

Abstract
Globally, nearly 2 billion people are infected with the intracellular protozoan Toxoplasma gondii1. This persistent infection can cause severe disease in immunocompromised people and is epidemiologically linked to major mental illnesses2 and cognitive impairment3. There are currently no options for curing this infection. The lack of effective therapeutics is due partly to a poor understanding of essential pathways that maintain this long-term infection. Although it is known that Toxoplasma replicates slowly within intracellular cysts demarcated with a cyst wall, precisely how it sustains itself and remodels organelles in this niche is unknown. Here we identify a key role for proteolysis within the parasite lysosomal organelle (the vacuolar compartment or VAC) in turnover of autophagosomes and persistence during neural infection. We found that disrupting a VAC-localized cysteine protease compromised VAC digestive function and markedly reduced chronic infection. Death of parasites lacking the VAC protease was preceded by accumulation of undigested autophagosomes in the parasite cytoplasm. These findings suggest an unanticipated function for parasite lysosomal degradation in chronic infection and identify an intrinsic role for autophagy in the T. gondii parasite and its close relatives. This work also identifies a key element of Toxoplasma persistence and suggests that VAC proteolysis is a prospective target for pharmacologic development.
The Toxoplasma lysosomal VAC contains two papain family cysteine proteases termed cathepsin protease L (CPL; toxodb accession number TGME49_321530) and cathepsin protease B (CPB; TGME49_249670)4–6. Upregulated expression of CPL and CPB in chronically infected mice7 suggests an active role for VAC proteolysis in Toxoplasma persistence. To test this prospect, we focused on CPL because maturation of CPB is dependent on its activity6, thus ablating CPL effectively impairs both proteases. Genetic ablation of CPL in Prugniaud (P) strain parasites, which express cytosolic GFP during chronic infection8, was confirmed by PCR, immunofluorescence, and immunoblotting (Supplementary Figs 1–3). Consistent with findings in a different strain background9, PΔcpl acute stage parasites (tachyzoites) presented a dilated VAC suggestive of organellar dysfunction, a phenotype that was rescued by genetic complementation (Fig. 1a, b). Although CPL deficient parasites appeared to differentiate normally to chronic stage bradyzoites within cultured intracellular cysts (Fig. 1c), after differentiation they progressively lost stage specific expression of GFP (Fig. 1d,e) and viability (Fig. 1f). Re-expression of active CPL restored cyst viability, whereas cysts expressing a catalytically inactive CPL (Fig. 1f) or treated with a CPL inhibitor (LHVS9; Fig. 1g and Supplementary Fig. 4) died. Taken together, these findings suggest a key role for VAC proteolytic activity for maintaining viability of cultured Toxoplasma cysts.
Figure 1. VAC proteolytic activity is required for Toxoplasma viability and persistence in vitro.
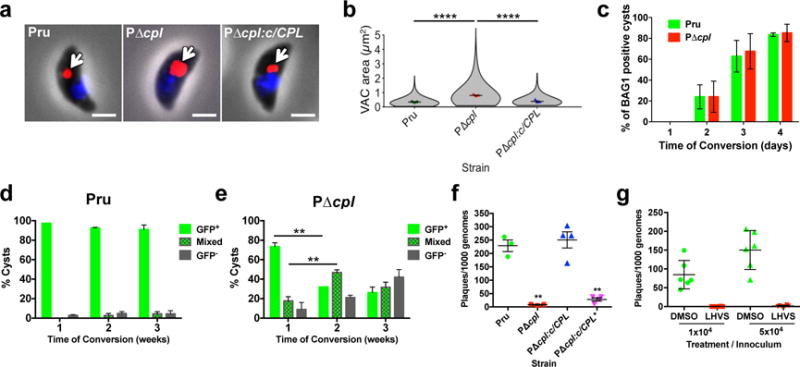
