

ABSTRACT
The dormant phenotype acquired by Mycobacterium tuberculosis during infection poses a major challenge in disease treatment, since these bacilli show tolerance to front-line drugs. Therefore, it is imperative to find novel compounds that effectively kill dormant bacteria. By screening 4,400 marine natural product samples against dual-fluorescent M. tuberculosis under both replicating and nonreplicating conditions, we have identified compounds that are selectively active against dormant M. tuberculosis. This validates our strategy of screening all compounds in both assays as opposed to using the dormancy model as a secondary screen. Bioassay-guided deconvolution enabled the identification of unique pharmacophores active in each screening model. To confirm the activity of samples against dormant M. tuberculosis, we used a luciferase reporter assay and enumerated CFU. The structures of five purified active compounds were defined by nuclear magnetic resonance (NMR) and mass spectrometry. We identified two lipid compounds with potent activity toward dormant and actively growing M. tuberculosis strains. One of these was commercially obtained and showed similar activity against M. tuberculosis in both screening models. Furthermore, puupehenone-like molecules were purified with potent and selective activity against dormant M. tuberculosis. In conclusion, we have identified and characterized antimycobacterial compounds from marine organisms with novel activity profiles which appear to target M. tuberculosis pathways that are conditionally essential for dormancy survival.
KEYWORDS: dormancy, tuberculosis, marine natural products, drug screening, Mycobacterium tuberculosis, drug discovery, natural antimicrobial products
INTRODUCTION
Tuberculosis (TB) is one of the leading causes of death by an infectious disease worldwide. The large pool of individuals latently infected with Mycobacterium tuberculosis (∼2 billion) and the lack of an effective vaccine hinder attempts to eradicate this disease (1). Furthermore, a long multidrug regimen is required to treat TB, which leads to poor patient compliance and increasing occurrence of multidrug-resistant (MDR) M. tuberculosis strains. A prolonged treatment is required due to dormancy-induced drug tolerance developed by M. tuberculosis during infection in response to conditions within the host (2, 3). A considerable body of work has validated the causality between hypoxic granuloma environments and persistent tubercle bacilli (4–6). Also, a recent study has demonstrated that M. tuberculosis becomes drug tolerant as a consequence of stress cues it experiences while residing in activated macrophages (7). These issues highlight a dire need for new scaffolds with novel modes of action effective against MDR and phenotypically drug-tolerant M. tuberculosis.
Efforts to find new effective treatment options have uncovered promising lead compounds; however, only two new drugs have been approved specifically for the treatment of TB in the past 4 decades (8–12). Recent studies have shown the value of screening compound libraries under in vivo-like conditions, since M. tuberculosis undergoes profound metabolic shifts during infection, which may alter target vulnerability. Inducing M. tuberculosis dormancy phenotypes in vitro can enrich the discovery of compounds targeting novel and conditionally essential pathways (13–18). The Wayne model of gradual oxygen depletion is the most well-characterized in vitro dormancy model. However, this is difficult to translate into high-throughput screening, since it requires 2 sequential stages of oxygen depletion in sealed tubes (19). An alternative multistress dormancy model (MSD) described by Deb et al. produced phenotypically dormant M. tuberculosis after 9 days of incubation under conditions of low oxygen, low pH, and nutrient limitation. Bacilli in this model exhibited all of the classical dormancy traits, including lipid droplet accumulation, loss of acid fastness, upregulation of the dosR regulon, and, most importantly, tolerance to front-line drugs (20). In this study, we have successfully adapted this method for high-throughput screening and used it to identify marine natural products (MNPs) which kill dormant M. tuberculosis.
The search for antimycobacterials from natural sources is a historically validated approach. This is emphasized by the fact that 9 out of 12 classes of currently available antibiotics are natural product-derived scaffolds (21). Although screening libraries of pure synthetic compounds may yield hits more efficiently, these libraries are often redundant, leading to the rediscovery of similar scaffolds with known modes of action (22). This observation has incentivized renewed efforts in recent years toward screening of the more chemically diverse natural product libraries (23). The chemical diversity may be amplified for marine natural products due to the vast biodiversity encountered in marine environments (24). Also, bioactive secondary metabolites produced by marine organisms to fight parasitic microorganisms are often very potent, since they rapidly become diluted in the environment (25). Many antimicrobial compounds with activity against M. tuberculosis have been isolated from marine organisms, although very few have moved beyond initial hit identification (23, 26).
In this study, a library of MNPs was screened under replicating and dormant conditions as our primary screening approaches. To our knowledge, this is the only large-scale screen of MNPs against M. tuberculosis, not only under replicating conditions, but also in the context of dormancy. This screen uncovered unique hits which would have otherwise been missed in a conventional in vitro screen. Use of the dormancy model as a primary screening approach was key to the discovery of fractions distinctively active against nonreplicating M. tuberculosis. Rather than crude extracts, as is typical for natural product screens, this MNP library contained 4,400 peak fractions purified by medium-pressure liquid chromatography (MPLC) (∼75% pure). The purity of the samples facilitated the deconvolution and isolation of active components. Five pure compounds were isolated from select hits, and their structures and activities were characterized. Four out of these 5 compounds were active against dormant M. tuberculosis, with 2 puupehenone class metabolites being highly selective for these phenotypically tolerant bacteria. Further investigation of dormancy-active hits could reveal novel druggable targets in dormant bacilli vulnerable to inhibition, ultimately leading to novel therapeutics able to reduce treatment time for tuberculosis.
RESULTS
Screening MNPs against M. tuberculosis. (i) Replicating M. tuberculosis.
The Harbor Branch Oceanographic Institute (HBOI) at Florida Atlantic University MNP library containing 4,400 peak fractions was screened at 20 μg/ml in a dual-fluorescent whole-cell assay for inhibition of growth of replicating M. tuberculosis. Initially, 114 peak fractions were deemed active based on an arbitrary threshold of 50% inhibition relative to 10 μM rifampin (RIF). The close correlation between the mCherry and green fluorescent protein (GFP) readouts ruled out the possibility of false positives due to quenching of reporter protein fluorescence. The average Z scores and signal-to-background ratio among all the plates were 0.82 and 63, respectively, emphasizing the robustness of the assay. During validation of hits by repeating the primary assay, 52 hits of the initial 114 hits which were near the 50% cutoff had inconsistent results and therefore were not further characterized in this study. The 62 remaining in vitro hits were tested for cytotoxicity against J774 macrophages at the screening concentration (20 μg/ml), and 26 fractions were noncytotoxic (<20% cytotoxic relative to dimethyl sulfoxide [DMSO] controls) (Table 1 and Fig. 1).
TABLE 1.
Primary screening assay results
| Assay | Z score | S/Ba | Total no. of hits/4,400 hits | Hit rate (%) | Noncytotoxic hits |
|---|---|---|---|---|---|
| Primary in vitro assay | 0.82 | 63 | 95 | 2.1 | 26 |
| Primary dormancy assay | 0.81 | 23 | 35 | 0.77 | 19 |
| Dual active | NAb | NA | 19 | 0.42 | 7 |
S/B, signal-to-background ratio.
NA, -not applicable.
FIG 1.
