglobin, the absorbance of the supernatants at 405 nm was recorded. Assays were carried out twice in triplicate, and the degree of hemolysis was expressed in percentage relative to the hemolysis caused by Triton X-100.
Cell culture and in vitro cytotoxicity assays.
Cytotoxicity of CLP-4 was determined using the CellTiter-Glo 2.0 assay kit (Promega, Madison, WI). Assays were performed in flat-bottom polystyrene 96-well plates with 10,000 HEPM cells (ATCC CRL-1486) per well grown in Eagle's minimum essential medium (EMEM) containing 10% fetal bovine serum (FBS) and 1× penicillin-streptomycin. After overnight incubation at 37°C in a humidified atmosphere with 5% CO2, medium was removed and replaced with fresh medium containing 2% FBS and CLP-4 at concentrations of 0 to 256 μg/ml. CLP-4 was dissolved in DMSO, and appropriate dilutions were made in buffer to a final concentration of 1% (vol/vol) DMSO. Plates were incubated again at 37°C in a humidified atmosphere with 5% CO2. The known cytotoxic drug adriamycin (Doxorubicin; Sigma-Aldrich, St. Louis, MO) at 10 μM was used as a positive control. After incubation for 24 h, 48 h, and 72 h, medium was removed, and 100 ml of PBS buffer was added, followed by 100 ml of CellTiter-Glo 2.0 reagent. Plates were incubated for 10 min at room temperature (protected from light) before luminescence readout.
LIVE/DEAD staining and confocal microscopy.
Biofilms were established in Nunc Lab-Tek 8-well chambered cover slides (Thermo Fisher Scientific, Grand Island, NY) and treated with CLP-4, chlorhexidine, or erythromycin, as described for the biofilm susceptibility assays. Biofilm bacteria were labeled with 200 μl of LIVE/DEAD BacLight bacterial viability stain (L7012; Molecular Probes, Eugene, OR) containing a mixture of 10.02 μM SYTO 9 green fluorescent nucleic acid stain (excitation, 480 nm; emission, 500 nm) and 0.06 μM propidium iodide red fluorescent nucleic acid stain (excitation, 490 nm; emission, 635 nm) and then incubated in the dark at room temperature for 30 min. Excess staining was removed by gently washing the biofilms with sterile deionized water, and confocal imaging was performed using a Leica TCS SP5 inverted microscope (Leica Lasertechnik GmbH, Heidelberg, Germany). z-stacks were acquired using a 40× oil immersion objective lens with optical sections of 1-μm thickness at a total magnification of ×400. The pinhole size was set at 1 Airy unit, and bidirectional laser scanning was set with line averaging of 2 at a speed of 500 Hz. Confocal z-stacks were obtained at 4 random locations for each sample and visualized using the maximum intensity projection or volume-rendering mode using FIJI version 1.5.1d (55).
ACKNOWLEDGMENTS
We thank Justin Merritt, Jeffrey A. Banas, and Celine Levesque for providing clinical and laboratory isolates of S. mutans used in this study.
We declare no conflicts of interest.
REFERENCES
- 1.World Health Organization. 2012. Oral health. Fact sheet no. 318. World Health Organization, Geneva, Switzerland. http://www.who.int/mediacentre/factsheets/fs318/en/.