a, Extracellular tachyzoites were stained with α-CPB (red) to mark the VAC and DAPI (blue) to identify the nucleus. An arrow indicates the localization of the VAC. Scale bar, 2 μm. b, Violin plots of VAC size. Data are mean ± SEM of 3 biological replicates, with the following number of parasites evaluated: Pru, 317, 153, 154; PΔcpl, 271, 177, 176; PΔcpl:c/CPL, 293, 153, 153. Shape indicates distribution of the pooled data. ****, p<0.0001 Mann Whitney test. c, In vitro differentiation based on expression of the bradyzoite specific antigen BAG1. Cultures were differentiated for the indicated time, fixed, stained with α-BAG1 and enumerated by fluorescence microscopy. Bars are mean ± SEM of 2 biological replicates. Experiment 1 assessed for Pru 205, 214, 204, and 208 parasitophorous vacuoles (PV) on days 1, 2, 3, and 4, respectively; and for PΔcpl, 209, 211, 213, and 202 PV, respectively. Experiment 2 assessed for Pru 212, 202, 208, and 202 PV on days 1, 2,3, and 4, respectively; and for PΔcpl 205, 207, 214, and 208 PV, respectively. Viability of Pru (d) or PΔcpl (e) bradyzoites based on stage specific expression of cytosolic GFP under the LHD2 promoter8. Cultures were differentiated, and enumerated by fluorescence microscopy. Green bars indicate cysts that are uniformly GFP positive. Checkerboard bars specify cysts that contain GFP positive and GFP negative bradyzoites. Grey bars indicate cysts that are uniformly GFP negative. Data are mean ± SD of 3 biological replicates each with 100 cysts evaluated. **, p<0.001, two-tailed paired student’s t-test. f, Viability of CPL proficient and deficient strains based on plaquing efficiency. Strains were differentiated for 4 weeks, mechanically liberated from cysts, quantified by qPCR and inoculated onto fresh cell monolayers for plaque formation. PΔcpl:c/CPL* is complemented with catalytically inactive CPL. Bars are mean ± SEM of 3 (Pru, PΔcpl) or 4 (PΔcpl:c/CPL, PΔcpl:c/CPL*) biological replicates each with two technical replicates. **, p<0.01, Mann Whitney test. g, Viability of Pru bradyzoites differentiated for 1 week before treatment with solvent (DMSO) or 1 μM LHVS for 4 weeks. Viability was measure as in (f). n=2 biological replicates each with 3 technical replicates.
Next we sought to define the contribution of VAC proteolysis to infection in mice, which are a convenient model and an important natural host for T. gondii. Consistent with earlier work10, survival of PΔcpl infected mice was higher than WT-infected mice, indicating a role for CPL in acute virulence (Fig. 2a). Nevertheless, mice infected with 106 WT or PΔcpl showed a similar pattern of parasite burden in the first 8 days of infection (Fig. 2b)10, comparable kinetics of parasite entry into the brain (Fig. 2c) and lungs (Supplementary Fig. 5), and similar cytokine profiles (Supplementary Fig. 5). Notably, mice infected with PruΔcpl had substantially fewer cysts despite comparable kinetics of acute infection (Fig. 2a, Supplementary Fig. 6). Further, although mice infected with the highest dose (107) of PΔcpl yielded cysts, these cysts contained markedly enlarged and misshapen bradyzoites (Fig. 2d).
Figure 2. VAC proteolytic function is required for Toxoplasma persistence in infected mice.