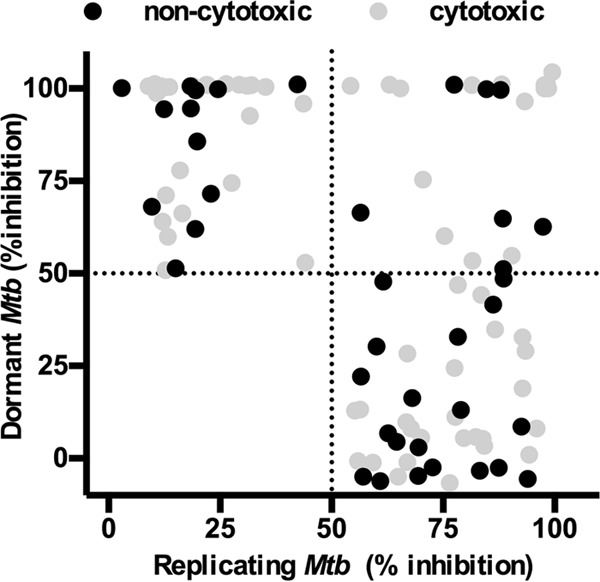
Activities of peak fractions against dormant and active Mtb-RG. The activities of peak fractions screened at 20 μg/ml are shown. One hundred percent inhibition is defined as the inhibition observed in the presence of 12 μg/ml rifampin. Peak fractions were considered cytotoxic when they resulted in >20% killing of J774 macrophages.
(ii) Dormant M. tuberculosis.
In the primary dormancy screen, the average Z scores and signal-to-background ratios were 0.81 and 23, respectively. Fifty-four fractions had activity above the defined threshold, 35% of which were also active against replicating M. tuberculosis. Further testing of dormancy-active hits revealed that 19 out of 54 hit fractions were noncytotoxic (<20% toxicity to J774), 7 of which were also active against replicating M. tuberculosis (Fig. 1 and Table 1). All the noncytotoxic dormancy hits (19 peak fractions) were validated by CFU without a regrowth stage in 3 independent experiments (see Fig. S2 in the supplemental material). Approximately 50% of the hits had activity below the threshold by CFU; these included fractions that had shown potent activity with the fluorescent readout (data not shown). These data indicated that fractions which inhibited only replicating M. tuberculosis were also detected in the primary dormancy assay. The reason for this outcome could be inhibition of replicating bacilli during the regrowth stage due to carryover drug in the wells. Furthermore, the MICs for control drugs obtained with the fluorescent regrowth assay were similar between replicating and dormant M. tuberculosis (RIF dormant Mtb-Lux strain [MICD], 0.2 μM, and replicating Mtb-Lux [MICR], 0.5 μM; isoniazid [INH] MICD, 0.2 μM and MICR, 0.2 μM) (Fig. 2A and B). In contrast, complete killing of dormant bacteria was not observed by CFU enumeration at any concentration of RIF or INH (Fig. 2C and D). Thus, the fluorescent regrowth results significantly underrepresented the tolerant phenotype observed for dormant M. tuberculosis in this study. To minimize this, an alternative assay was optimized for characterization of dormancy-active hits which did not require a regrowth step.
FIG 2.

Optimization of MSD model screening assay. All panels show dose-response curves for positive-control drugs RIF (A, C, and E) and INH (B, D, and F) against replicating and dormant M. tuberculosis using three distinct screening assays and readouts. (A and B) Dual-fluorescent assay. Mtb-RG cultures were treated with serial dilutions of RIF and INH for 5 days. Then, 1 volume of buffered 2× 7H9 was added to each well, and surviving bacilli were allowed to regrow for 7 days under normoxic conditions before fluorescence was read. (C and D) CFU assay. Mtb-RG (replicating and dormant) was treated for 5 days with RIF and INH before serial dilution and plating for CFU counting. (E and F) Luminescence assay. Mtb-Lux (replicating and dormant) was treated for 2 days with RIF and INH, after which luminescence was read. Note, there is no regrowth stage for CFU and luminescence assays (C to F). Open squares, dormant culture; closed squares, replicating culture in complete Dubos medium; conc., concentration.
Use of Mtb-Lux in dormancy assay eliminates the need for a regrowth stage.
The secondary dormancy method was developed based on a loss of luminescent signal. Dormant and replicating Mtb-Lux cultures were diluted to an optical density at 600 nm (OD600) of 0.2 and treated for 3 defined time points (2, 4, and 6 days) before the luminescence was read. The 2-day time point was used for all further experiments since an optimum signal-to-background ratio was observed at that time (data not shown). The MIC of INH against dormant Mtb-Lux (MICD; 285 μM) was >500-fold higher than that of replicating Mtb-Lux (MICR; 0.3 μM); similarly, a >100-fold difference was shown for RIF (MICD, 4 μM; MICR, 0.02 μM) (Fig. 2E and F). Eliminating the regrowth stage by using luciferase shortened the assay by 10 days. Moreover, luminescent data closely reflected the highly drug-tolerant nature of bacteria in our dormancy model (Fig. S3).
Characterization of pure compound activity.
Bioactivity-guided fractionation of primary hits and characterization of pure compound activities were carried out using the luminescent assay described above. Three peak fractions (047.F07, 031.C02, and 050.F04) from different marine organisms out of the 26 hits obtained were investigated, and 5 active compounds were isolated from these samples. The compound structures are shown in Fig. 3, and their biological activity profiles are shown in Table 2 and Fig. 4 and 5.
FIG 3.
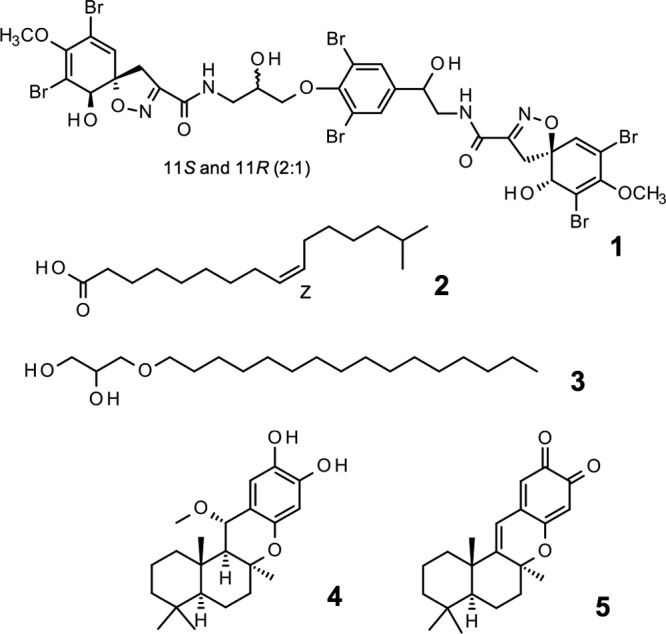
The structures of active molecules as defined by NMR. Hit peak fractions had to undergo at least 2 rounds of HPLC to isolate pure active compounds. Compound 1, fistularin-3/11-epi-fistularin-3 (purified from 047.F07); compound 2, 15-methyl-9(Z)-hexadecenoic acid (purified from 047.F07); compound 3, (hexadecyloxy)propane,1,2-diol (purified from 031.C02); compound 4, 15-α-methoxypuupehenol (purified from 050.F04); compound 5, puupehedione (purified from 050.F04).
TABLE 2.
Activity profile of MNP-derived pure compoundsa
| Compound | Formula | Molecular mass (kDa) | MICR (μg/ml)a | MICD (μg/ml)b | MICR/MICD | IC50 (μg/ml) | SIRc | SIDd | Source |
|---|---|---|---|---|---|---|---|---|---|
| 1 | C31H30Br6N4O11 | 1,114.02 | 8.5 | Inactive | NA | >200 | >23.5 | NA | HBOI.047.F07 |
| 2 | C19H40O3 | 316.53 | 60.8 | 22.5 | 2.7 | >200 | >3.3 | >8.5 | HBOI.047.F07 |
| 3 | C16H30O2 | 254.41 | 28.5 | 7.9 | 3.6 | >200 | >7.0 | >31.1 | HBOI.031.C02 |
| 4 | C22H32O4 | 360.49 | 11.3 | 0.5 | 21.8 | 8 | 0.7 | 15.5 | HBOI.050.F04 |
| 5 | C21H26O3 | 326.44 | 87.6 | 15.4 | 5.6 | 50.4 | 0.6 | 6.2 | HBOI.050.F04 |
MICR, MIC against replicating Mtb-Lux.
