

ABSTRACT
A novel fibronectin-based protein (Adnectin) HIV-1 inhibitor was generated using in vitro selection. This inhibitor binds to human CD4 with a high affinity (3.9 nM) and inhibits viral entry at a step after CD4 engagement and preceding membrane fusion. The progenitor sequence of this novel inhibitor was selected from a library of trillions of Adnectin variants using mRNA display and then further optimized for improved antiviral and physical properties. The final optimized inhibitor exhibited full potency against a panel of 124 envelope (gp160) proteins spanning 11 subtypes, indicating broad-spectrum activity. Resistance profiling studies showed that this inhibitor required 30 passages (151 days) in culture to acquire sufficient resistance to result in viral titer breakthrough. Resistance mapped to the loss of multiple potential N-linked glycosylation sites in gp120, suggesting that inhibition is due to steric hindrance of CD4-binding-induced conformational changes.
KEYWORDS: Adnectin, CD4, N17, gp41, HIV inhibitor, human immunodeficiency virus
INTRODUCTION
Due to the facility with which HIV mutates and develops resistance to antiviral drugs, continued success in the treatment of HIV infection necessitates the development of novel agents with unique mechanisms of action. As the armamentarium of anti-HIV agents grows, one crucial avenue of study is the development of long-acting agents to improve compliance and quality of life. To that end, small-molecule inhibitors are currently being tested in advanced clinical trials as intramuscular depot injections (1, 2). Biologics (proteins, peptides, aptamers, and the like) also provide the promise of novel mechanisms of action, with the added advantage that they can often be long-acting, either inherently, as with monoclonal antibodies (MAbs) (3–5), or through the addition of a pharmacokinetic-enhancing protein or peptide (6–9). However, one current limitation of biologics administered through conventional routes is typically poor penetration through the plasma membrane of the cell, which constrains the targets of such drugs to those located extracellularly.
CD4 has been an attractive therapeutic target since its discovery as the primary receptor for HIV infection (10, 11), and its location on the T-cell surface makes it readily accessible to biologics. One obvious approach for utilizing CD4-targeted biologics to treat HIV infection is to block the binding of CD4 by the viral envelope protein gp160, which is generally required for subsequent coreceptor engagement and eventual fusion of the cell and viral membranes (12). However, due to the significant overlap of the gp160 binding site and the major histocompatibility complex class II (MHC-II) binding site on domain 1 (D1) of CD4 (13–16), the likelihood is high that a molecule that binds CD4 and blocks gp160 would also block the essential immune functions that the MHC-II/CD4 interaction affords.
Ibalizumab, an anti-CD4 antibody that is currently in phase 3 clinical trials, blocks HIV entry through an alternate mechanism that still allows gp160 (and, presumably, MHC-II) to bind to CD4 (17). Ibalizumab binds to CD4 primarily through contacts with domain 2 and is thought to block HIV entry at an undefined step subsequent to coreceptor engagement (18). As the loss of glycosylation in the V5 loop of gp160 confers resistance to ibalizumab, a steric clash between gp120 and ibalizumab when in complex with CD4 is thought to play a role in the inhibitory mechanism (19). Although the mechanism of ibalizumab inhibition of HIV entry is not completely understood, ibalizumab inhibition of HIV entry illustrates that epitopes on CD4 other than the gp160 binding site can be targeted safely and effectively for the treatment of HIV infection.
In an effort to identify novel anti-CD4 agents with broad-spectrum anti-HIV activity that do not interfere with the essential immune functions of CD4, we utilized a versatile fibronectin-based scaffold known as an Adnectin. Adnectins are small proteins (10 to 12 kDa) that are derived from the 10th type III domain of human fibronectin (10Fn3) (20–23). Despite limited sequence homology, Adnectins exhibit strong structural similarity to the VH domain of antibodies, with a stable β-sheet core supporting flexible loops that tolerate significant variation, akin to antibody complementarity-determining regions (CDRs) (24, 25). Adnectins with defined binding specificities can be readily selected from libraries of trillions of variants utilizing various established in vitro selection technologies, including mRNA display (26, 27), yeast surface display (28), and phage display (29). Moreover, the lack of disulfides in typical Adnectins enables their expression in high-throughput bacterial systems, facilitating rapid screening of the selection output for leads with desired properties.
The versatility of Adnectins and their potential as therapeutics against a wide variety of therapeutic targets, including vascular endothelial growth factor receptor 2 (VEGFR2) (30–34), epidermal growth factor receptor (35–37), insulin-like growth factor 1 receptor (34, 37), interleukin-23 (35, 38), proprotein convertase subtilisin/kexin type 9 (39), and myostatin (40), have been previously demonstrated. Here, we report the application of the mRNA display selection technology to the discovery and optimization of CD4-binding Adnectins with potent, broad-spectrum anti-HIV-1 activity.
RESULTS
Selected CD4-binding Adnectins that exhibit anti-HIV activity.
The native 10Fn3 domain from human fibronectin (20, 22) was used as the template for construction of novel libraries of Adnectin variants. Two different libraries were created, and each contained trillions of different members with invariant framework sequences and patches of randomized sequence. Libraries differed in the location and number of randomized amino acid residues (Fig. 1; in this publication, numbering and loop definitions follow those described previously [35]). One library, called NP1, contains randomized BC, DE, and FG loops, which are the same three loops used in earlier Adnectin libraries (22, 26), but extends the randomization onto several of the neighboring β-strand surface residues. In addition, the N-terminal segment is extended and randomized. The other library, called WS1, contains randomized CD and FG loops, which are on opposite ends of the 4-strand β-sheet, and also randomizes the intervening surface residues on the C, D, F, and G β-strands.
FIG 1.

Adnectin library designs. (A) Depiction of randomized regions of the 10Fn3 domain within the classic (left), NP1 (center), and WS1 (right) libraries. The classic and NP1 libraries have the BC loop in blue, the DE loop in green, and the FG loop in red. Flanking and N-terminal randomized residues in the NP1 library are shown in purple. The WS1 structure is turned so that the B-C-F-G β-sheet is facing front. In the WS1 structure, randomized residues in the CD strands and loop are green, while those in the FG strands and loop are purple. The structure was taken from the protein with PDB accession number 1FNF. (B) Comparison of the sequences of the WS1 and NP1 libraries with the sequence of the wild-type (WT) 10Fn3 domain. Highlighted sequences in wild-type 10Fn3 indicate the residues that are randomized in the classic library. The color coding is as described in the legend to panel A. Highlighted X's indicate randomized residues in the new libraries. Dashes indicate sequence gaps to facilitate visual alignment across the libraries.
Libraries were subjected to multiple rounds of selection using mRNA display (41) for binding to the ectodomain of human CD4 fused to human IgG1-Fc. The entire selection pathway is outlined in Fig. 2. After each round, the population of selected Adnectins was amplified into a new sublibrary, which was then used in the next selection round. Enrichment of binders from the randomized Adnectin population was monitored by quantitative PCR (qPCR). As seen in Fig. 3, the percentage of the population binding in early rounds was low and variable, while significant enrichment of CD4-binding Adnectins was achieved by round 6 of the primary selection for both libraries, with up to 20% of the sublibrary binding to the target. Adnectin DNAs from rounds 5 and 6 from each selection were cloned and sequenced. Over 800 unique sequences were identified; however, many of these clones exhibited some relatedness to each other and thus could be grouped into a few dozen families. Cloned DNA sequences were expressed in Escherichia coli, and proteins were partially purified using a high-throughput immobilized-metal ion affinity chromatography (IMAC) method similar to that described previously (42). The partially purified Adnectins were first assessed by size exclusion chromatography (SEC), and proteins that exhibited ≤50% aggregation progressed to an enzyme-linked immunosorbent assay (ELISA) that detected binding of the molecules to the CD4-Fc target. Adnectins that exhibited a signal greater than twice the background in the ELISA were further screened for anti-HIV-1 activity using a cell-cell fusion assay (43).
FIG 2.

Pathway of selection to an optimized anti-CD4 Adnectin. Each step used in the selection and optimization process is outlined in the flowchart.
FIG 3.
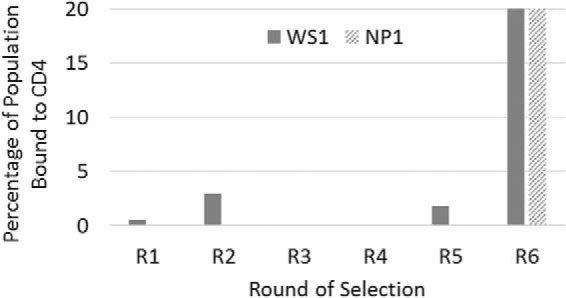
Enrichment of binding populations by mRNA display. Each bar represents one of the libraries that was subjected to selections for binding to human CD4. The percentage of each population that bound to CD4, as determined by quantitative PCR, is plotted as a function of the selection round. In many of the rounds, the binding percentage was a small fraction of 1% and thus does not produce a visible bar in the graph. By round 6, the binding populations derived from both the NP1 and WS1 libraries exceeded 20%.
Using this screening paradigm, two representatives from different Adnectin sequence families were chosen for further characterization. One of these clones, 2886_C08, was derived from the WS1 library, while the other, 3517_G01, was derived from the NP1 library (Table 1). These candidate molecules were further purified and analyzed for their biophysical and antiviral properties. Antiviral potency against NL4-3 virus was examined in cell culture and in a cell-cell fusion assay. In addition, specific binding to the CD4-Fc target and to CD4+ T cells was measured, as were thermal stability and the extent of monomers in the purified lot (Table 2). The last two properties are important parameters for the developability of these polypeptides as potential therapeutics. Both Adnectins demonstrated potent antiviral activity and good physical stability. 3517_G01 demonstrated higher-affinity binding to CD4 than 2886_C08, using both recombinant soluble CD4-Fc (Biacore surface plasmon resonance [SPR]) and CD4-expressing human peripheral blood mononuclear cells (PBMCs). This stronger binding affinity correlated with a lower antiviral 50% effective concentration (EC50) for 3517_G01. However, despite these advantages, 3517_G01 was expressed relatively poorly in the E. coli system and, in both antiviral assays, failed to reach 100% viral inhibition, achieving a maximum percent inhibition (MPI) of only 60% to 80% (Table 2 and Fig. 4). 2886_C08, however, was expressed well in E. coli and was able to achieve an MPI of 100% with increasing concentrations (Fig. 4).
TABLE 1.