- 2.Kolenbrander PE, Palmer RJ Jr, Rickard AH, Jakubovics NS, Chalmers NI, Diaz PI. 2006. Bacterial interactions and successions during plaque development. Periodontol 2000 42:47–79. doi: 10.1111/j.1600-0757.2006.00187.x. [DOI] [PubMed] [Google Scholar]
- 3.Loesche WJ. 1986. Role of Streptococcus mutans in human dental decay. Microbiol Rev 50:353–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loesche WJ. 1996. Chapter 99 Microbiology of dental decay and periodontal disease. In Baron S. (ed), Medical microbiology, 4th ed University of Texas Medical Branch, Galveston, TX. [PubMed] [Google Scholar]
- 5.Haffajee AD, Socransky SS, Patel MR, Song X. 2008. Microbial complexes in supragingival plaque. Oral Microbiol Immunol 23:196–205. doi: 10.1111/j.1399-302X.2007.00411.x. [DOI] [PubMed] [Google Scholar]
- 6.Ajdić D, Pham VT. 2007. Global transcriptional analysis of Streptococcus mutans sugar transporters using microarrays. J Bacteriol 189:5049–5059. doi: 10.1128/JB.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paster BJ, Olsen I, Aas JA, Dewhirst FE. 2006. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000 42:80–87. doi: 10.1111/j.1600-0757.2006.00174.x. [DOI] [PubMed] [Google Scholar]
- 8.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J Bacteriol 192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson M. 1996. Susceptibility of oral bacterial biofilms to antimicrobial agents. J Med Microbiol 44:79–87. doi: 10.1099/00222615-44-2-79. [DOI] [PubMed] [Google Scholar]
- 10.Brown MR, Allison DG, Gilbert P. 1988. Resistance of bacterial biofilms to antibiotics: a growth-rate related effect? J Antimicrob Chemother 22:777–780. doi: 10.1093/jac/22.6.777. [DOI] [PubMed] [Google Scholar]
- 11.Mah TF, O'Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 12.Lewis K. 2008. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131. [DOI] [PubMed] [Google Scholar]
- 13.Montgomery EH, Kroeger DC. 1984. Use of antibiotics in dental practice. Dent Clin North Am 28:433–453. [PubMed] [Google Scholar]
- 14.Anderson MH. 2003. A review of the efficacy of chlorhexidine on dental caries and the caries infection. J Calif Dent Assoc 31:211–214. [PubMed] [Google Scholar]
- 15.Riley P, Lamont T. 2013. Triclosan/copolymer containing toothpastes for oral health. Cochrane Database Syst Rev (12):CD010514. doi: 10.1002/14651858.CD010514.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitten FA, Kramer A. 2001. Efficacy of cetylpyridinium chloride used as oropharyngeal antiseptic. Arzneimittelforschung 51:588–595. [DOI] [PubMed] [Google Scholar]
- 17.Emilson CG, Lindquist B, Wennerholm K. 1987. Recolonization of human tooth surfaces by Streptococcus mutans after suppression by chlorhexidine treatment. J Dent Res 66:1503–1508. doi: 10.1177/00220345870660091801. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Godoy F, Garcia-Godoy F, DeVizio W, Volpe AR, Ferlauto RJ, Miller JM. 1990. Effect of a triclosan/copolymer/fluoride dentifrice on plaque formation and gingivitis: a 7-month clinical study. Am J Dent 3(Spec No):S15–S26. [PubMed] [Google Scholar]
- 19.Sweeney LC, Dave J, Chambers PA, Heritage J. 2004. Antibiotic resistance in general dental practice–a cause for concern? J Antimicrob Chemother 53:567–576. doi: 10.1093/jac/dkh137. [DOI] [PubMed] [Google Scholar]
- 20.Babich H, Wurzburger BJ, Rubin YL, Sinensky MC, Blau L. 1995. An in vitro study on the cytotoxicity of chlorhexidine digluconate to human gingival cells. Cell Biol Toxicol 11:79–88. doi: 10.1007/BF00767493. [DOI] [PubMed] [Google Scholar]
- 21.Pucher JJ, Daniel JC. 1992. The effects of chlorhexidine digluconate on human fibroblasts in vitro. J Periodontol 63:526–532. doi: 10.1902/jop.1992.63.6.526. [DOI] [PubMed] [Google Scholar]
- 22.Zuckerbraun HL, Babich H, May R, Sinensky MC. 1998. Triclosan: cytotoxicity, mode of action, and induction of apoptosis in human gingival cells in vitro. Eur J Oral Sci 106:628–636. doi: 10.1046/j.0909-8836.1998.eos106204.x. [DOI] [PubMed] [Google Scholar]