MICD, MIC against dormant Mtb-Lux.
SIR, SI for replicating Mtb-Lux.
SID, SI for dormant Mtb-Lux.
FIG 4.

Dose-dependent activity of select pure MNP compounds against dormant and replicating Mtb-Lux. Cultures adapted under multiple-stress conditions for 9 days or grown in 7H9 were used in the 16-point dose-response curves (200 μg/ml to 0.006 μg/ml). Dormant (open squares) and replicating (closed squares) Mtb-Lux were treated for 2 days with compounds 2 (A), 3 (B), 4 (C), and 5 (D), after which the luminescence was read.
FIG 5.

Activity of pure MNP compounds against dormant and replicating Mtb-Lux validated by CFU. Dormant (white bars) and replicating (black bars) Mtb-Lux were treated for 5 days with compounds 2 (A), 3 (B), 4 (C), and 5 (D), after which samples were taken for plating on 7H10 agar medium. Colonies were counted after a 3-week incubation. UNT day 0, untreated culture inoculum; UNT day 5, untreated culture plated after 5 days of incubation under the same conditions as treated cultures. One-way ANOVA was used to compare data points. Asterisks denote data points which are significantly different (P < 0.05) from the control (UNT D0 for replicating M. tuberculosis; UNT D5 for dormant M. tuberculosis).
The peak fraction (047.F07) isolated from a sponge in the family Aplysinidae inhibited replicating Mtb-RG and moderately inhibited dormant Mtb-RG in the primary screens. The original active fraction was small, and therefore, an additional organism was extracted and chromatographed to yield a series of 20 fractions which were tested against dormant and replicating Mtb-Lux to determine the active compound(s). Out of the 20 subfractions obtained, subfraction 047.F07-B8 had potent activity against replicating M. tuberculosis. Compound 1 was purified from this subfraction and was inactive against dormant Mtb-Lux, which is consistent with previous results obtained for the 047.F07-B8 subfraction (data not shown). The MICR (8.5 μg/ml) of compound 1 was determined by luminescent assays described above (Table 2 and Fig. S4). The structure of compound 1 (determined by nuclear magnetic resonance [NMR], detailed in the supplemental methods in File S1 in the supplemental material, as well as chemical database searches) was found to be an inseparable mixture of fistularin-3 and 11-epi-fistularin 3 (27) (Fig. 3). Since antitumor activity of fistularin-3 was previously reported (28), we tested its cytotoxicity against HepG2 in addition to J774 cells, and the 50% inhibitory concentration (IC50) was >200 μg/ml. Another subfraction (047.F07-B10) from the same Aplysinidae-derived peak fraction (047.F07) maintained potent inhibitory activity against dormant Mtb-Lux by luminescence and CFU readouts at 20 μg/ml (data not shown). Moreover, this subfraction was less inhibitory against replicating M. tuberculosis. The dormancy-active subfraction was a mixture of lipids and required two rounds of preparative high-pressure liquid chromatography (HPLC) before defining the structure of the major bioactive component as 15-methyl-9(Z)-hexadecenoic acid (compound 2; see File S1 for details) (Fig. 3) (29). The MICR and MICD for this compound were 60.8 μg/ml and 22.5 μg/ml, respectively. Cytotoxicity against J774 macrophages was not observed at the highest concentration tested (200 μg/ml), yielding a selectivity index (SI) of >8.5, relative to its MICD (Table 2 and Fig. 4A).
The peak fraction 031.C02, isolated from the soft coral Pterogorgia citrina, was active against dormant Mtb-RG in the primary screen and had low activity against growing bacteria. Further investigation of this peak fraction suggested it contained a single compound identified as 3-(hexadecyloxy)propane,1,2-diol (compound 3; see File S1 for details) (Fig. 3) (30, 31). This compound was tested in the secondary luminescent assays. The MICR and MICD of compound 3 were 28.5 μg/ml and 7.9 μg/ml, respectively, with an SI (SID = IC50/MICD) of at least 31 (Table 2 and Fig. 4B). Moreover, the activity of commercially acquired compound 3 correlated with that of the marine-derived nature product (data not shown).
The Petrosia (Strongylophora) sp.-derived peak fraction (050.F04) potently inhibited dormant Mtb-RG in the primary screen as well as by CFU enumeration (Fig. S2). Further deconvolution yielded 11 subfractions, 5 of which maintained activity against dormant Mtb-Lux. Further analysis of these subfractions suggested they contained mixtures of related hydroquinone-like structures. Compounds 4 and 5, isolated from 050.F04, were identified as 15-α-methoxypuupehenol and puupehedione, respectively (see File S1 for details) (Fig. 3) (32, 33). We have demonstrated their potent and selective activity against dormant M. tuberculosis using the described luminescent assay. Table 2 and Fig. 4C show that the MICD of compound 4 (0.5 μg/ml) is 21-fold lower than its MICR (11.3 μg/ml). Even though the SI of this compound was low in a comparison of the IC50 to MICR, the SI was 15 in comparison to MICD, which highlights its potential as a scaffold-targeting dormant M. tuberculosis. Interestingly, compound 5 maintained the dormant selectivity; however, it was less potent in all the assays, including the cytotoxicity assay (Table 2 and Fig. 4D). Compounds 4 and 5 are very similar but differ in the oxidation state of the ortho-quinol ring and in the addition of methanol across the C-11=C-15 double bond found in compound 5 (Fig. 3). This results in a molecule that is less planar than others in the puupehenone class of terpene quinones and may contribute to its activity against the dormant form of M. tuberculosis versus that of others in the class.
Pure compounds show bactericidal activity against M. tuberculosis.
To investigate the mode of action of each compound, samples were taken from duplicate wells in the luminescent assays after 5 days of treatment and plated on 7H10 oleic acid-albumin-dextrose-catalase (OADC). Four concentrations were chosen for plating (50, 12.5, 3.1, and 0.8 μg/ml) covering a broad portion of the 16-point dose-response curves. Untreated controls were plated on day 0 (inoculum) and after 5 days of incubation. A decrease in CFU per milliliter (<1 log) was observed after 5 days for the untreated dormant Mtb-Lux. Therefore, the day 5 untreated control was considered a fairer comparison with treated dormant samples. However, replicating treated bacteria were compared to day 0 controls. These data are shown in Fig. 5. Compound 1 was inactive in the primary dormancy assay and showed no activity by CFU in this assay. Furthermore, it was bacteriostatic against replicating Mtb-Lux at all four concentrations tested (Fig. S5). As expected, compound 2 was bactericidal against dormant but not replicating Mtb-Lux at 50 μg/ml (>2-fold MICD), given that no colonies were recovered for dormant samples treated at this concentration (Fig. 5A). Although complete killing was not observed for dormant bacteria treated with compound 3, a 5-log decrease in CFU per milliliter relative to untreated controls was noticed for dormant Mtb-Lux at 50 and 12 μg/ml (8- and 2-fold the MICD, respectively). Compound 3 showed only moderate bactericidal activity (2-log less than the untreated control) against replicating Mtb-Lux at 50 μg/ml (Fig. 5B). No colonies were recovered from dormant Mtb-Lux samples treated with 50 and 12 μg/ml of compound 4. Furthermore, a 3-log decrease in CFU per milliliter was observed for dormant samples treated with 3.1 μg/ml (6-fold the MICD) relative to untreated controls and a 1-log decrease for those treated with 0.8 μg/ml (<2-fold the MICD). Replicating Mtb-Lux treated with this same compound was only killed (2-log difference) at 50 μg/ml (5-fold the MICR), suggesting that this compound is mostly bacteriostatic against replicating M. tuberculosis but bactericidal against dormant M. tuberculosis (Fig. 5C). These results validate the selectivity of compounds 2, 3, 4, and 5 against dormant M. tuberculosis, suggesting a unique mode of action in which bacterial killing by these compounds, especially compound 4, is observed only for dormant but not metabolically active bacteria.