Protein sequences of selected anti-CD4 Adnectins
| Library and Adnectin | Sequencea |
|---|---|
| WS1 library | MASTSGVSDVPRDLEVVAATPTSLLISWDAPAVTVXXYXIXYXXXXXXXXXQXFXVPGSKSTATISGLKPGVXYXIXVXAXTXXXXXXXXXXXIXIXYRTE------- |
| 2886_C08 | ...................................HS.H.Q.WPLGWYQRY.V.S...............E.E.Q.R.Y.E.GGGGSQQSFGW.Q.G....GSGS#-- |
| 4945_C06 | .----..............................QS.H.Q.WPLGSYQRY.V.S.................E.Q.R.Y.E.GGADSDQSMGW.Q.G....GDKPSQ# |
| 4945_G05 | .----..............................HS.H.Q.WPLGWYQRY.V.S.................E.Q.R.Y.E.RSGLADESFGW.Q.G....GDKPSQ# |
| 4945_G06 | .----..............................HS.H.Q.WPLGSYQRY.V.S.................E.Q.R.Y.E.GGADSDQSMGW.Q.G....GDKPSQ# |
| 6940_B01 | .G#-G..............................HS.H.Q.WPLGSYQRY.V.S.................E.Q.R.Y.E.GGADSDQSFGW.Q.G...PES----- |
| NP1 library | MASTSGXXXXXXXXDLEVVAATPTSLXIXWXXXXXXXXXXRITYGETGGNSPVQEFTVXXXXXXATISGLKPGVDYTITVYAXXXXXXXXXXXXXXXISINYRTE----- |
| 3517_G01 | ......SSSYLMPS............Y.H.YPIASTIINF..................PGSQVH..................VHYEHKYSELWMGHP........GSGS# |
| 4911_E03 | ......SASYLIPS............S.Y.YPVASTIINF....V.............PGSKST..................VHYEQKYSEYWIGHP........GSGS# |
| 4911_A07 | ......SSPYLMPY............F.R.YGSASSIVKF..................GGTQLH..................VHFEHKYSELWIGHP........GSGS# |
| 4910_A08 | ......YTSYPIPY............Y.H.YWIAATIISF..................PAGQDH..................VHYEEEYSEFWTGHP........GSGS# |
Adnectin sequences derived from the two original libraries are shown below their respective library's sequence. X, residues allowed to vary in the original libraries; number signs, His6 tags; periods, identity with the original library residue; hyphens, a deleted amino acid compared to the original library sequence.
TABLE 2.
Properties of anti-CD4 Adnectins
| Adnectin | Origin library | Cell-cell fusion EC50 (nM) | Replicating virus EC50 (nM) | Affinity at 37°C (nM)a | Cell-binding EC50 (nM)b | Melting temp (°C)c | Monomers (%)d |
|---|---|---|---|---|---|---|---|
| 2886_C08 | WS1 | 105 | 48 | 12 | 7.9 | 49.5 | 97 |
| 4945_C06 | WS1 | 21 | 7.8 | 1.3 | 2.2 | 63.0 | 97 |
| 4945_G05 | WS1 | 36 | 11 | 4.0 | 4.4 | 51.3 | 93 |
| 4945_G06 | WS1 | 7.1 | 4.9 | 2.0 | 2.3 | 55.8 | 98 |
| 6940_B01 | WS1 | 6.9 | 8.5 ± 2.5f | 3.9 ± 0.7f | 2.1 | 77.7 | 96 |
| 3517_G01 | NP1 | 12g | 6g | 0.3 | 0.7 | NDe | 96 |
| 4911_E03 | NP1 | 0.7g | >400g | <0.01h | 0.4 | 58.4 | 99 |
| 4911_A07 | NP1 | 1.6g | >5,500g | <0.01h | 1.0 | 46.5 | 86 |
| 4910_A08 | NP1 | 0.7g | 255 | <0.01h | 1.2 | 55.7 | 97 |
| Ibalizumab | None | 0.2 | 0.2 ± 0.1f | 0.3 ± 0.1f | 0.1 | ND | 95 |
Affinity (equilibrium dissociation constant) measurements made by Biacore SPR.
Binding to CD4-expressing human PBMCs was determined by FACS; no binding to CD8+/CD4− PBMCs was detected for any Adnectin at concentrations up to 1 μM.
Differential scanning calorimetry (DSC) was used to determine the temperature at which the protein unfolds.
Size exclusion chromatography was used to determine the percent monomers.
ND, not determined.
n = 1 for most measurements made during screening; for the activity and affinity of the final lead Adnectin (6940_B01) and ibalizumab, the average of at least three independent measurements ± 1 standard deviation is reported.
Inhibition curves do not surpass 80%.
Off-rates were too slow to measure as they were out of the range of the instrument.
FIG 4.
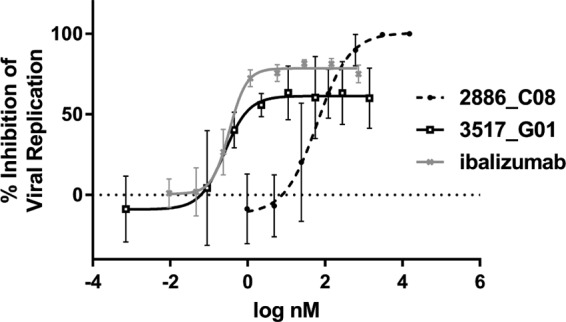
Comparison of inhibition profiles. Overlay of representative inhibition curves for parental Adnectins in the replicating virus assay, with ibalizumab being used as a comparator. The data shown are the averages from 3 experiments ± 1 standard deviation. Best-fit curves were generated with a 5-parameter equation. 3517_G01 was more potent than 2886_C08 in terms of EC50, yet it did not achieve 100% inhibition.
Epitope mapping reveals distinct binding modes for the two Adnectin families.
To explore further the unique properties of these two Adnectins, an epitope mapping experiment was performed. A panel of 5 commercial murine antibodies to human CD4 with known epitopes (see Table S2 in the supplemental material) was used to probe the binding epitope of the Adnectins via a Biacore SPR sandwich approach. A Biacore chip derivatized with protein A was used to capture the various anti-CD4 antibodies, which were in turn used to capture soluble human CD4 (sCD4) consisting of all four ectodomains. The two Adnectins were then flowed over these CD4-antibody complexes, and binding was determined by an increase in the number of response units (RU) on the active flow cell (with antibody plus CD4) compared to that in the reference flow cell (with antibody alone). 2886_C08 was able to bind to CD4 already in complex with all 5 of the antibodies tested, achieving binding levels at or near the theoretical maximum (Fig. 5). Therefore, this Adnectin does not share an epitope with any of the antibodies tested. 3517_G01, however, bound to sCD4 in complex with only 4 of the 5 antibodies, as binding was not detected when sCD4 was immobilized by antibody L120. This antibody is known to bind to domain 4 of CD4, the most membrane-proximal ectodomain (44), and presumably blocks at least a major part of the binding site for 3517_G01. Interestingly, although they appear to share an epitope on CD4, this Adnectin is potent for inhibition of HIV-1 replication, whereas the L120 antibody has little or no antiviral activity (17).
FIG 5.
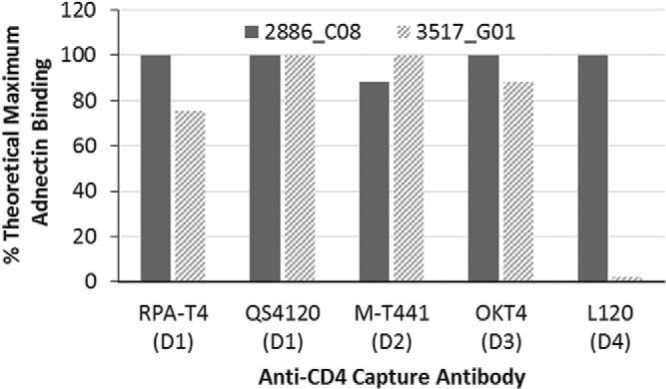
Mapping epitopes of anti-CD4 Adnectins by SPR. Binding of anti-CD4 Adnectins to CD4-antibody complexes. The indicated anti-CD4 antibodies (the domain [D] of CD4 to which the antibody binds is given in parentheses) were captured onto the chip surface via protein A and used to capture soluble CD4. Anti-CD4 Adnectins were flowed over the CD4-antibody complexes to detect cobinding. On the basis of the number of response units (RU) of CD4 bound to each antibody, a theoretical maximum number of RU for binding was calculated for each Adnectin. The actual level of Adnectin binding to CD4-antibody complexes is presented as a percentage of this theoretical maximum. A high percentage of binding denotes that the Adnectin binds an epitope distinct from that the epitope to which the indicated antibody binds.
Optimization selections improve anti-CD4 Adnectin properties.
While the initial selections were successful, in that they yielded Adnectins with a strong affinity for CD4 and potent antiviral activity, further improvements in the functional and biophysical properties of these Adnectins could be achieved through more stringent optimization selections. These selections entailed making new libraries based on the 2886_C08 and 3517_G01 sequences by partially rerandomizing the originally randomized regions to explore the sequence space in more depth. The resulting optimization libraries averaged ∼50% amino acid sequence variation in the directed regions (the CD and FG loops in the WS1 library and the BC, DE, and FG loops plus the N terminus in the NP1 library; see the supplemental material).
Selections were carried out essentially as described above. The concentration of the CD4-Fc target used was decreased as the selection progressed to provide additional stringency and to select for the strongest binders (Table 3). Beginning in round 5, to select for clones with the slowest off-rates, a modified protocol was used, in which the population was allowed to first bind to 10 nM biotinylated human CD4 (D1 to D4) for 30 min. An excess of CD4-Fc was then added, and incubation continued. As complexes of mRNA-displayed Adnectin and biotinylated CD4 dissociate, released Adnectin would be most likely to bind to the CD4-Fc in excess rather than the biotinylated CD4. At various time points, intact Adnectin-biotinylated CD4 complexes were captured onto streptavidin beads; thus, those tight-binding Adnectins with slower off-rates from CD4 were collected. These off-rate selections were repeated for two additional rounds, with the incubation time with the excess CD4-Fc being increased in order to increase the stringency of selection. As before, populations from various rounds were selected for cloning and sequencing on the basis of the observed increase in the degree of target binding, as measured by qPCR (Table 3). Cloned Adnectins were then expressed in E. coli and assessed for biophysical behavior, CD4 binding, and antiviral activity in a cell-cell fusion assay.
TABLE 3.
Optimizing the two lead Adnectin families for stronger CD4 binding
| Round | CD4 concn (nM) | Off-rate selection time point (min) | % of population bound to CD4 |
|
|---|---|---|---|---|
| 2886_C08 library | 3517_G01 library | |||
| 1 | 100 | NAa | 0.1 | 0.1 |
| 2 | 100 | NA | 0.1 | 0.1 |
| 3 | 10 | NA | 0.5 | 1.2 |
| 4 | 1 | NA | 0.1 | 0.5 |
| 5 | 10 | 30 | 3.9 | 4.9 |
| 6 | 10 | 90 | 2.5 | 4.4 |
| 7 | 10 | 300 | 4.8 | 1.2 |
NA, not applicable.