- 23.Grove C, Liebenberg W, du Preez JL, Yang W, de Villiers MM. 2003. Improving the aqueous solubility of triclosan by solubilization, complexation, and in situ salt formation. J Cosmet Sci 54:537–550. [PubMed] [Google Scholar]
- 24.Zanatta FB, Antoniazzi RP, Rosing CK. 2010. Staining and calculus formation after 0.12% chlorhexidine rinses in plaque-free and plaque covered surfaces: a randomized trial. J Appl Oral Sci 18:515–521. doi: 10.1590/S1678-77572010000500015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beckloff N, Laube D, Castro T, Furgang D, Park S, Perlin D, Clements D, Tang H, Scott RW, Tew GN, Diamond G. 2007. Activity of an antimicrobial peptide mimetic against planktonic and biofilm cultures of oral pathogens. Antimicrob Agents Chemother 51:4125–4132. doi: 10.1128/AAC.00208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo L, McLean JS, Yang Y, Eckert R, Kaplan CW, Kyme P, Sheikh O, Varnum B, Lux R, Shi W, He X. 2015. Precision-guided antimicrobial peptide as a targeted modulator of human microbial ecology. Proc Natl Acad Sci U S A 112:7569–7574. doi: 10.1073/pnas.1506207112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 28.Hassan M, Kjos M, Nes IF, Diep DB, Lotfipour F. 2012. Natural antimicrobial peptides from bacteria: characteristics and potential applications to fight against antibiotic resistance. J Appl Microbiol 113:723–736. doi: 10.1111/j.1365-2672.2012.05338.x. [DOI] [PubMed] [Google Scholar]
- 29.Bionda N, Fleeman RM, de la Fuente-Nunez C, Rodriguez MC, Reffuveille F, Shaw LN, Pastar I, Davis SC, Hancock RE, Cudic P. 2016. Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur J Med Chem 108:354–363. doi: 10.1016/j.ejmech.2015.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batoni G, Maisetta G, Brancatisano FL, Esin S, Campa M. 2011. Use of antimicrobial peptides against microbial biofilms: advantages and limits. Curr Med Chem 18:256–279. doi: 10.2174/092986711794088399. [DOI] [PubMed] [Google Scholar]
- 31.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 32.Guilhelmelli F, Vilela N, Albuquerque P, Derengowski Lda S, Silva-Pereira I, Kyaw CM. 2013. Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front Microbiol 4:353. doi: 10.3389/fmicb.2013.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wimley WC. 2010. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem Biol 5:905–917. doi: 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bionda N, Pastar I, Davis SC, Cudic P. 2014. In vitro and in vivo activities of novel cyclic lipopeptides against staphylococcal biofilms. Protein Pept Lett 21:352–356. doi: 10.2174/09298665113206660101. [DOI] [PubMed] [Google Scholar]
- 35.Kajimura Y, Kaneda M. 1996. Fusaricidin A, a new depsipeptide antibiotic produced by Bacillus polymyxa KT-8. Taxonomy, fermentation, isolation, structure elucidation and biological activity. J Antibiot (Tokyo) 49:129–135. doi: 10.7164/antibiotics.49.129. [DOI] [PubMed] [Google Scholar]
- 36.Kajimura Y, Kaneda M. 1997. Fusaricidins B, C and D, new depsipeptide antibiotics produced by Bacillus polymyxa KT-8: isolation, structure elucidation and biological activity. J Antibiot (Tokyo) 50:220–228. doi: 10.7164/antibiotics.50.220. [DOI] [PubMed] [Google Scholar]
- 37.Nomura T, Kuroda J, Fukai T, Konishi M, Uno J, Kurusu K. 2000. LI-F antibiotics, a family of antifungal cyclic depsipeptides produced by Bacillus polymyxa L-1129. Heterocycles 53:1533–1549. doi: 10.3987/COM-00-8922. [DOI] [Google Scholar]
- 38.Bionda N, Stawikowski M, Stawikowska R, Cudic M, Lopez-Vallejo F, Treitl D, Medina-Franco J, Cudic P. 2012. Effects of cyclic lipodepsipeptide structural modulation on stability, antibacterial activity, and human cell toxicity. ChemMedChem 7:871–882. doi: 10.1002/cmdc.201200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurusu K, Ohba K, Arai T, Fukushima K. 1987. New peptide antibiotics LI-F03, F04, F05, F07, and F08, produced by Bacillus polymyxa. I. Isolation and characterization. J Antibiot (Tokyo) 40:1506–1514. doi: 10.7164/antibiotics.40.1506. [DOI] [PubMed] [Google Scholar]