DISCUSSION
In the current TB treatment regimen, bactericidal drugs are effective at rapidly reducing the bacterial burden in sputum (8), as evidenced by sputum culture conversion within the first 3 months of treatment (34). Nonetheless, patients will face at least 3 to 4 more months of antibiotics to achieve a relapse-free cure. This severely delayed clearance of TB is thought to be due to subpopulations of dormant or persister M. tuberculosis that arise during infection (2). Dormancy renders the bacteria phenotypically tolerant to front-line drugs, such as RIF and INH (20, 35). However, in contrast to its poor activity in vitro, pyrazinamide (PZA) provides potent sterilizing activity in vivo without relapse. As a result, it is credited with shortening TB treatment from 12 to 6 months (36). The discovery of PZA's unique activity was serendipitous rather than through intentional screening for compounds active against dormant bacilli (36). Recent studies have used more focused approaches designed to uncover lead compounds which distinctively inhibit nonreplicating M. tuberculosis (14, 37). With heritable resistance to PZA on the rise (38), novel molecules that are bactericidal against dormant M. tuberculosis will be important components of much-needed new cocktails for shorter treatment without relapse.
Despite continuous efforts to improve TB treatment, the core drug regimen has not changed in the past 4 decades (39). The focus on screening drugs against replicating M. tuberculosis in rich media has proved unsuccessful at filling the need for compounds that can shorten treatment time. Alternatively, research into the physiology of M. tuberculosis during in vivo infection has revealed subpopulations of bacilli in distinct metabolic states and microenvironments within a single host (40). This knowledge is being exploited to uncover essential processes critical to the survival of nonreplicating persisters which are vulnerable to inhibition (41). These recent realizations in the field are also transforming the way we approach drug discovery. Some groups are conducting chemical screens against conditionally essential targets of M. tuberculosis. Whole-cell screening under dormancy-inducing conditions provides a more direct path to discovering novel scaffolds with bactericidal activity against phenotypically tolerant M. tuberculosis. Considering this, the main goal of our study was to identify compounds which could effectively kill dormant bacteria. A two-pronged approach was utilized, which combined the chemical diversity of MNPs with physiologically relevant in vitro screening conditions.
Some dormancy screening models involve exposure of M. tuberculosis to a single environmental cue, such as starvation or hypoxia (42). A streptomycin-dependent M. tuberculosis strain has also been employed for detecting inhibition of persister bacteria (43). Large-scale screens have been performed combining in vivo-like stresses to induce M. tuberculosis dormancy (15, 17, 37). Interestingly, one study showed that using different individual stress conditions to generate nonreplicating dormant M. tuberculosis yielded different hit compounds (14). These data demonstrate the importance of combining the key stresses that trigger persistence to maximize the hits obtained in a dormant screen. Our use of a screening model which mimics several host-like conditions to induce a dormant phenotype enabled the discovery of compounds which may have otherwise gone undetected in a typical in vitro screen. In our screen, 12 out of 19 noncytotoxic dormant hits were selectively active against nonreplicating M. tuberculosis at 20 μg/ml (Fig. 1). This unique hit list allowed us to find pure compounds that were bactericidal against dormant bacteria (Fig. 5). We will pursue hit-to-lead development of the scaffolds for TB treatment. Perhaps more importantly, they may serve as chemical biology tools for the discovery of new druggable targets essential for the viability of dormant M. tuberculosis.
Even though our primary dormancy screen has proven effective at finding dormancy-active hits, we also noted false-positive hits, which further validation revealed were only inhibitory against replicating bacteria (Fig. S2). We speculated this was due to the regrowth stage required for this assay, during which carryover compounds could be inhibiting M. tuberculosis replication. This was evident in the discordant results for RIF and INH seen in the fluorescent readout, which showed minimal differences between replicating and dormant bacilli versus the dramatic drug tolerance of dormant M. tuberculosis measured by CFU enumeration. Similar assay artifacts have been noted by others. A previously reported solution included using activated charcoal in agar to sequester drugs from the medium, which may not be optimal for high-throughput screening (16). In this study, luciferase was selected as a reporter because it is highly sensitive, ATP dependent, and more amenable to high-throughput screening. This readout provides results more rapidly since it does not require a regrowth stage or bacterial growth on agar (44). The modified luminescent dormancy assay could rely on a loss of signal due to cell death after 2 days instead of a signal increase due to growth of uninhibited bacteria during 5 days, which is required for fluorescent assays. Due to the lack of bacterial replication under MSD conditions, by design, this assay will preferentially detect bactericidal drugs. Here, we describe a robust method in which luminescent M. tuberculosis CDC1551 bacteria are exposed to 3 of the more relevant dormancy-inducing conditions, resulting in a severe drug tolerance phenotype.
Screening of natural products has a long and successful history of yielding effective antimicrobial compounds (21). However, the limitations of this strategy, such as laborious deconvolution of complex mixtures, discourages high-throughput screens. Moreover, the use of host-like conditions to test natural products is rarely observed, especially in large primary screens. Due to attrition of drugs during the development pipeline, large-scale screening is necessary to allow prioritization of scaffolds with properties amenable to synthesis and medicinal chemistry optimization. A few recent studies have used nonreplicating M. tuberculosis to test small numbers of samples (45, 46). These small-scale reports illustrate the potential of naturally derived compounds against nonreplicating M. tuberculosis and highlight the need for larger-scale screens of natural products under these conditions. In our study, the use of both replicating and dormant M. tuberculosis to test a library of MNPs enabled the identification of 38 noncytotoxic active peak fractions, half with activity against phenotypically tolerant bacteria. Three of the hits were deconvoluted, leading to the isolation and activity profiling of 5 molecules.
The chemical diversity of MNPs was evidenced in our study upon purifying multiple active compounds from single-hit peak fractions. Unexpectedly, the activity of the Aplysinidae-derived fraction (047.F07) against dormant and replicating M. tuberculosis was a consequence of 2 different scaffolds as opposed to a single dual-active compound. Interestingly, 2 compounds (compounds 1 and 2) had a significantly different activity profile (Table 2). Although the parent fraction (047.F07) had only moderate dormancy activity, the dormancy-active component (compound 2) was more potent than previously thought, suggesting its enrichment upon deconvolution. Compound 2 was active against both replicating and dormant M. tuberculosis, although it was slightly more inhibitory toward dormant M. tuberculosis (2.7-fold) (Fig. 4). Conversely, fistularin-3/11-epi-fistularin (compound 1) was inhibitory against replicating but not dormant M. tuberculosis. The activity of this compound was previously described in a small screen of marine sponge extracts, with a reported MIC of 7.1 μM against replicating M. tuberculosis H37Rv, which is similar to the findings in this study (26, 47).
An advantage of screening semipure peak fractions instead of crude lysates is the facilitated deconvolution of pure compounds which follows the screening process. The finding of a hit peak fraction (031.C02) containing a single compound in our library illustrated this very well. This hit fraction yielded a dormancy-active lipid molecule (compound 3), known as 3-(hexadecyloxy)propane,1,2-diol. Compound 3 was almost 4-fold more inhibitory against dormant than replicating M. tuberculosis (Fig. 4). To our knowledge, this is the first report of this compound having inhibitory activity against M. tuberculosis. This compound and the lipid isolated from peak fraction 047.F07 are both polar lipids and might act as detergents. However, their cytotoxicity against J774 cells was above 200 μg/ml, suggesting a specific mechanism of action for these lipids rather than detergent activity.