For the 3517_G01 lineage, selection identified three progeny molecules (4911_E03, 4911_A07, and 4910_A08) that were then purified and characterized; their sequences and properties are summarized in Tables 1 and 2, respectively. Compared to the potency and the maximal percent inhibition of their 3517_G01 parent, these Adnectin variants showed improved potency in the cell-cell fusion assay by a factor of 7 to 17, but their maximal percent inhibition still did not surpass 80%. Unexpectedly, in contrast to the cell-cell fusion assay results, their potency against fully replicating virus compared to that of 3517_G01 became severely compromised (at least 40-fold). The reason for this disparity in cell-cell fusion versus replicating virus potency is not understood at this time. As optimization of 3517_G01 failed to produce variants with sufficiently improved virological properties, further development of this lineage was terminated.
Optimization of the 2886_C08 lineage produced multiple variants of 2886_C08 with improved potency in the cell-cell fusion assay and against NL4-3 replication-competent virus. The sequences and properties of three of these Adnectins (4945_C06, 4945_G05, and 4945_G06) are described in Tables 1 and 2, respectively. These variants exhibited 3- to 9-fold improvements in affinity for recombinant CD4 with concomitant 2- to 4-fold improvements in cell binding and 4- to 10-fold improvements in antiviral potency, while they retained good biophysical properties.
One of the optimized variants of 2886_C08, 4945_G06, was identified after the final round of off-rate selection. The sequence of 4945_G06 differed from the sequence of its parent by 6 amino acids in the presumed CD4-binding region, and it had length/sequence differences at the N and C termini. These changes resulted in an approximately 10-fold enhancement of antiviral activity (EC50 = 4.9 nM), a 6-fold enhancement of affinity for CD4 by SPR (equilibrium dissociation constant = 2 nM), and a 4-fold enhancement of binding to cell-associated CD4 on PBMCs (EC50 = 2.2 nM). In addition, the thermal stability of 4945_G06 increased by more than 6°C (55.8°C) compared to that of the parent, 2886_C08. Because of its superior potency, 4945_G06 was chosen from among all the optimized variants of 2886_C08 for additional characterization.
Additional epitope mapping indicates a distributed epitope for the 2886_C08 Adnectin family.
As the previous epitope mapping experiment could not positively identify the binding site for this family of related Adnectins, a competitive SPR assay was designed. 4945_G06 was covalently coupled to a Biacore T-series CM5 chip and used to capture the sCD4 protein. Then, gp120 or one of an expanded panel of various anti-CD4 monoclonal antibodies with known binding epitopes was flowed over the captured CD4 and analyzed for binding. If the binding site for 4945_G06 overlapped the binding site for gp120 or a known MAb, binding to the captured CD4 would be inhibited. Seven different MAbs covering binding sites on each of the 4 domains of CD4 were examined (Table S2). A positive control, 4945_G06 itself, was inhibited from binding to CD4 in this assay, as expected, but none of the 7 MAbs were inhibited from binding to CD4, and neither was gp120. Thus, this CD4 Adnectin family binds to a unique site on CD4 not blocked by any of these 7 antibodies or by gp120. The ability of gp120 to bind to CD4 already in complex with the Adnectin suggests that the Adnectin exerts inhibitory activity by a mechanism other than blocking viral attachment to CD4.
To gain additional insight into the binding epitope, experiments were conducted with partial CD4 proteins. Fragments of CD4 containing only domains 1 and 2 (D1D2) or only domains 3 and 4 (D3D4) were expressed and purified. A Biacore chip was derivatized via amine coupling with 4945_G06, anti-CD4 antibody OKT4 (which binds domain 3) (44), and ibalizumab (which binds domain 2) (18) on separate flow cells (Fig. S2), and the CD4 fragments were then flowed over these surfaces. The observed number of binding RU was then expressed as a percentage of the possible number of RU by normalizing the responses of the D1D2 and D3D4 fragments to the response of the D1D4 fragment, accounting for molecular weight differences (Table 4). As expected, both the ibalizumab and OKT4 antibodies bound only to those CD4 fragments containing their respective epitopes. 4945_G06 bound very poorly to the D1D2 and D3D4 fragments, suggesting that the high-affinity binding site for this Adnectin either is at the junction of domains 2 and 3 or contains noncontiguous binding sites residing on both D1D2 and D3D4. Additional work toward identifying and characterizing this epitope is ongoing.
TABLE 4.
Binding of anti-CD4 Adnectin to CD4 fragments
| Immobilized ligand | % maximal bindinga |
||
|---|---|---|---|
| D1D4 | D1D2 | D3D4 | |
| 4945_G06 | 100 | 1 | 9 |
| OKT4 | 100 | 0 | 69 |
| Ibalizumab | 100 | 43 | 0 |
Binding was normalized to that of D1D4.
4945_G06 exhibits specificity for human CD4.
Binding studies were performed to determine the binding specificity of the 4945_G06 Adnectin. Flow cytometry analysis of PBMCs from different species was used to determine the relative strength of 4945_G06 binding to these cells and, by inference, the CD4 protein from that species. Isolated PBMCs were exposed to the 4945_G06 Adnectin (which contains a His6 tag) at various concentrations, and Adnectin that was bound to cells was detected with a fluorescently labeled anti-His tag antibody. The binding EC50 for human PBMCs was found to be 2.3 ± 0.8 nM (Fig. 6), which agrees well with the affinity measured by SPR (Table 2). Interestingly, 4945_G06 is highly specific for binding to human CD4, as binding to PBMCs from a cynomolgus monkey was >500-fold weaker than that to human PBMCs, while no significant binding to PBMCs from a marmoset, African green monkey, or mouse was detected at concentrations up to 10 μM (Fig. 6). In each of these experiments, no binding to CD8+/CD4− PBMCs was observed, indicating no nonspecific interaction with other cell surface proteins (data not shown). Thus, 4945_G06 has a stronger specificity for CD4 from humans than CD4 from other nonhuman primates.
FIG 6.
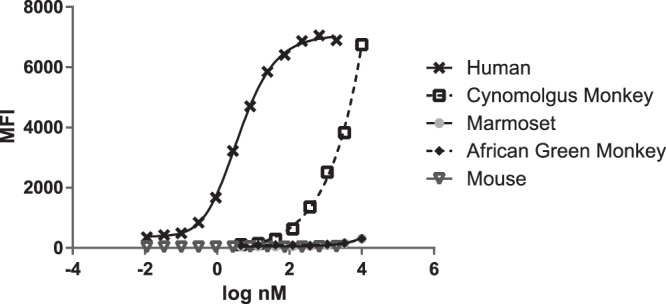
Binding of 4945_G06 to CD4-expressing peripheral blood mononuclear cells (PBMCs) of various species. PBMCs were obtained from whole blood from each of the indicated species. 4945_G06 was incubated in a titration series with cells, and binding was detected with a fluorescently labeled anti-His tag antibody by FACS. CD3+/CD8− cells (presumed to be CD4+) were gated using species-matched antibodies (see Materials and Methods). Strong binding to human PBMCs and weak binding (>500-fold less) to cynomolgus monkey PBMCs were observed, with no binding to marmoset, African Green monkey, or murine PBMCs being observed. The average signal from duplicates is plotted, with 4-parameter best-fit curves being shown. MFI, mean fluorescence intensity.
Additional sequence adjustments further improve anti-CD4 Adnectin properties.
Although 4945_G06 has improved antiviral and biophysical properties compared to the parent 2886_C08, additional modifications were required. For instance, 4945_G06 contains a methionine residue at position 87, which poses an oxidation risk and affects the developability of this Adnectin as a biologic inhibitor. Reversion of this methionine to phenylalanine, the residue present at this position in the parent 2886_C08, was found to be well tolerated and was thus incorporated. In addition, the His6 tag was moved to the N terminus, and slight adjustments to the N- and C-terminal sequences that would allow linkage with other biologic molecules, if desired, were made. Finally, an E97P substitution was made. This substitution has been found in other instances to enhance Adnectin thermal stability (D. Lipovšek, personal communication). When all these changes were incorporated into the final Adnectin, 6940_B01, the thermal stability increased by almost 22°C to 77.7°C with a minimal loss of CD4-binding and antiviral activity (Table 2).
The anti-CD4 Adnectin exhibits broad-spectrum activity.
6940_B01 was tested in a cell-cell fusion assay against a panel of 124 envelopes (gp160) from clinical HIV-1 isolates spanning 11 subtypes (Table S3). Each of the cloned envelopes was transiently expressed (45), and the EC50 for the activity of 6940_B01 against each envelope was determined. A fold change (FC) in potency, defined as the ratio of the EC50 against that envelope to the EC50 against a reference envelope derived from the LAI strain of HIV-1, which was assessed in the same experiment, was then calculated for each envelope. The result of this analysis is shown in Fig. 7. All the envelopes tested exhibited fold changes in potency of less than 5, indicating that all 124 envelopes, regardless of subtype, were susceptible to the anti-CD4 Adnectin 6940_B01. Additionally, 6940_B01 was tested against a panel of 6 clinical isolates of various subtypes and coreceptor specificities (Table S4). EC50s ranged from 0.6 to 6.5 nM, indicating full susceptibility to 6940_B01.
FIG 7.
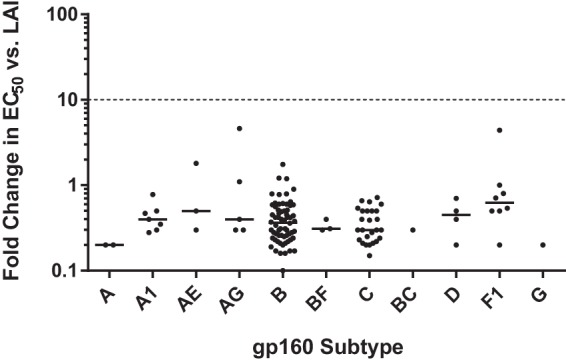
Broad-spectrum activity of 6940_B01 against clinical envelopes. The activity of 6940_B01 against 124 gp160 variants from 11 subtypes was evaluated in a transient-expression cell-cell fusion assay. Potency is reported as the fold change in EC50 (log scale) relative to the EC50 against gp160 from LAI virus. The solid line for each group indicates the median value. Values greater than 1 indicate a weaker potency (a higher EC50) against a given strain than against LAI. Given the intrinsic assay variability, only fold change values greater than 10 (dashed line) or less than 0.1 were considered significant. Actual fold change values are listed in Table S3 in the supplemental material.
Resistance selection with 6940_B01.