- 40.Kennedy S, Davis C, Abrams WR, Billings PC, Nagashunmugam T, Friedman H, Malamud D. 1998. Submandibular salivary proteases: lack of a role in anti-HIV activity. J Dent Res 77:1515–1519. doi: 10.1177/00220345980770070601. [DOI] [PubMed] [Google Scholar]
- 41.Söder PO. 1972. Proteolytic activity in the oral cavity: proteolytic enzymes from human saliva and dental plaque material. J Dent Res 51:389–393. doi: 10.1177/00220345720510022601. [DOI] [PubMed] [Google Scholar]
- 42.CLSI. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 43.Pankey GA, Sabath LD. 2004. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin Infect Dis 38:864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 44.McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Li W. 2002. Chemistry and biology of the ramoplanin family of peptide antibiotics. Biopolymers 66:261–284. doi: 10.1002/bip.10296. [DOI] [PubMed] [Google Scholar]
- 45.Streit JM, Jones RN, Sader HS. 2004. Daptomycin activity and spectrum: a worldwide sample of 6737 clinical Gram-positive organisms. J Antimicrob Chemother 53:669–674. doi: 10.1093/jac/dkh143. [DOI] [PubMed] [Google Scholar]
- 46.Boman HG. 1995. Peptide antibiotics and their role in innate immunity. Annu Rev Immunol 13:61–92. doi: 10.1146/annurev.iy.13.040195.000425. [DOI] [PubMed] [Google Scholar]
- 47.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 48.Wood TK, Knabel SJ, Kwan BW. 2013. Bacterial persister cell formation and dormancy. Appl Environ Microbiol 79:7116–7121. doi: 10.1128/AEM.02636-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leung V, Ajdic D, Koyanagi S, Levesque CM. 2015. The formation of Streptococcus mutans persisters induced by the quorum-sensing peptide pheromone is affected by the LexA regulator. J Bacteriol 197:1083–1094. doi: 10.1128/JB.02496-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu WB, Yin CY, Zhou Y, Ye BC. 2012. Prediction of the mechanism of action of fusaricidin on Bacillus subtilis. PLoS One 7:e50003. doi: 10.1371/journal.pone.0050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu WB, Ye BC. 2016. High-level iron mitigates fusaricidin-induced membrane damage and reduces membrane fluidity leading to enhanced drug resistance in Bacillus subtilis. J Basic Microbiol 56:502–509. doi: 10.1002/jobm.201500291. [DOI] [PubMed] [Google Scholar]
- 52.Knappe D, Henklein P, Hoffmann R, Hilpert K. 2010. Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob Agents Chemother 54:4003–4005. doi: 10.1128/AAC.00300-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Terleckyj B, Willett NP, Shockman GD. 1975. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect Immun 11:649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol 28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 55.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ajdić D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A 99:14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clarke JK. 1924. On the bacterial factor in the ætiology of dental caries. Br J Exp Pathol 5:141–147. [Google Scholar]
- 58.Bratthall D. 1970. Demonstration of five serological groups of streptococcal strains resembling Streptococcus mutans. Odontol Revy 21:143–152. [PubMed] [Google Scholar]
- 59.Xie Z, Okinaga T, Niu G, Qi F, Merritt J. 2010. Identification of a novel bacteriocin regulatory system in Streptococcus mutans. Mol Microbiol 78:1431–1447. doi: 10.1111/j.1365-2958.2010.07417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banas JA, Zhu M, Dawson DV, Cao H, Levy SM. 2016. PCR-based identification of oral streptococcal species. Int J Dent 2016:3465163. doi: 10.1155/2016/3465163. [DOI] [PMC free article] [PubMed] [Google Scholar]