In this study, we isolated two terpene quinones with high selectivity against dormant M. tuberculosis from a sponge in the Petrosia (formerly Strongylophora) genus. Terpene quinones, including puupehenone metabolites, have been extensively studied for their antimicrobial and cytotoxic properties (32, 48–52). Nonetheless, this is the first report demonstrating that they selectively inhibit dormant drug-tolerant M. tuberculosis. The puupehenone derivatives identified here showed antituberculosis activity similar to that previously reported for puupehenone itself (26). The MICD of 15-α-methoxypuupehenol, 0.5 μg/ml, was more than 20-fold lower than its MICR of 11.5 μg/ml (Fig. 4 and Table 2). Furthermore, bactericidal activity was observed for this compound against dormant, but not replicating, M. tuberculosis at only 2-fold the MICD (Fig. 5). The methanol adduct of puupehenone had been previously isolated from the sponge Hyrtios sp. (32). Another analog, puupehedione, was isolated from the same organism. Consistent with previous studies, this compound had minor activity against replicating M. tuberculosis (26). However, a 6-fold selectivity for dormant M. tuberculosis was still observed with puupehedione (Table 2).
Target-based studies with puupehenone metabolites have shown that they inhibit NADH oxidase activity in submitochondrial particles and human 5-lipoxigenase (53, 54). Weinstein et al. observed a bactericidal effect for M. tuberculosis's type II NADH oxidase (NDH-2) inhibitors in a murine model (55). The essentiality of an NDH-2 has been demonstrated in replicating bacteria (56). More importantly, however, inhibitors of NDH-2 proteins, such as thioridazine, have increased bactericidal activity toward quiescent M. tuberculosis compared to replicating M. tuberculosis (57). Considering this, we are currently investigating NADH oxidases as possible mycobacterial targets of these dormancy-selective puupehenone metabolites. Obtaining sufficient quantities of natural product (NP) compounds to execute follow-up studies is often a limiting factor with natural product-derived scaffolds. However, the synthesis of puupehenone and its analogs has been shown, enabling a path forward for these molecules (50, 58, 59).
Characterization of the molecular targets for these antimycobacterial MNPs with intriguingly selective activity against dormant M. tuberculosis will help elucidate essential mechanisms underlying the survival of dormant M. tuberculosis during latent infections. The use of alternate in vitro dormancy models to confirm the activity of compounds detected in our assay under a variety of other dormancy-inducing conditions will also provide further evidence that they are likely to work in vivo. Animal models of persistent infection, such as the Kramnik mouse model, will further validate their unique activity profile and determine whether these compounds are capable of shortening treatment time (60, 61).
In conclusion, our findings underscore the significance of screening under in vivo-like conditions, not only to discover novel drugs but also chemical tools to understand the physiologically relevant pathways required to initiate and sustain M. tuberculosis persistence. Future efforts will uncover the mode of action of compounds isolated in this study. Deconvolution of all the dormant-active hit peak fractions is under way and will continue to provide molecules with unique activities against tubercle bacilli under various host-like conditions.
MATERIALS AND METHODS
MNP collection.
The marine natural product (MNP) library from the Harbor Branch Oceanographic Institute (HBOI) at Florida Atlantic University used in this study was generated by extraction of frozen specimens (either exhaustive extraction with ethanol or using an accelerated solvent extractor [Dionex]). Extracts were analyzed by HPLC, and appropriate chromatographic stationary phases were selected for large-scale chromatography. The majority of the fractionations were conducted by medium-pressure liquid chromatography on either the Teledyne Isco CombiFlash Companion or CombiFlash RF+ purification system. Some samples were fractionated using vacuum column chromatography. The library was supplied in 96-well plates for assay. The MNP library was resuspended in 100% dimethyl sulfoxide (DMSO) at 2 mg/ml. Working solutions were prepared at 100 μg/ml in a 2.5% DMSO solution and screened at final concentrations of 20 μg/ml in 0.5% DMSO. Pure compounds were resuspended at 10 mg/ml in 100% DMSO. Refer to File S1 for a detailed description of source materials, purification, and identification of compounds.
Plasmid and reporter strain construction.
Plasmid pVVRG was constructed by utilizing a PCR-based FastCloning method (62) in which a 1,084-bp hsp60-GFP cassette was inserted into the pVV16 smyc:mCherry plasmid (63). Vector primers pVV_FC_F (5′-TTCAGGCCTGGTATGAGTCAGC-3′) and pVV_FC_R (5′-GCTGGATGATGGGGCGA-3′) and insert primers hspGFP_FC_F (5′-GCTGGATGATGGGGCGAGGTGACCACAACGACGC-3′) and hspGFP_FC_R (5′-GCTGACTCATACCAGGCCTGAACTATAGTTCATCCATGCCATGTGTAA-3′) were used. The resulting pVVRG plasmid was confirmed by sequencing and introduced into M. tuberculosis CDC1551 by electroporation. The transformants were selected on 7H10 plates supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC) and 50 μg/ml kanamycin. The resulting Mtb-RG expresses mCherry and GFP constitutively from the smyc promoter and hsp60 promoter, respectively.
Bacterial strains and growth conditions.
M. tuberculosis CDC1551-derived strains containing reporter plasmids were used in this study (Table 3). Screening assays were performed using fluorescent and luminescent reporter strains: (i) the Mtb-RG strain harbors plasmid pVVRG, as described above; and (ii) the Mtb-Lux strain contains pMV306hsp+LuxG13 (a gift from Brian Robertson & Siouxsie Wiles; Addgene plasmid 26161), which was constructed for constitutive expression of the entire lux operon (44) (Table 3). All strains were routinely cultured in Middlebrook 7H9 broth medium supplemented with 0.05% Tween 80 and 10% OADC. Either 50 μg/ml kanamycin or hygromycin was added for maintenance of the reporter plasmids. For experiments requiring a CFU readout, serially diluted bacteria were plated on quad plates containing Middlebrook 7H10 agar supplemented with 10% OADC and 100 μg/ml cycloheximide. For the dormancy screening assays, complete Dubos broth (Difco) medium was prepared according to the manufacturer's instructions, including 10% Dubos medium with albumin supplement (20).
TABLE 3.
Strains and plasmids used in this study
| Name | Descriptiona | Source |
|---|---|---|
| Plasmids | ||
| pVV16-smyc:mCherry | Episomal plasmid, mCherry driven by smyc promoter, Hygr Kanr | This study |
| pVVRG | Episomal plasmid expressing constitutively GFP and mCherry, Hygr Kanr | This study |
| pMV306hsp+LuxG13 | Integration vector constitutively expressing luxCDABE operon, Kanr | Addgene plasmid 26161 |
| Strains | ||
| M. tuberculosis CDC1551 | Wild-type M. tuberculosis | This study |
| Mtb-RG | M. tuberculosis strain expressing mCherry and GFP | This study |
| Mtb-Lux | M. tuberculosis strain expressing lux operon | This study |
Hygr, hygromycin resistance; Kanr, kanamycin resistance.
MNP library screening. (i) Overview of screening scheme.