In an effort to select viruses resistant to 6940_B01 in cell culture, MT-2 cells were infected with NL4-3 virus in the initial presence of a 2× EC50 (∼17 nM) of 6940_B01. Cell cultures were observed until they exhibited signs of virus breakthrough (cytopathic effect [CPE] and/or syncytium formation), at which time a sample of the supernatant was used to infect new MT-2 cells in the presence of double the 6940_B01 concentration from the previous passage. The process was continued with successive doublings until the concentration of drug reached 10.7 μM, where it was then kept constant through multiple passages. NL4-3 virus was also passaged concurrently without 6940_B01 selection as a control arm. Virus growth was observed to be slower in the 6940_B01 selection sample, and it was not until passage 30 (151 days in culture) that virus seemed to grow fast enough to warrant harvesting. Examination of the passage 30 virus population revealed an 11.5-fold reduced susceptibility to 6940_B01. Population sequencing of the virus stocks identified 4 amino acid changes in gp160 compared to the sequence of the control virus (Table 5). A recombinant NL4-3 virus containing a gp160 gene with all four of these substitutions exhibited a 7-fold loss of susceptibility to 6940_B01. All of these amino acid changes resided in gp120, and each resulted in the loss of a potential N-linked glycosylation site (PNGS). For the anti-CD4 MAb and anti-HIV agent ibalizumab, resistance also mapped to the loss of PNGSs, and structural studies suggest that the loss of these PNGSs removed a steric block that prevented the drug-bound CD4-gp120 complex from undergoing the conformational change required for coreceptor binding (18, 46, 47). Our results identifying resistance through the loss of PNGSs suggest that the mechanism of action of 6940_B01 may be similar to that of ibalizumab, although the locations of the PNGSs that are lost differ (48), and the results described above establish that the binding sites for the two molecules do not overlap.
TABLE 5.
Amino acid substitutions after prolonged selection in the presence of 6940_B01
| Virus | Passage no. | No. of days in culture | Fold reduced susceptibility | Amino acid at the following positiona: |
|||
|---|---|---|---|---|---|---|---|
| S143 | N197 | N301 | S465 | ||||
| Control (no drug) | 30 | 151 | 1 | S | N | N | S |
| Selected virus population | 30 | 151 | 11.5 | Rb | Db | Kb | Pb |
| Recombinant virus | 7 | R | D | K | P | ||
The amino acid sequence and the position numbering are based upon the HXB2 sequence starting with gp120.
Results in the loss of a potential N-linked glycosylation site.
Retrospectively, virus populations from approximately every third passage were sequenced and examined for susceptibility to 6940_B01. As can be seen from Fig. 8, although the loss of 1 of the potential N-linked glycosylation sites occurred by passage 9, these viruses did not exhibit a significant change in susceptibility to 6940_B01. At passage 18, various levels of the 3 additional lost PNGSs were mutated in a percentage of the population, and the fold change in susceptibility to 6940_B01 increased to ∼6-fold. At passage 27, all 4 PNGS mutations were fixed and susceptibility decreased 11.5- to 17-fold compared to that of the wild type. Thus, decreased susceptibility to 6940_B01 mapped to the loss of multiple potential glycosylation sites within gp120.
FIG 8.
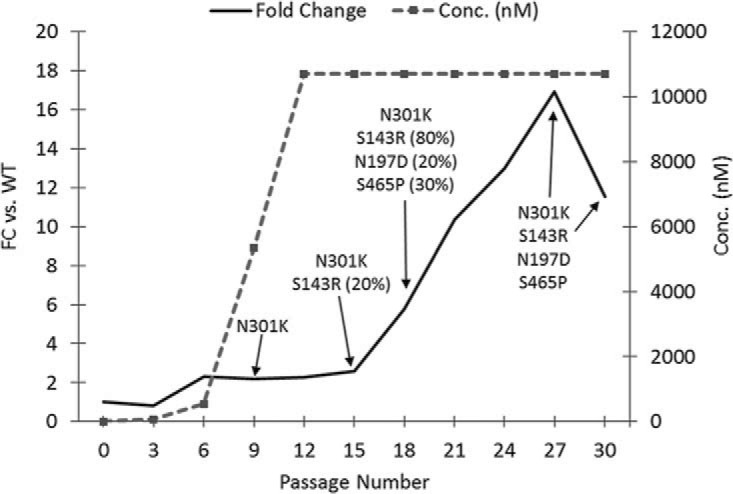
Selection of resistance to 6940_B01. NL4-3 virus was passaged in the presence of increasing concentrations of 6940_B01 (dashed line). Breakthrough virus from every third passage was analyzed for decreased susceptibility to 6940_B01 (solid line), and the gp160 gene was population sequenced to identify selected changes. Percentages represent the estimated percentage of genes within the population that contained the substitution. If there is no number next to a sequence, the prevalence of this mutation is believed to be 100%.
DISCUSSION
As emphasis on the development of longer-acting anti-HIV agents increases, biologic molecules have gained greater interest for both their therapeutic and vaccine potential. For instance, the anti-CCR5 antibody PRO 140 and the anti-CD4 antibody ibalizumab are MAbs targeted for therapy (49), while broadly neutralizing antibodies to gp120 are being developed for multiple uses (50, 51). In addition to antibodies, a variety of peptides (52–54), aptamers (55–57), and alternative scaffolds (58) have also been explored for HIV-1 inhibition. In this study, we report on the discovery of the Adnectin 6940_B01 as a novel CD4-binding protein with potent and broad antiviral properties. The selection and subsequent optimization of this Adnectin utilizing mRNA display technology have resulted in a high-affinity, thermally stable, and highly specific molecule.
Initially, multiple Adnectin library designs were utilized in the selection process, with the goal that each might be capable of interacting with CD4 in a unique way, thus providing a wide variety of epitopes and interaction modes and increasing the probability of finding an Adnectin with the desired therapeutic profile. This strategy resulted in leads that showed antiviral activity and represented multiple sequence families. Leads from two of these families, represented by 2886_C08 and 3517_G01, warranted further characterization and optimization on the basis of their biophysical and antiviral behavior.
These two Adnectin families displayed very different properties. First, they bound to different epitopes on CD4, as determined by binding competition studies with antibodies of known epitope. The 3517_G01 family likely binds to domain 4 (the most membrane-proximal domain), on the basis of its competition with antibody L120 (59) for binding to CD4, while the 2886_C08 family showed no competition with any of the antibodies tested. Second, the 3517_G01 family failed to reach 100% inhibition of viral replication, despite an EC50 more potent than that of the 2886_C08 family. It is possible that inhibition of HIV-1 by both of these families is due to steric hindrance of CD4-bound gp160 conformational changes, as suggested by a resistance profile similar to that of the anti-CD4 antibody ibalizumab (at least for the 2886_C08 family). If so, the 3517_G01 family may exhibit suboptimal maximal percent inhibition (MPI) of HIV-1 inhibition by allowing more freedom of movement of the CD4-bound gp160 than the 2886_C08 family, as a result of either binding farther away from CD4-bound gp160 or the particular geometry with which it binds, even though it has a greater affinity for CD4. This suboptimal MPI phenotype has also been observed with ibalizumab for some viral strains and could be mitigated through the addition of glycosylation sites to the light chain of ibalizumab, which is thought to increase the steric clash between the antibody and gp120 during the entry process (19). Lastly, the optimized variants of 3517_G01 showed a disconnect in the potency determined by the cell-cell fusion assay from that determined by the replicating virus assay, while the potency of the 2886_C08 family showed good agreement in these systems. The cause of the potency disconnect between the cell-cell fusion and replicating virus assays for the 3517_G01 family is unclear but could relate to the different cell types used in the two assay systems or to differences in the efficiency or kinetics of cell-cell fusion from that of virus-cell fusion. Moreover, the behavior of the 3517_G01 family provides reason for caution against reliance on a single in vitro measure of activity for characterization of entry inhibitors.
These distinctions in virologic profiles between the 3517_G01 and 2886_C08 Adnectin families may indicate different mechanisms of action. However, both the disconnect in potency assay results and the inability to reach 100% inhibition of replication, which may increase the likelihood of resistance development in vivo, caused us to halt further characterization work on the 3517_G01 family. Further optimization of the 2886_C08 family was performed through more stringent selections and site-specific modifications to identify the final inhibitor candidate, 6940_B01. This optimized Adnectin exhibits potent inhibitory activity against a broad array of HIV-1 envelopes and has physical properties favorable for development (Table 2). Specifically, it possesses a high affinity for CD4 (3.9 ± 0.7 nM) that is similar in magnitude to its EC50 against NL4-3 virus (8.5 ± 2.5 nM). In addition, it retains activity against 124 different envelopes encompassing many different subtypes in a cell-cell fusion assay. The high thermal stability (77.7°C) and monomericity (96%) of the protein are properties that should aid in its manufacture and maintenance of its integrity during storage.
The Adnectin 6940_B01 could exert its anti-HIV activity through a variety of mechanisms, the most obvious of which would be to block the binding of gp120 to CD4. This mechanism is unlikely, however, given that recombinant gp120 can cobind recombinant CD4 that is already in complex with a progenitor Adnectin from this family. Also, induction of CD4 downregulation from the cell surface is an unlikely mechanism, as fluorescence-activated cell sorting (FACS) studies of MT-2 cells during prolonged incubation with micromolar concentrations of Adnectin showed no change in the staining intensity of anti-CD4 antibodies (data not shown). It remains possible that the Adnectin could impair the binding of whole virions to cells or adversely impact the kinetics of the gp120-CD4 interaction, but on the basis of the resistance profiling data, the mechanism of action of 6940_B01 likely involves some steric block to entry events after CD4 binding that can be alleviated by the loss of glycosylation sites in gp120, akin to the mechanism of the anti-CD4 antibody ibalizumab (19). Indeed, viruses selected for resistance to this Adnectin were also resistant to ibalizumab (data not shown), although the converse was not necessarily the case; this asymmetry in resistance may be due to different binding sites on CD4 or may indicate some other distinction in the mechanisms of these anti-CD4 entry inhibitors. This distinction is further borne out by the locations of the lost potential glycosylation sites. The lost glycosylation sites in gp120 during selection of resistance to 6940_B01 were scattered throughout gp120 (S143 in the V1 loop, N197 in the V1-V2 stem, N301 in the V3 loop, S465 in the V5 loop), while glycosylation sites in the V5 loop appear to be primary determinants of ibalizumab resistance (48).