The HBOI MNP library containing 4,400 peak fractions was initially screened in both primary screening assays (7H9 replicating and multistress dormancy) using the Mtb-RG fluorescent readout. All hit fractions from both screens were tested against mammalian cells to filter out cytotoxic fractions. Peak fractions were prioritized for deconvolution based on their activity against dormant M. tuberculosis, quantity of source sample available, and apparent chemical novelty of extract constituents. Bioactivity-guided fractionation of select hits allowed the isolation of pure compounds. Finally, the compound structures were defined, and the activities of pure compounds were characterized using secondary in vitro replicating and dormancy assays (luminescent readout with Mtb-Lux) (Fig. S1).
(ii) Primary in vitro screening assay.
Bacterial cultures at mid-log phase were diluted in Middlebrook 7H9 OADC broth to an optical density at 600 nm (OD600) of 0.05. The culture and drugs were transferred to solid black 384-well plates (Corning) using a Precision robotic liquid handler (BioTek). All 4,400 peak fractions were screened at 20 μg/ml in a final volume of 30 μl. GFP and mCherry fluorescence was read after a 6-day incubation at 37°C using a Synergy H4 plate reader (BioTek) at wavelengths of excitation (Ex) 480 nm/emission (Em) 528 nm and Ex 580 nm/Em 620 nm, respectively. Each 384-well screening plate contained three control columns: (i) 0.5% DMSO, (ii) 10 μM rifampin (RIF), and (iii) 10 μM isoniazid (INH).
(iii) Secondary in vitro screening assay.
Hit validation and MIC experiments were carried out using luminescence as an alternative readout. Luminescent assays with Mtb-Lux differed only in the starting OD600 (0.01) and use of white instead of black 384-well plates.
To determine the MIC against replicating M. tuberculosis (MICR), 16-point dose-response curves were carried out in triplicate using 200 μg/ml as the initial concentration in a 2-fold dilution series. The controls and serial dilution curves were all prepared to contain a final DMSO concentration of 2%.
(iv) Primary dormancy screening assay.
In order to detect compounds active against dormant M. tuberculosis, the multistress dormancy (MSD) model was used in this study to screen the entire MNP library (4,400 peak fractions) (20). Mtb-RG cultures in log phase were pelleted and resuspended in MSD medium (10% complete Dubos [pH 5.0] containing 0.018% tyloxapol and no glycerol) and incubated in a hypoxia chamber (37°C, 5% O2, 10% CO2) for 9 days prior to the dormancy screening assay.
MSD-adapted Mtb-RG was diluted in the same medium to an OD600 of 0.1 and added to 384-well plates with compounds at 20 μg/ml in a 30-μl final volume. Eight-point dose-response curves for RIF (0.5 μM to 0.003 μM) and INH (1 μM to 0.015 μM) were included in the assay as controls to confirm bacterial tolerance to front-line drugs. These positive controls, as well as the 0.5% DMSO negative control, were done under both MSD and replicating conditions (complete Dubos medium, normoxic atmosphere) to compare drug susceptibilities. The plates were incubated with drugs for 5 days in the hypoxia chamber, after which 30 μl of 2× 7H9 OADC buffered with 100 mM morpholinepropanesulfonic (MOPS) (RPI Intl.) was added for regrowth of the surviving dormant bacteria under normoxic conditions. In addition, controls were plated in duplicate on quad plates for CFU counting. Fluorescence was read following a 7-day regrowth period using a Synergy H4 plate reader (BioTek). RIF and INH at 10 μM and 0.5% DMSO controls were also included in the screening plates for Z score calculation. Hit peak fractions were validated with a CFU readout by plating samples immediately after treatment phase.
(v) Secondary dormancy assay.
Mtb-Lux was used for screening of deconvoluted fractions, validation, and dose-response curves. Cultures were grown and adapted in the dormancy model for 9 days, as described above. Dormant cultures at an OD600 of 0.2 were treated with compounds in white 384-well plates (30 μl total volume per well). The luminescent signal was read after 2, 4, and 6 days of treatment using a Synergy H4 plate reader (BioTek). The MICs of compounds against dormant M. tuberculosis (MICD) were determined using the same dilution series described for the in vitro assay. Sixteen-point dose-response curves were prepared for RIF and INH starting at 12 μM and 500 μM, respectively. These control drugs were used against dormant bacteria and replicating Mtb-Lux in complete Dubos medium for comparison of MICs. Appropriate DMSO-free untreated controls were also included.
(vi) Cytotoxicity counterscreening assay.
J774A.1 macrophages and HepG2 cells were cultured from a freezer stock in Dulbecco's modified Eagle medium (DMEM; Gibco) supplemented with 10% heat-inactivated fetal calf serum (Atlanta Biologicals), 1 mM sodium pyruvate (Mediatech, Inc.), 2 mM l-glutamine (Mediatech, Inc.), and 1% PenStrep (100 U/ml penicillin, 100 μg/ml streptomycin; Gibco). On the day prior to performing the assay, 2.5 × 104 cells/well were seeded in black 384-well plates in a final volume of 30 μl per well. Six hours later, the compounds (20 μg/ml) and the control drugs (0.5% DMSO and 2% Triton X) were added to each well. Cell survival was determined 24 h later based on the reduction of resazurin (64). A stock solution of resazurin was prepared in water at 140 μg/ml and added to each well at a final concentration of 20 μg/ml. Fluorescence was measured following a 4-h incubation at 37°C by excitation at 560 nm and emission at 590 nm using a Synergy H4 plate reader (BioTek). Fractions exhibiting greater than 20% inhibition of J774 cells were considered cytotoxic and eliminated from the study. After deconvolution of noncytotoxic fractions, compounds isolated with lower than 20% cytotoxicity against J774 cells were used for dose-response curves. To determine the IC50 of pure compounds, 12-point dose-response curves were prepared starting at 200 μg/ml. The untreated controls for these experiments were all prepared using a final 2% DMSO solution.
Data analysis.
Z factors and percent inhibition were calculated using the respective formulas. “X” is the drug sample value, μp and μn are the averages of positive (10 μM RIF) and negative (0.5% DMSO) controls, respectively, and σp and σn are standard deviations of the same controls.
The dose-response curves were analyzed using normalized data considering the highest and lowest output values in the curve to be 100% and 0% growth, respectively. A Gompertz model and a nonlinear regression–normalized response curve fit were used to determine the MIC and IC50, respectively, in GraphPad Prism 5 (65), considering the MIC to be 99% killing. The selectivity index (SI) was calculated as IC50/MIC.
Statistical analysis of the CFU results obtained for the pure compounds and control drugs was also done using GraphPad Prism 5. One-way analysis of variance (ANOVA) and Tukey's multiple comparisons posttest were performed to compare treatment groups with each other and with the respective untreated controls. Differences were considered significant at a P value of <0.05.
All the data from hit validation and characterization of pure compounds were averaged from at least 2 independent experiments, with the standard error of the mean (SEM) shown in the figures.
Supplementary Material
ACKNOWLEDGMENTS
C.R.F., K.H.R., and A.E.W. designed the experiments and wrote the manuscript. C.R.F., R.G., and S.G. performed all the biological assays. The chemical aspects of the study were performed by A.E.W., J.R., and P.W. M.C.D. and J.K.R. determined the classification of the marine organisms used. All authors contributed to review and editing of the manuscript.
A.E.W. and K.H.R. received support from NIAID grants 1R21AI105977 and 4R33AI105977. K.H.R. also received support from University of Central Florida grant ORC2012.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00743-17.
REFERENCES
- 1.WHO. 2016. Global tuberculosis report. World Health Organization, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/250441/1/9789241565394-eng.pdf?ua=1. [Google Scholar]
- 2.Gengenbacher M, Kaufmann SHE. 2012. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev 36:514–532. doi: 10.1111/j.1574-6976.2012.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wayne LG, Sohaskey CD. 2001. Nonreplicating persistence of Mycobacterium tuberculosis. Annu Rev Microbiol 55:139–163. doi: 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- 4.Lenaerts A, Barry CE III, Dartois V. 2015. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev 264:288–307. doi: 10.1111/imr.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan C, Barry CE III. 2015. TB drug development: immunology at the table. Immunol Rev 264:308–318. doi: 10.1111/imr.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaler CR, Horvath CN, Jeyanathan M, Xing Z. 2013. Within the enemy's camp: contribution of the granuloma to the dissemination, persistence and transmission of Mycobacterium tuberculosis. Front Immunol 4:30. doi: 10.3389/fimmu.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Tan S, Huang L, Abramovitch RB, Rohde KH, Zimmerman MD, Chen C, Dartois V, VanderVen BC, Russell DG. 2016. Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J Exp Med 213:809–825. doi: 10.1084/jem.20151248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Ambrosio L, Centis R, Sotgiu G, Pontali E, Spanevello A, Migliori GB. 2015. New anti-tuberculosis drugs and regimens: 2015 update. ERJ Open Res 1:00010–2015. doi: 10.1183/23120541.00010-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, Vargas-Vasquez DE, Gao M, Awad M, Park SK, Shim TS, Suh GY, Danilovits M, Ogata H, Kurve A, Chang J, Suzuki K, Tupasi T, Koh WJ, Seaworth B, Geiter LJ, Wells CD. 2012. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 366:2151–2160. doi: 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- 10.Skripconoka V, Danilovits M, Pehme L, Tomson T, Skenders G, Kummik T, Cirule A, Leimane V, Kurve A, Levina K, Geiter LJ, Manissero D, Wells CD. 2013. Delamanid improves outcomes and reduces mortality in multidrug-resistant tuberculosis. Eur Respir J 41:1393–1400. doi: 10.1183/09031936.00125812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thakare R, Soni I, Dasgupta A, Chopra S. 2015. Delamanid for the treatment of pulmonary multidrug-resistant tuberculosis. Drugs Today (Barc) 51:117–123. doi: 10.1358/dot.2015.51.2.2245645. [DOI] [PubMed] [Google Scholar]
- 12.Diacon AH, Pym A, Grobusch MP, de los Rios JM, Gotuzzo E, Vasilyeva I, Leimane V, Andries K, Bakare N, De Marez T, Haxaire-Theeuwes M, Lounis N, Meyvisch P, De Paepe E, van Heeswijk RP, Dannemann B, TMC207-C208 Study Group. 2014. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N Engl J Med 371:723–732. doi: 10.1056/NEJMoa1313865. [DOI] [PubMed] [Google Scholar]
- 13.VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, Wang T, Locher CP, Russell DG. 2015. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog 11:e1004679. doi: 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant SS, Kawate T, Nag PP, Silvis MR, Gordon K, Stanley SA, Kazyanskaya E, Nietupski R, Golas A, Fitzgerald M, Cho S, Franzblau SG, Hung DT. 2013. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem Biol 8:2224–2234. doi: 10.1021/cb4004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warrier T, Martinez-Hoyos M, Marin-Amieva M, Colmenarejo G, Porras-De Francisco E, Alvarez-Pedraglio AI, Fraile-Gabaldon MT, Torres-Gomez PA, Lopez-Quezada L, Gold B, Roberts J, Ling Y, Somersan-Karakaya S, Little D, Cammack N, Nathan C, Mendoza-Losana A. 2015. Identification of novel anti-mycobacterial compounds by screening a pharmaceutical small-molecule library against nonreplicating Mycobacterium tuberculosis. ACS Infect Dis 1:580–585. doi: 10.1021/acsinfecdis.5b00025. [DOI] [PubMed] [Google Scholar]
- 16.Gold B, Roberts J, Ling Y, Quezada LL, Glasheen J, Ballinger E, Somersan-Karakaya S, Warrier T, Warren JD, Nathan C. 2015. Rapid, semiquantitative assay to discriminate among compounds with activity against replicating or nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:6521–6538. doi: 10.1128/AAC.00803-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold B, Warrier T, Nathan C. 2015. A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis. Methods Mol Biol 1285:293–315. doi: 10.1007/978-1-4939-2450-9_18. [DOI] [PubMed] [Google Scholar]
- 18.Bassett IM, Lun S, Bishai WR, Guo H, Kirman JR, Altaf M, O'Toole RF. 2013. Detection of inhibitors of phenotypically drug-tolerant Mycobacterium tuberculosis using an in vitro bactericidal screen. J Microbiol 51:651–658. doi: 10.1007/s12275-013-3099-4. [DOI] [PubMed] [Google Scholar]
- 19.Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64:2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deb C, Lee CM, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, Pawar S, Rogers L, Kolattukudy PE. 2009. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman DJ, Cragg GM. 2016. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 22.Harvey AL, Edrada-Ebel R, Quinn RJ. 2015. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov 14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 23.Farah SI, Abdelrahman AA, North EJ, Chauhan H. 2016. Opportunities and challenges for natural products as novel antituberculosis agents. Assay Drug Dev Technol 14:29–38. doi: 10.1089/adt.2015.673. [DOI] [PubMed] [Google Scholar]
- 24.Montaser R, Luesch H. 2011. Marine natural products: a new wave of drugs? Future Med Chem 3:1475–1489. doi: 10.4155/fmc.11.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes CC, Fenical W. 2010. Antibacterials from the sea. Chemistry 16:12512–12525. doi: 10.1002/chem.201001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El Sayed KA, Bartyzel P, Shen X, Perry TL, Zjawiony JK, Hamann MT. 2000. Marine natural products as antituberculosis agents. Tetrahedron 56:949–953. doi: 10.1016/S0040-4020(99)01093-5. [DOI] [Google Scholar]
- 27.Rogers EW, de Oliveira MF, Berlinck RGS, Konig GM, Molinski TF. 2005. Stereochemical heterogeneity in verongid sponge metabolites. Absolute stereochemistry of (+)-fistularin-3 and (+)-11-epi-fistularin-3 by microscale LCMS-Marfey's analysis. J Nat Prod 68:891–896. [DOI] [PubMed] [Google Scholar]
- 28.Gopichand Y, Schmitz FJ. 1979. Marine natural products: fistularin-1, -2 and -3 from the sponge Aplysina fistularis forma fulva. Tetrahedron Lett 20:3921–3924. doi: 10.1016/S0040-4039(01)86465-0. [DOI] [Google Scholar]
- 29.Zheng CJ, Sohn MJ, Chi SW, Kim WG. 2010. Methyl-branched fatty acids, inhibitors of enoyl-ACP reductase with antibacterial activity from Streptomyces sp. A251. J Microbiol Biotechnol 20:875–880. doi: 10.4014/jmb.1001.01004. [DOI] [PubMed] [Google Scholar]
- 30.Heilbron IM, Owen WM. 1928. The unsaponifiable matter from the oils of elsamobranch fish. Part IV. The establishment of the structure of selachyl and batyl as monoglyceryl ethers. J Chem Soc :942–947. [Google Scholar]
- 31.Magnusson CD, Haraldsson GG. 2011. Ether lipids. Chem Phys Lipids 164:315–340. doi: 10.1016/j.chemphyslip.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Bourguet-Kondracki ML, Lacombe F, Guyot M. 1999. Methanol adduct of puupehenone, a biologically active derivative from the marine sponge Hyrtios species. J Nat Prod 62:1304–1305. doi: 10.1021/np9900829. [DOI] [PubMed] [Google Scholar]