While no conclusive assignment of a binding epitope can be made on the basis of the current data, members of the 2886_C08 Adnectin family, of which 6940_B01 is a member, appear to bind CD4 via an epitope that is distinct from the ibalizumab epitope in D2 (18, 60) and that either includes the D2-D3 junction or is distributed between the D1D2 and D3D4 segments. Comparison of the amino acid sequences of the CD4 extracellular domains from binding and nonbinding species (see Fig. S3 in the supplemental material) reveals that amino acid substitutions in nonbinding species are distributed throughout the 4 extracellular domains, including the region around the D2-D3 junction, which provides little insight for distinguishing between these possibilities. In either case, binding of the Adnectin has the potential to alter the flexibility and spatial relationships among the CD4 ectodomains, which could in turn diminish the ability of CD4 to interact productively with HIV-1 virions. Indeed, small-angle X-ray scattering (SAXS) studies of CD4 in solution (61) have indicated that gp120 binding induces a bifold collapse of the D1D2 half of CD4 against the D3D4 half by bending at the D2-D3 junction, which could be important for allowing contact of CD4-bound gp120 with the coreceptor. While the results of subsequent studies have contradicted these results (18), it remains likely that some degree of flexibility in CD4 is required to permit bound gp120 to undergo its own structural rearrangements prior to or even after coreceptor contact, which may be precluded by binding of this Adnectin family. Additional studies to define more precisely both the binding epitope and the mechanism of viral inhibition are ongoing.
Regardless of the exact inhibition mechanism, virus exposed to the optimized lead representative of this family, 6940_B01, required 30 passages (151 days) in culture and the loss of multiple PNGSs to acquire sufficient resistance to result in titer breakthrough. Additionally, a survey of 124 clinical envelopes indicated broad susceptibility to 6940_B01 across major subtypes and recombinant strains. Importantly, 6940_B01 was able to achieve 100% inhibition of all strains within this diverse panel, unlike the 3517_G01 Adnectin family. Thus, the 6940_B01 anti-CD4 Adnectin represents a novel HIV-1 biologic entry inhibitor with a high potency, a broad spectrum of activity, and physical properties amenable to clinical development.
MATERIALS AND METHODS
Recombinant CD4 proteins.
Two different recombinant human CD4 molecules were used as targets during selection for a binding Adnectin. A version of CD4 that consisted of amino acids 26 to 390 of the extracellular domain of human CD4 (R&D Systems) was biotinylated on free amines with N-hydroxysulfosuccinimide-(polyethylene glycol)4-biotin (Pierce) according to the manufacturer's specifications. In addition, an Fc fusion (CD4-D1-D4-Fc) was designed by fusing the C terminus of extracellular domains 1 to 4 (residues 26 to 389 of the human CD4 sequence) to the hinge of human IgG1-Fc at position 232 (according to the numbering of Kabat et al. [62]) with an additional C233S mutation. CD4-D1-D4-Fc was expressed via transient transfection of HEK293 cells and purified by standard protein A chromatography.
Additional soluble recombinant fragments of human CD4 were purchased (D1D2; ectodomains 1 and 2 [catalog number 7002-2; ImmunoDX]) or expressed with C-terminal His6 tags in a baculovirus system and purified by nickel affinity chromatography (D3D4 [amino acids 207 to 386] and D1D4 [amino acids 26 to 394]).
Adnectin library design and construction.
Adnectin libraries were constructed via PCR using overlapping oligonucleotides synthesized with phosphoramidite trimers encoding defined mixtures of amino acids in specific locations, similar to methods described previously (26, 27, 63, 64). Two novel libraries, details of which are provided in Fig. 1 and in the supplemental material, were employed for this selection.
Primary selection of Adnectins binding human CD4 by mRNA display.
The overall selection strategy is outlined in Fig. 2. The first round of mRNA display selection was carried out essentially as described elsewhere (26, 41, 65) with CD4-D1-D4-Fc as the target. Briefly, transcription of the library DNA into mRNA was accomplished with T7 RNA polymerase (New England BioLabs). Two nanomoles of purified library mRNA (RNeasy kit; Qiagen) was reacted with 3 nmol of the puromycin linker and then translated in 2 ml of rabbit reticulocyte lysate (Ambion). mRNA-Adnectin fusions were purified with oligo(dT) cellulose beads and then reverse transcribed (SuperScript II reverse transcriptase; Thermo Fisher Scientific, Waltham, MA) to yield 5.4 pmol of mRNA-Adnectin fusions hybridized to their respective cDNAs to eliminate the RNA secondary structure. Prior to selection, 0.4 ml of protein G-coated magnetic beads (Invitrogen) was washed with buffer comprised of phosphate-buffered saline (PBS) with 1 mM CaCl2, 0.5 mM MgCl2, and 0.025% Tween 20 and blocked overnight using a similar buffer with the addition of 1 mg/ml bovine serum albumin (BSA) and 0.1 mg/ml sheared salmon sperm DNA (Ambion). One-third of the library of mRNA-Adnectin fusions was allowed to equilibrate with 100 nM human CD4-D1-D4-Fc for 45 min. This was followed by capture with protein G-coated magnetic beads for 30 min. The beads were washed, and cDNA encoding bound Adnectins was eluted with 100 mM KOH. The eluted cDNA was then amplified by PCR with oligonucleotides 0 and 14 (see Table S1 in the supplemental material). The resulting PCR product after round 1 was then quantitated using qPCR. The fraction of the library that bound CD4-D1-D4-Fc after one round was determined by comparing the qPCR measurements of protein-mRNA fusion molecules before and after the selection step. cDNA from round 1 was then transcribed with T7 RNA polymerase to feed the next selection round.
Rounds 2 to 6 were carried out on a smaller scale. The protocol was similar to that described above, except that 150 pmol of library mRNA was translated in 0.2 ml of rabbit reticulocyte lysate. In addition, a negative selection (preclear) step was first carried out prior to each positive selection round by exposing the library of mRNA-protein fusions to 100 nM an unrelated VEGFR2-Fc fusion protein prebound to protein G-coated magnetic beads for 30 min. This step was intended to deplete any Adnectins that bound to Fc or to the protein G beads. After carrying out the preclear three times, the portion of the library that did not bind to the unrelated Fc-tagged protein was then allowed to proceed into the positive selection round against CD4-D1-D4-Fc. The selection conditions were identical to those described above for round 1, except that equilibration with 100 nM target occurred for 30 min during these rounds.
The PCR products obtained after rounds 5 and 6 were amplified with cloning primers that added an N-terminal NcoI restriction site and a C-terminal His6 tag and BamHI restriction site. The resulting PCR product was cleaved with NcoI and BamHI and ligated into the NcoI and BamHI sites of pET9d (Novagen). The ligated plasmids were transformed into E. coli strain BL21(DE3) pLysS and were plated on kanamycin- and chloramphenicol-containing LB agar plates. E. coli colonies, each expressing a single Adnectin clone, were grown in 96-well plates for sequencing, expression, and assays.
Optimization and off-rate selection methods.
Once initial lead Adnectins with desirable antiviral properties were identified, new libraries were produced on the basis of those sequences (see the supplemental material) and subjected to more stringent selections aimed at finding variants with lower off-rates. These optimization selections followed a streamlined version of the primary selection protocol that substituted a combined in vitro transcription and translation system consisting of purified E. coli components (PURExpress; New England BioLabs) for the previously separate transcription and translation reactions. The first round of selection was carried out using 20 pmol of library DNA as the input (approximately 1.2 × 1013 in library size) and 400 pmol of the puromycin linker in 80 μl of in vitro transcription/translation volume. At this point, mRNA-protein fusions hybridized to their respective cDNAs were selected against 100 nM CD4-D1-D4-Fc as described above for the primary selections. The stringency was increased during rounds 2 to 4 by evaluating binding to 100 nM, 10 nM, and 1 nM CD4-D1-D4-Fc, respectively.
For the 5th round, the protocol was further modified to focus selection on variants with lower off-rates. The Adnectin libraries were bound to 10 nM CD4-D1-D4-biotin at room temperature for 30 min. At that time, a 100-fold molar excess of CD4-D1-D4-Fc was added to the binding mixture. Adnectins that dissociated from the biotinylated target would likely rebind to the nonbiotinylated target. After a 30-min incubation with CD4-D1-D4-Fc, the Adnectins that remained bound to CD4-D1-D4-biotin (and which presumably had lower off-rates) were captured with streptavidin-coated magnetic beads. These beads were washed, eluted, and measured by qPCR as before. This eluted cDNA was also fed into the 6th round of selection, wherein the time of incubation with CD4-D1-D4-Fc was increased to 90 min. In the 7th round of selection, the time of incubation with CD4-D1-D4-Fc was further increased to 300 min. Following amplification by PCR, the round 6 and 7 Adnectin variants were cloned, sequenced, and expressed as described above for the primary selections. The complete selection pathway is summarized in Fig. 2.
Expression and purification of Adnectins.
A complete description of high-throughput Adnectin expression and purification methods is provided in the supplemental material. Briefly, each clone was transformed into E. coli BL21(DE3) pLysS and grown in cultures of MagicMedia expression medium (Life Technologies). After centrifugation, bacterial pellets were chemically lysed. Lysates were clarified by filtration, and Adnectins were purified via their His6 tags, using HisPur cobalt spin plates (Thermo Fisher Scientific). Estimates of yield (typically, 10 to 200 μg in 100 μl) and purity (typically >80%) were obtained using a LabChip 90 (Caliper) or LabChip GX (PerkinElmer) capillary electrophoresis system. For larger-scale expression (5 to 10 mg), additional column chromatography steps, such as size exclusion chromatography (SEC) or anion exchange chromatography, were utilized to achieve a purity of >95%.
Purified Adnectins were analyzed for monomericity by SEC using an Agilent high-performance liquid chromatography instrument fitted with a Superdex 75 5/150 column (GE Healthcare) with 100 mM sodium phosphate, 100 mM sodium sulfate, 150 mM sodium chloride, pH 6.8, as the mobile phase at a flow rate of 0.3 ml/min. Melting point determinations by differential scanning calorimetry (DSC) were conducted with a MicroCal VP-capillary DSC instrument (Malvern) with protein diluted to 0.5 mg/ml in PBS and a scan rate of 1°C/min.
ELISA for identifying CD4-binding Adnectins.
Nunc MaxiSorp 384-well plates were coated overnight with 2 μg/ml of anti-His6 antibody mAb050 (R&D Systems) in PBS (20 μl per well). Coated plates were blocked in 20 μl per well casein-PBS (Thermo Fisher Scientific) for 1 to 2 h at room temperature. After the blocked plate was washed 3 times in PBS-Tween 20 (PBST), Adnectins were added (15 μl per well semipurified Adnectin diluted 1:10 in casein-PBS) and the mixture was incubated for 1 h at room temperature. For initial screening, the concentration of each Adnectin varied but was generally in excess of 1 μM. A nonbinding Adnectin (a 10Fn3 domain with the native RGD integrin-binding motif mutated to SGE) was included as a negative control to determine the assay background signal. The plates were washed 3 times in PBST, and then 10 nM CD4-D1-D4-Fc diluted in casein-PBS was added (15 μl per well). The plates were incubated for 1 h at room temperature and washed 3 times in PBST, and then goat anti-human IgG-Fc conjugated with horseradish peroxidase (Pierce), diluted 1:15,000 in casein, was added (15 μl per well). After a further incubation for 1 h at room temperature, the plates were washed 3 times in PBST and developed with 15 μl per well room temperature tetramethylbenzidine substrate (Invitrogen). The reaction was stopped at 7 min or less with 15 μl per well 2 N sulfuric acid. The plates were read (by determination of the optical density at 450 nm) in a SpectraMax plate reader (Molecular Devices).