- 33.Hamann MT, Scheuer PJ, Kelly-Borges M. 1993. Biogenetically diverse, bioactive constituents of a sponge, order Verongida: bromotyramines and sesquiterpene-shikimate derived metabolites. J Org Chem 58:6565–6569. doi: 10.1021/jo00076a012. [DOI] [Google Scholar]
- 34.Kanda R, Nagao T, Tho NV, Ogawa E, Murakami Y, Osawa M, Saika Y, Doi K, Nakano Y. 2015. Factors affecting time to sputum culture conversion in adults with pulmonary tuberculosis: a historical cohort study without censored cases. PLoS One 10:e0142607. doi: 10.1371/journal.pone.0142607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garton NJ, Waddell SJ, Sherratt AL, Lee SM, Smith RJ, Senner C, Hinds J, Rajakumar K, Adegbola RA, Besra GS, Butcher PD, Barer MR. 2008. Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med 5:e75. doi: 10.1371/journal.pmed.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Mitchison D. 2003. The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis 7:6–21. [PubMed] [Google Scholar]
- 37.Gold B, Smith R, Nguyen Q, Roberts J, Ling Y, Lopez Quezada L, Somersan S, Warrier T, Little D, Pingle M, Zhang D, Ballinger E, Zimmerman M, Dartois V, Hanson P, Mitscher LA, Porubsky P, Rogers S, Schoenen FJ, Nathan C, Aube J. 2016. Novel cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis. J Med Chem 59:6027–6044. doi: 10.1021/acs.jmedchem.5b01833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitfield MG, Soeters HM, Warren RM, York T, Sampson SL, Streicher EM, van Helden PD, van Rie A. 2015. A global perspective on pyrazinamide resistance: systematic review and meta-analysis. PLoS One 10:e0133869. doi: 10.1371/journal.pone.0133869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zumla A, Nahid P, Cole ST. 2013. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 40.Dhar N, McKinney J, Manina G. 2016. Phenotypic heterogeneity in Mycobacterium tuberculosis. Microbiol Spectr 4:. doi: 10.1128/microbiolspec.TBTB2-0021-2016. [DOI] [PubMed] [Google Scholar]
- 41.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. 2011. The challenge of new drug discovery for tuberculosis. Nature 469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 42.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. 2007. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vocat A, Hartkoorn RC, Lechartier B, Zhang M, Dhar N, Cole ST, Sala C. 2015. Bioluminescence for assessing drug potency against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 59:4012–4019. doi: 10.1128/AAC.00528-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreu N, Zelmer A, Fletcher T, Elkington PT, Ward TH, Ripoll J, Parish T, Bancroft GJ, Schaible U, Robertson BD, Wiles S. 2010. Optimisation of bioluminescent reporters for use with mycobacteria. PLoS One 5:e10777. doi: 10.1371/journal.pone.0010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arai M, Sobou M, Vilcheze C, Baughn A, Hashizume H, Pruksakorn P, Ishida S, Matsumoto M, Jacobs WR Jr, Kobayashi M. 2008. Halicyclamine A, a marine spongean alkaloid as a lead for anti-tuberculosis agent. Bioorg Med Chem 16:6732–6736. doi: 10.1016/j.bmc.2008.05.061. [DOI] [PubMed] [Google Scholar]
- 46.Siricilla S, Mitachi K, Wan B, Franzblau SG, Kurosu M. 2015. Discovery of a capuramycin analog that kills nonreplicating Mycobacterium tuberculosis and its synergistic effects with translocase I inhibitors. J Antibiot (Tokyo) 68:271–278. doi: 10.1038/ja.2014.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Oliveira MF, de Oliveira JH, Galetti FC, de Souza AO, Silva CL, Hajdu E, Peixinho S, Berlinck RG. 2006. Antimycobacterial brominated metabolites from two species of marine sponges. Planta Med 72:437–441. doi: 10.1055/s-2005-916239. [DOI] [PubMed] [Google Scholar]
- 48.Gordaliza M. 2010. Cytotoxic terpene quinones from marine sponges. Mar Drugs 8:2849–2870. doi: 10.3390/md8122849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Copp BR. 2003. Antimycobacterial natural products. Nat Prod Rep 20:535–557. doi: 10.1039/b212154a. [DOI] [PubMed] [Google Scholar]
- 50.Kraus GA, Nguyen T, Bae J, Steadham E. 2004. Synthesis and antitubercular activity of tricyclic analogs of puupehenone. Tetrahedron 60:4223–4225. doi: 10.1016/j.tet.2004.03.043. [DOI] [Google Scholar]
- 51.Xu WH, Ding Y, Jacob MR, Agarwal AK, Clark AM, Ferreira D, Liang ZS, Li XC. 2009. Puupehanol, a sesquiterpene-dihydroquinone derivative from the marine sponge Hyrtios sp. Bioorg Med Chem Lett 19:6140–6143. doi: 10.1016/j.bmcl.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagiwara K, Garcia Hernandez JE, Harper MK, Carroll A, Motti CA, Awaya J, Nguyen HY, Wright AD. 2015. Puupehenol, a potent antioxidant antimicrobial meroterpenoid from a Hawaiian deep-water Dactylospongia sp. sponge. J Nat Prod 78:325–329. doi: 10.1021/np500793g. [DOI] [PubMed] [Google Scholar]
- 53.Ciavatta ML, Lopez Gresa MP, Gavagnin M, Romero V, Melek D, Manzo E, Guo YW, van Soest R, Cimino G. 2007. Studies on puupehenone metabolites of a Dysdea sp.: structure and biology activity. Tetrahedron 63:1380–1384. doi: 10.1016/j.tet.2006.11.088. [DOI] [Google Scholar]
- 54.Robinson SJ, Hoobler EK, Riener M, Loveridge ST, Tenney K, Valeriote FA, Holman TR, Crews P. 2009. Using enzyme assays to evaluate the structure and bioactivity of sponge-derived meroterpenes. J Nat Prod 72:1857–1863. doi: 10.1021/np900465e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinstein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. 2005. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci U S A 102:4548–4553. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Awasthy D, Ambady A, Narayana A, Morayya S, Sharma U. 2014. Roles of the two type II NADH dehydrogenases in the survival of Mycobacterium tuberculosis in vitro. Gene 550:110–116. doi: 10.1016/j.gene.2014.08.024. [DOI] [PubMed] [Google Scholar]
- 57.Rao SPS, Alonso S, Rand L, Dick T, Pethe K. 2008. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quideau S, Lebon M, Lamidey AM. 2002. Enantiospecific synthesis of the antituberculosis marine sponge metabolite (+)-puupehenone. The arenol oxidative activation route. Org Lett 4:3975–3978. [DOI] [PubMed] [Google Scholar]
- 59.Barrero AF, Alvarez-Manzaneda EJ, Mar Herrador M, Valdivia MV, Chahnoun R. 1998. Synthesis of monoterpenic analogues of puupehenone and puupehedione. Tetrahedron 39:2425–2428. doi: 10.1016/S0040-4039(98)00216-0. [DOI] [Google Scholar]
- 60.Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, Bishai WR, Pomper MG, Nuermberger EL, Jain SK. 2012. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis 205:595–602. doi: 10.1093/infdis/jir786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kramnik I, Dietrich WF, Demant P, Bloom BR. 2000. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 97:8560–8565. doi: 10.1073/pnas.150227197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li CK, Wen AY, Shen BC, Lu J, Huang Y, Chang YC. 2011. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol 11:92. doi: 10.1186/1472-6750-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee W, VanderVen BC, Fahey RJ, Russell DG. 2013. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem 288:6788–6800. doi: 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Brien J, Wilson I, Orton T, Pognan F. 2000. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 65.Lambert RJ, Pearson J. 2000. Susceptibility testing: accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J Appl Microbiol 88:784–790. doi: 10.1046/j.1365-2672.2000.01017.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.