Cells and viruses.
MT-2 cells, HEK293T cells, CEM-NKR-CCR5-Luc cells, and the proviral DNA clone of NL4-3 virus were obtained from the NIH AIDS Research and Reference Reagent Program. B6 cells, which contain a long terminal repeat-driven luciferase reporter, were generated at the DuPont Pharmaceutical Company (66). HeLa C14 cells expressing CD4, CXCR4, and CCR5 and containing an integrated copy of a tetracycline-responsive inducible luciferase reporter gene were constructed at Bristol-Myers Squibb (43). MT-2 cells were propagated in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 10 mM HEPES buffer (pH 7.55), and 2 mM l-glutamine. HEK293T cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated FBS, 10 mM HEPES buffer (pH 7.5), and 2 mM l-glutamine. Unless otherwise noted, all cell culture reagents came from Gibco or Invitrogen, subsidiaries of Thermo Fisher Scientific (Waltham, MA). The CEM-NKR-CCR5-Luc cells were maintained in medium containing RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 100 units/ml penicillin (Pen), 0.1 mg/ml streptomycin (Strep), and 0.8 mg/ml Geneticin. Recombinant B6 cells were propagated in RPMI 1640 medium plus 10% FBS, 1% Pen-Strep, and 0.6 mg/ml Geneticin. HeLa C14 cells were cultured in DMEM plus 10% FBS (Sigma, St. Louis, MO), 0.1 mg/ml hygromycin B, 0.2 mg/ml Geneticin, 1.5 μg/ml puromycin (EMD Biochemical, Billerica, MA), and 0.4 mg/ml zeocin. NLRepRluc virus, in which a section of the nef gene from the proviral clone of NL4-3 virus was replaced with the Renilla luciferase gene, was constructed at Bristol-Myers Squibb (67). The replication-competent virus or defined variants of this virus were harvested 3 days after transfection of HEK293T cells with the modified proviral clones. Transfections were performed using the Lipofectamine Plus reagent (Invitrogen), according to the manufacturer's instruction. The titers of the recombinant viruses in MT-2 cells were determined using luciferase enzyme activity as a marker.
Cell-cell fusion assay.
Populations of envelope genes were obtained from previous studies of HIV attachment inhibitors (45, 68). The envelope genes were cloned into the pCMV-HA expression vector (Clontech) using the In-Fusion HD EcoDry technology (Clontech) and were used in the assay to evaluate a wide spectrum of subtypes. Two populations of cells, designated effector and target cells, were used in the cell-cell fusion assay. The effector cells (HeLa or HEK293T cells) were prepared by cotransfecting cells with the envelope-expressing plasmid, along with the pTET-Off plasmid (Clontech), both at 50 ng/well in 6-well plates, for 1 day prior to the assay. HeLa C14 cells expressing CD4, CXCR4, and CCR5 and containing an integrated copy of a Tat-inducible luciferase reporter gene were used as target cells. The effector and target cells were trypsinized, mixed at a ratio of 1:2, and seeded into a 96-well or 384-well plate in the presence of serial dilutions of the inhibitor protein. After 18 h of incubation, luciferase signals were generated with the Steady-Glo luciferase reagent (Promega), allowing the extent of cell fusion to be quantitated. Inhibition of cell fusion was determined by XLfit analysis of the luciferase signals. The results from at least 2 experiments (each conducted in triplicate) were averaged to establish the EC50s. Assay results for envelopes from clinical isolates were always normalized against those for an LAI envelope and expressed as the fold change (FC). On the basis of assay variability, a FC of >10 was considered significant in this assay. Statistical analysis was performed using GraphPad Prism software.
Replicating virus assay.
MT-2 cells were cultured in RPMI 1640 medium plus 10% FBS, 1% Pen-Strep, 1% HEPES, and 1% l-glutamine. Inhibitors were serially diluted 4- or 5-fold stepwise, and 10 concentrations were plated in quadruplicate into a white 384-well assay plate with a clear bottom (catalog number 3707; Corning). Recombinant viruses containing the luciferase reporter gene were used to infect 8,000 MT-2 cells/well at a multiplicity of infection (MOI) of 0.002. After 3 to 4 days of incubation in a tissue culture incubator (37°C, 5% CO2), cells were processed and quantitated for virus growth by the amount of expressed luciferase as described previously (67). Data were analyzed using a 5-parameter fitting method (XLfit).
Resistance selection.
Two million MT-2 cells were infected with NL4-3 virus in the presence of 2× the EC50 of the inhibitor at an MOI of 0.005 to 0.05. Syncytium formation was monitored as a marker for viral infection. Typically, more than 10% syncytium formation occurred within 3 to 4 days. A 1/1,000 to 1/100 volume of the infectious supernatant was then transferred to fresh MT-2 cells in the presence of the inhibitor at an increased concentration of twice the previous concentration, stepwise. When consistent virus breakthrough was observed, the potency of the inhibitor was evaluated against the viral supernatant in the B6 antiviral assay. A potency shift of more than 10-fold usually indicated the appearance of resistant virus. The infected cells or the viral supernatants were used to obtain the viral genomes by PCR or reverse transcription-PCR, followed by sequencing to identify the amino acid changes. Amino acid changes were then introduced into the wild-type NL4-3 viral genome using site-directed mutagenesis and cloning. The recombinant viruses were evaluated in a replicating virus assay for a shift in potency versus that against wild-type virus.
Potency of anti-CD4 Adnectin against viruses from resistance selections.
B6 cells were cultured in RPMI 1640 medium plus 10% FBS, 1% Pen-Strep, and 0.6 mg/ml Geneticin. Inhibitors were serially diluted 4- or 5-fold stepwise, and 10 concentrations were plated in quadruplicate into white 384-well assay plates with a clear bottom (Corning). HIV-1 NL4-3 isolates from various time points during resistance selection were used to infect 6,000 B6 cells per well at an MOI of 0.02. After 1 to 2 days of incubation in a tissue culture incubator (37°C, 5% CO2), cells were processed and virus growth was quantitated by determination of the amount of luciferase expressed. Data were analyzed using a 5-parameter fitting method (XLfit).
Determination of kinetics of Adnectin binding to CD4 by SPR.
A Biacore T-series CM5 chip (GE Healthcare) was derivatized with protein A (Pierce) via a standard amine-coupling kit (GE Healthcare) to a level of 2,600 response units (RU). CD4-D1-D4-Fc was diluted to 25 nM in running buffer consisting of 0.01 M HEPES, pH 7.4, 0.15 M NaCl, and 0.005% (vol/vol) Surfactant P20 (HBS-P+; GE Healthcare), and flowed over the protein A surface until 950 RU was captured. Adnectins diluted to various concentrations in running buffer were then flowed over the captured CD4-D1-D4-Fc surface at 37°C at a flow rate of 40 μl/min for 2 min. Dissociation was measured for 10 to 30 min. A surface consisting of only protein A was used for reference subtraction, and buffer-only samples were included for background subtraction. The protein A surface was regenerated between cycles with two injections of 10 mM glycine, pH 1.5. A 1:1 Langmuir binding model was fit to the double-referenced sensorgrams to determine kinetic parameters using Biacore T100 evaluation software, version 2.0.1 (GE Healthcare).
Simultaneous cobinding of anti-CD4 Adnectin, anti-CD4 antibodies, and HIV gp120 to CD4.
A Biacore 3000 CM5 chip (GE Healthcare) was derivatized with 1,700 RU of protein A/G (Pierce) via a standard amine-coupling kit (GE Healthcare). Antibodies to CD4 were captured onto this surface to approximately 1,000 RU in HBS-P+ running buffer (GE Healthcare). Soluble recombinant human CD4 containing amino acids 26 to 390 (R&D Systems) was diluted to 50 nM in running buffer and flowed over the captured antibodies to levels of 75 to 700 RU. Adnectins were then flowed over the antibody-CD4 complexes at 0.2 to 1 μM in running buffer, and further increases in the numbers of RU relative to those in flow cells without CD4 were measured.
In an alternative format, the anti-CD4 Adnectin 4945_G06 was diluted to 5 μM in 10 mM sodium acetate, pH 5.5, and immobilized on a Biacore 3000 CM5 chip or T-series CM5 chip (GE Healthcare) via a standard amine-coupling kit (GE Healthcare) to a level of approximately 2,000 RU on two flow cells. Recombinant human CD4 consisting of the four ectodomains (amino acids 26 to 394) was diluted to 100 nM in running buffer consisting of 0.01 M HEPES, pH 7.4, 0.15 M NaCl, 3 mM EDTA, and 0.005% (vol/vol) Surfactant P20 (HBS-EP; GE Healthcare) and flowed over the second (active) flow cell for 2 min at 5 μl/min (∼860 RU was captured). Then, anti-CD4 antibodies (Table S2) at 100 nM in running buffer or gp120 at 5 μg/ml was flowed successively over both flow cells for 3 min at 5 μl/min. Regeneration of the chip surface between cycles was accomplished with two injections of 10 mM glycine, pH 2.1, at 30 μl/min for 15 s each time. Cobinding of antibodies or gp120 to CD4 captured by 4945_G06 was determined by the accumulation of additional mass on the active flow cell (with CD4 plus 4945_G06) compared to that on the reference flow cell (with 4945_G06 only).
SPR binding of CD4 fragments.
A Biacore T-series CM5 chip (GE Healthcare) was derivatized via a standard amine-coupling kit (GE Healthcare) as follows: flow cell 1 received 2,450 RU of a nonbinding Adnectin negative control, flow cell 2 received 2,720 RU of 4945_G06, flow cell 3 received 5,270 RU of antibody OKT4, and flow cell 4 received 11,060 RU of ibalizumab. See Table S2 for antibody information.
Soluble recombinant fragments of human CD4 consisting of ectodomains 1 and 2 (D1D2), ectodomains 3 and 4 (D3D4), or ectodomains 1 to 4 (D1D4) were diluted to 1 μM in running buffer (HBS-EP; GE Healthcare). Diluted fragments were flowed over the chip surfaces at 15 μl/min with 2 min of contact time and 3 min of dissociation time. Fragment identity and integrity were confirmed by the binding pattern observed for the two antibodies OKT4 and ibalizumab, whose epitopes are known to be contained within domain 3 and domain 2, respectively (Fig. S2).
Binding to CD4-expressing cells.
The cross-reactivity of the anti-CD4 Adnectin was assessed on human, cynomolgus monkey, marmoset, and African green monkey peripheral blood mononuclear cells (PBMCs) (all nonhuman primate tissues were sourced from BioReclamation) and murine PBMCs. Cells (2 × 105 per condition) were incubated with a titration series of anti-CD4 Adnectin for 2 h at 4°C. Cells were washed and fixed with 2% formaldehyde for 10 min at 4°C. After two additional rounds of washing, cells were stained with separate antibody mixes depending on their origin. Human cells were stained with Alexa Fluor 488-labeled anti-human CD3 (clone SP34-2; Becton, Dickinson), phycoerythrin (PE)-labeled anti-human CD8 (Becton, Dickinson), and Alexa Fluor 647-labeled anti-His antibody (prepared at Bristol-Myers Squibb). Nonhuman primate cells were stained with PE-labeled anti-nonhuman primate CD3 (Miltenyi), fluorescein isothiocyanate (FITC)-labeled anti-nonhuman primate CD8 (Becton, Dickinson), and Alexa Fluor 647-labeled anti-His (prepared at Bristol-Myers Squibb). Murine cells were stained with FITC-labeled anti-mouse CD3 (Becton, Dickinson), PE-labeled anti-mouse CD8 (Becton, Dickinson), and Alexa Fluor 647-labeled anti-His (prepared at Bristol-Myers Squibb). Anti-CD4 antibodies were not used in the experiment due to their potential to block Adnectin binding. Each cell-antibody mix was prepared in a total volume of 100 μl of PBS plus 0.1% BSA. All samples were incubated for 45 min at 4°C. Cells were washed twice and fixed in 2% formaldehyde (diluted in 1× PBS), and data were acquired on a BD FACSCanto II flow cytometer.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elliott Ethridge for selection support; Geoff Loizeaux, Luke Turecheck, Holly Schmidt, Ping Zhang, and Andrew Douglas for protein expression and purification; Pallavi Gambhire, Kevin Smith, and Samantha Povlich for protein analytics; and Bree Goldstein, Radhika Nayak, and Irina Zhan for molecular biology.
D.W., Z.L., S.Z., M.C., and M.K. were employees of Bristol-Myers Squibb at the time of data generation and are current employees of ViiV Healthcare; all employees own stock/stock options in GlaxoSmithKline, the majority owner of ViiV Healthcare. Y.S., C.P., T.M., and J.D. are current employees of Bristol-Myers Squibb; all employees own stock/stock options in Bristol-Myers Squibb. D.F. was an employee of Bristol-Myers Squibb at the time of data generation.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00508-17.
REFERENCES
- 1.Trezza C, Ford SL, Spreen W, Pan R, Piscitelli S. 2015. Formulation and pharmacology of long-acting cabotegravir. Curr Opin HIV AIDS 10:239–245. doi: 10.1097/COH.0000000000000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams PE, Crauwels HM, Basstanie ED. 2015. Formulation and pharmacology of long-acting rilpivirine. Curr Opin HIV AIDS 10:233–238. doi: 10.1097/COH.0000000000000164. [DOI] [PubMed] [Google Scholar]
- 3.Bruno CJ, Jacobson JM. 2010. Ibalizumab: an anti-CD4 monoclonal antibody for the treatment of HIV-1 infection. J Antimicrob Chemother 65:1839–1841. doi: 10.1093/jac/dkq261. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson JM, Lalezari JP, Thompson MA, Fichtenbaum CJ, Saag MS, Zingman BS, D'Ambrosio P, Stambler N, Rotshteyn Y, Marozsan AJ, Maddon PJ, Morris SA, Olson WC. 2010. Phase 2a study of the CCR5 monoclonal antibody PRO 140 administered intravenously to HIV-infected adults. Antimicrob Agents Chemother 54:4137–4142. doi: 10.1128/AAC.00086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson JM, Thompson MA, Lalezari JP, Saag MS, Zingman BS, D'Ambrosio P, Stambler N, Rotshteyn Y, Marozsan AJ, Maddon PJ, Morris SA, Olson WC. 2010. Anti-HIV-1 activity of weekly or biweekly treatment with subcutaneous PRO 140, a CCR5 monoclonal antibody. J Infect Dis 201:1481–1487. doi: 10.1086/652190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strohl WR. 2015. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 29:215–239. doi: 10.1007/s40259-015-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swanson RV. 2014. Long live peptides—evolution of peptide half-life extension technologies and emerging hybrid approaches. Drug Discovery World 15:57–61. [Google Scholar]
- 8.Schlapschy M. 2013. PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng Des Sel 26:489–501. doi: 10.1093/protein/gzt023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jevševar S, Kusterle M, Kenig M. 2012. PEGylation of antibody fragments for half-life extension, p 233–246. In Proetzel G, Ebersbach H (ed), Antibody methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 10.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 11.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 12.Melikyan GB. 2008. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology 5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. 2001. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci U S A 98:10799–10804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin Y, Wang XX, Mariuzza RA. 2012. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc Natl Acad Sci U S A 109:5405–5410. doi: 10.1073/pnas.1118801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moebius U, Clayton LK, Abraham S, Harrison SC, Reinherz EL. 1992. The human immunodeficiency virus gp120 binding site on CD4: delineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resolution CD4 atomic structure. J Exp Med 176:507–517. doi: 10.1084/jem.176.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore JP, Sattentau QJ, Klasse PJ, Burkly LC. 1992. A monoclonal antibody to CD4 domain 2 blocks soluble CD4-induced conformational changes in the envelope glycoproteins of human immunodeficiency virus type 1 (HIV-1) and HIV-1 infection of CD4+ cells. J Virol 66:4784–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman MM, Seaman MS, Rits-Volloch S, Hong X, Kao CY, Ho DD, Chen B. 2010. Crystal structure of HIV-1 primary receptor CD4 in complex with a potent antiviral antibody. Structure 18:1632–1641. doi: 10.1016/j.str.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song R, Oren DA, Franco D, Seaman MS, Ho DD. 2013. Strategic addition of an N-linked glycan to a monoclonal antibody improves its HIV-1-neutralizing activity. Nat Biotechnol 31:1047–1052. doi: 10.1038/nbt.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipovsek D. 2011. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel 24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sachdev E, Gong J, Rimel B, Mita M. 2015. Adnectin-targeted inhibitors: rationale and results. Curr Oncol Rep 17:35. doi: 10.1007/s11912-015-0459-8. [DOI] [PubMed] [Google Scholar]
- 22.Koide A, Bailey CW, Huang X, Koide S. 1998. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol 284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 23.Koide S, Koide A, Lipovsek D. 2012. Target-binding proteins based on the 10th human fibronectin type III domain (10Fn3). Methods Enzymol 503:135–156. doi: 10.1016/B978-0-12-396962-0.00006-9. [DOI] [PubMed] [Google Scholar]
- 24.Main AL, Harvey TS, Baron M, Boyd J, Campbell ID. 1992. The three-dimensional structure of the tenth type III module of fibronectin: an insight into RGD-mediated interactions. Cell 71:671–678. doi: 10.1016/0092-8674(92)90600-H. [DOI] [PubMed] [Google Scholar]
- 25.Dickinson CD, Veerapandian B, Dai XP, Hamlin RC, Xuong NH, Ruoslahti E, Ely KR. 1994. Crystal structure of the tenth type III cell adhesion module of human fibronectin. J Mol Biol 236:1079–1092. doi: 10.1016/0022-2836(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 26.Xu L, Aha P, Gu K, Kuimelis RG, Kurz M, Lam T, Lim AC, Liu H, Lohse PA, Sun L, Weng S, Wagner RW, Lipovsek D. 2002. Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol 9:933–942. doi: 10.1016/S1074-5521(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 27.Getmanova EV, Chen Y, Bloom L, Gokemeijer J, Shamah S, Warikoo V, Wang J, Ling V, Sun L. 2006. Antagonists to human and mouse vascular endothelial growth factor receptor 2 generated by directed protein evolution in vitro. Chem Biol 13:549–556. doi: 10.1016/j.chembiol.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Lipovsek D, Lippow SM, Hackel BJ, Gregson MW, Cheng P, Kapila A, Wittrup KD. 2007. Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: molecular convergence with single-domain camelid and shark antibodies. J Mol Biol 368:1024–1041. doi: 10.1016/j.jmb.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Ibilcieta D, Bokov M, Cherkasov V, Sveshnikov P, Hanson SF. 2008. Simple method for production of randomized human tenth fibronectin domain III libraries for use in combinatorial screening procedures. Biotechniques 44:559–562. doi: 10.2144/000112726. [DOI] [PubMed] [Google Scholar]
- 30.Ackermann M, Carvajal IM, Morse BA, Moreta M, O'Neil S, Kossodo S, Peterson JD, Delventhal V, Marsh HN, Furfine ES, Konerding MA. 2011. Adnectin CT-322 inhibits tumor growth and affects microvascular architecture and function in Colo205 tumor xenografts. Int J Oncol 38:71–80. [PubMed] [Google Scholar]
- 31.Dineen SP, Sullivan LA, Beck AW, Miller AF, Carbon JG, Mamluk R, Wong H, Brekken RA. 2008. The Adnectin CT-322 is a novel VEGF receptor 2 inhibitor that decreases tumor burden in an orthotopic mouse model of pancreatic cancer. BMC Cancer 8:352. doi: 10.1186/1471-2407-8-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mamluk R, Carvajal IM, Morse BA, Wong H, Abramowitz J, Aslanian S, Lim AC, Gokemeijer J, Storek MJ, Lee J, Gosselin M, Wright MC, Camphausen RT, Wang J, Chen Y, Miller K, Sanders K, Short S, Sperinde J, Prasad G, Williams S, Kerbel R, Ebos J, Mutsaers A, Mendlein JD, Harris AS, Furfine ES. 2010. Anti-tumor effect of CT-322 as an Adnectin inhibitor of vascular endothelial growth factor receptor-2. MAbs 2:199–208. doi: 10.4161/mabs.2.2.11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tolcher AW, Sweeney CJ, Papadopoulos K, Patnaik A, Chiorean EG, Mita AC, Sankhala K, Furfine E, Gokemeijer J, Iacono L, Eaton C, Silver BA, Mita M. 2011. Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted Adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clin Cancer Res 17:363–371. doi: 10.1158/1078-0432.CCR-10-1411. [DOI] [PubMed] [Google Scholar]
- 34.Ackermann M, Morse BA, Delventhal V, Carvajal IM, Konerding MA. 2012. Anti-VEGFR2 and anti-IGF-1R-Adnectins inhibit Ewing's sarcoma A673-xenograft growth and normalize tumor vascular architecture. Angiogenesis 15:685–695. doi: 10.1007/s10456-012-9294-9. [DOI] [PubMed] [Google Scholar]
- 35.Ramamurthy V, Krystek SR Jr, Bush A, Wei A, Emanuel SL, Das Gupta R, Janjua A, Cheng L, Murdock M, Abramczyk B, Cohen D, Lin Z, Morin P, Davis JH, Dabritz M, McLaughlin DC, Russo KA, Chao G, Wright MC, Jenny VA, Engle LJ, Furfine E, Sheriff S. 2012. Structures of Adnectin/protein complexes reveal an expanded binding footprint. Structure 20:259–269. doi: 10.1016/j.str.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 36.Iacob RE, Chen G, Ahn J, Houel S, Wei H, Mo J, Tao L, Cohen D, Xie D, Lin Z, Morin PE, Doyle ML, Tymiak AA, Engen JR. 2014. The influence of Adnectin binding on the extracellular domain of epidermal growth factor receptor. J Am Soc Mass Spectrom 25:2093–2102. doi: 10.1007/s13361-014-0973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emanuel SL, Engle LJ, Chao G, Zhu RR, Cao C, Lin Z, Yamniuk AP, Hosbach J, Brown J, Fitzpatrick E, Gokemeijer J, Morin P, Morse BA, Carvajal IM, Fabrizio D, Wright MC, Das Gupta R, Gosselin M, Cataldo D, Ryseck RP, Doyle ML, Wong TW, Camphausen RT, Cload ST, Marsh HN, Gottardis MM, Furfine ES. 2011. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. MAbs 3:38–48. doi: 10.4161/mabs.3.1.14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iacob RE, Krystek SR, Huang RY, Wei H, Tao L, Lin Z, Morin PE, Doyle ML, Tymiak AA, Engen JR, Chen G. 2015. Hydrogen/deuterium exchange mass spectrometry applied to IL-23 interaction characteristics: potential impact for therapeutics. Expert Rev Proteomics 12:159–169. doi: 10.1586/14789450.2015.1018897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitchell T, Chao G, Sitkoff D, Lo F, Monshizadegan H, Meyers D, Low S, Russo K, DiBella R, Denhez F, Gao M, Myers J, Duke G, Witmer M, Miao B, Ho SP, Khan J, Parker RA. 2014. Pharmacologic profile of the Adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J Pharmacol Exp Ther 350:412–424. doi: 10.1124/jpet.114.214221. [DOI] [PubMed] [Google Scholar]
- 40.Walker RG, Thompson TB. 2015. Fibronectin-based scaffold domain proteins that bind myostatin: a patent evaluation of WO2014043344. Expert Opin Ther Pat 25:619–624. doi: 10.1517/13543776.2015.1007954. [DOI] [PubMed] [Google Scholar]
- 41.Roberts RW, Szostak JW. 1997. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A 94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker MH, Chen Y, Danehy F, Dufu K, Ekstrom J, Getmanova E, Gokemeijer J, Xu L, Lipovsek D. 2005. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Eng Des Sel 18:435–444. doi: 10.1093/protein/gzi050. [DOI] [PubMed] [Google Scholar]
- 43.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A 100:11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merkenschlager M, Buck D, Beverley PC, Sattentau QJ. 1990. Functional epitope analysis of the human CD4 molecule. The MHC class II-dependent activation of resting T cells is inhibited by monoclonal antibodies to CD4 regardless whether or not they recognize epitopes involved in the binding of MHC class II or HIV gp120. J Immunol 145:2839–2845. [PubMed] [Google Scholar]
- 45.Zhou N, Nowicka-Sans B, Zhang S, Fan L, Fang J, Fang H, Gong YF, Eggers B, Langley DR, Wang T, Kadow J, Grasela D, Hanna GJ, Alexander L, Colonno R, Krystal M, Lin PF. 2011. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob Agents Chemother 55:729–737. doi: 10.1128/AAC.01173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun M, Pace CS, Yao X, Yu F, Padte NN, Huang Y, Seaman MS, Li Q, Ho DD. 2014. Rational design and characterization of the novel, broad and potent bispecific HIV-1 neutralizing antibody iMabm36. J Acquir Immune Defic Syndr 66:473–483. doi: 10.1097/QAI.0000000000000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toma J, Weinheimer SP, Stawiski E, Whitcomb JM, Lewis ST, Petropoulos CJ, Huang W. 2011. Loss of asparagine-linked glycosylation sites in variable region 5 of human immunodeficiency virus type 1 envelope is associated with resistance to CD4 antibody ibalizumab. J Virol 85:3872–3880. doi: 10.1128/JVI.02237-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pace CS, Fordyce MW, Franco D, Kao CY, Seaman MS, Ho DD. 2013. Anti-CD4 monoclonal antibody ibalizumab exhibits breadth and potency against HIV-1, with natural resistance mediated by the loss of a V5 glycan in envelope. J Acquir Immune Defic Syndr 62:1–9. doi: 10.1097/QAI.0b013e3182732746. [DOI] [PubMed] [Google Scholar]
- 49.Pace C, Markowitz M. 2015. Monoclonal antibodies to host cellular receptors for the treatment and prevention of HIV-1 infection. Curr Opin HIV AIDS 10:144–150. doi: 10.1097/COH.0000000000000146. [DOI] [PubMed] [Google Scholar]
- 50.Escolano A, Dosenovic P, Nussenzweig MC. 2017. Progress toward active or passive HIV-1 vaccination. J Exp Med 214:3–16. doi: 10.1084/jem.20161765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Z, Li S, Gu Y, Xia N. 2016. Antiviral therapy by HIV-1 broadly neutralizing and inhibitory antibodies. Int J Mol Sci 17:E1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von Recum HA, Pokorski JK. 2013. Peptide and protein-based inhibitors of HIV-1 co-receptors. Exp Biol Med (Maywood) 238:442–449. doi: 10.1177/1535370213480696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yi HA, Fochtman BC, Rizzo RC, Jacobs A. 2016. Inhibition of HIV entry by targeting the envelope transmembrane subunit gp41. Curr HIV Res 14:283–294. doi: 10.2174/1570162X14999160224103908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fumakia M, Yang S, Gu J, Ho EA. 26 August 2015. Protein/peptide-based entry/fusion inhibitors as anti-HIV therapies: challenges and future direction. Rev Med Virol. doi: 10.1002/rmv.1853. [DOI] [PubMed] [Google Scholar]
- 55.Prokofjeva M, Tsvetkov V, Basmanov D, Varizhuk A, Lagarkova M, Smirnov I, Prusakov K, Klinov D, Prassolov V, Pozmogova G, Mikhailov SN. 2017. Anti-HIV activities of intramolecular G4 and non-G4 oligonucleotides. Nucleic Acid Ther 27:56–66. doi: 10.1089/nat.2016.0624. [DOI] [PubMed] [Google Scholar]
- 56.Oliviero G, Stornaiuolo M, D'Atri V, Nici F, Yousif AM, D'Errico S, Piccialli G, Mayol L, Novellino E, Marinelli L, Grieco P, Carotenuto A, Noppen S, Liekens S, Balzarini J, Borbone N. 2016. Screening platform toward new anti-HIV aptamers set on molecular docking and fluorescence quenching techniques. Anal Chem 88:2327–2334. doi: 10.1021/acs.analchem.5b04268. [DOI] [PubMed] [Google Scholar]
- 57.Shum KT, Zhou J, Rossi JJ. 2013. Aptamer-based therapeutics: new approaches to combat human viral diseases. Pharmaceuticals (Basel) 6:1507–1542. doi: 10.3390/ph6121507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schweizer A, Rusert P, Berlinger L, Ruprecht CR, Mann A, Corthesy S, Turville SG, Aravantinou M, Fischer M, Robbiani M, Amstutz P, Trkola A. 2008. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog 4:e1000109. doi: 10.1371/journal.ppat.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Healey D, Dianda L, Moore JP, McDougal JS, Moore MJ, Estess P, Buck D, Kwong PD, Beverley PC, Sattentau QJ. 1990. Novel anti-CD4 monoclonal antibodies separate human immunodeficiency virus infection and fusion of CD4+ cells from virus binding. J Exp Med 172:1233–1242. doi: 10.1084/jem.172.4.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song R, Franco D, Kao CY, Yu F, Huang Y, Ho DD. 2010. Epitope mapping of ibalizumab, a humanized anti-CD4 monoclonal antibody with anti-HIV-1 activity in infected patients. J Virol 84:6935–6942. doi: 10.1128/JVI.00453-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashish, Juncadella IJ, Garg R, Boone CD, Anguita J, Krueger JK. 2008. Conformational rearrangement within the soluble domains of the CD4 receptor is ligand-specific. J Biol Chem 283:2761–2772. doi: 10.1074/jbc.M708325200. [DOI] [PubMed] [Google Scholar]
- 62.Kabat TTW, Bilofsky EAH, Reid-Miller M, Perry H. 1983. Sequence of proteins of immunological interest. National Institutes of Health, Bethesda, MD. [Google Scholar]
- 63.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. 2007. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol 373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 64.Koide A, Koide S. 2007. Monobodies: antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol Biol 352:95–109. [DOI] [PubMed] [Google Scholar]
- 65.Kurz M, Gu K, Lohse PA. 2000. Psoralen photo-crosslinked mRNA-puromycin conjugates: a novel template for the rapid and facile preparation of mRNA-protein fusions. Nucleic Acids Res 28:E83. doi: 10.1093/nar/28.18.e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nowicka-Sans B, Protack T, Lin Z, Li Z, Zhang S, Sun Y, Samanta H, Terry B, Liu Z, Chen Y, Sin N, Sit SY, Swidorski JJ, Chen J, Venables BL, Healy M, Meanwell NA, Cockett M, Hanumegowda U, Regueiro-Ren A, Krystal M, Dicker IB. 2016. Identification and characterization of BMS-955176, a second-generation HIV-1 maturation inhibitor with improved potency, antiviral spectrum, and Gag polymorphic coverage. Antimicrob Agents Chemother 60:3956–3969. doi: 10.1128/AAC.02560-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Z, Terry B, Olds W, Protack T, Deminie C, Minassian B, Nowicka-Sans B, Sun Y, Dicker I, Hwang C, Lataillade M, Hanna GJ, Krystal M. 2013. In vitro cross-resistance profile of nucleoside reverse transcriptase inhibitor (NRTI) BMS-986001 against known NRTI resistance mutations. Antimicrob Agents Chemother 57:5500–5508. doi: 10.1128/AAC.01195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou N, Nowicka-Sans B, McAuliffe B, Ray N, Eggers B, Fang H, Fan L, Healy M, Langley DR, Hwang C, Lataillade M, Hanna GJ, Krystal M. 2014. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother 69:573–581. doi: 10.1093/jac/dkt412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.