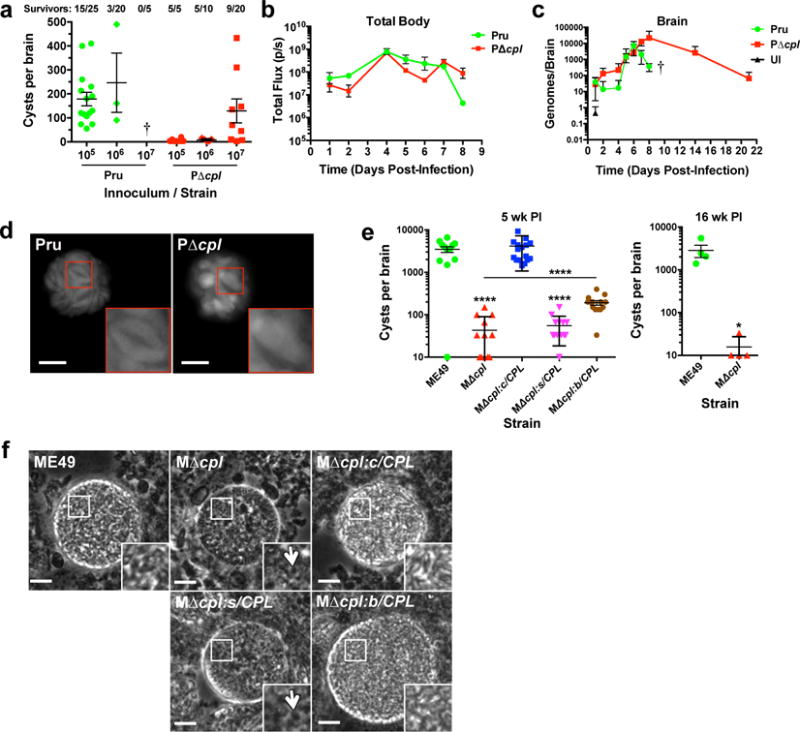
a, Burden of brain cysts in Pru and PruΔcpl infected C57BL/6 mice. Mice were infected with the indicated inoculum for 4 weeks, humanely euthanized and cysts were enumerated microscopically in brain homogenates. Each data point indicates cyst burden in one mouse. Data are mean ± SEM of data pooled from: Pru 105, 4 experiments; Pru 106, 3 experiments; Pru 107, 1 experiment; PΔcpl 105, 1 experiment; PΔcpl 106, 1 experiment; PΔcpl 107, 3 experiments. All of the mice in the Pru 107 group succumbed to the infection during the acute stage, as indicated by a cross. The number of mice that survived 4 weeks is indicated along with the total number of mice in each group. b, Parasite burden throughout the body of infected mice. C57BL/6 mice (4 per group) were infected intraperitoneally with 106 tachyzoites. Total body parasite burden was determined by bioluminescence imaging and measurement of light flux (photons/second) from parasites expressing firefly luciferase. Data are mean ± SEM from one experiment. c, Mice were infected as in panel b and parasite burden in the brain was measured by qPCR. Data are mean ± SEM from one experiment. A cross indicates that Pru infected mice succumbed to the infection prior to imaging on day 10. UI, uninfected. d, Live fluorescence microscopy of cysts in brain homogenates. Brain homogenates were prepared 4 weeks post-infection for fluorescence microscopy of cytosolic GFP in cysts. Insets show enlargements of boxed regions including two Pru bradyzoites and one PΔcpl bradyzoite. Scale bar, 10 μm. e, Burden of brain cysts in mice infected with ME49 WT or transgenic strains. Female CBA/J mice (20 mice per group) were infected intraperitoneally with 500 tachyzoites and humanely euthanized for cyst enumeration 5 weeks (left panel) or 16 weeks (right panel) post-infection. Five week PI data is pooled from two experiments. Sixteen week PI data is from one experiment. MΔcpl:c/CPL is complemented with CPL under its cognate promoter, MΔcpl:s/CPL is complemented with CPL under the SAG1 promoter for expression only in tachyzoites, MΔcpl:b/CPL is complemented with CPL under the BAG1 promoter for expression only in bradyzoites. Data are mean ± SEM. *, p<0.05; ****, p<0.0001, Mann Whitney test comparing MΔcpl to ME49, MΔcpl:s/CPL to ME49, or MΔcpl:b/CPL to MΔcpl (line). f, Phase contrast microscopy of ME49 WT or transgenic strain cysts in brain homogenates. Arrows indicate granular structures in CPL deficient cysts. Scale bar, 10 μm.
To independently validate these findings we ablated CPL in the highly cystogenic ME49 strain (MΔcpl; Supplementary Fig. 7). Mice infected with MΔcpl produced ~100-fold and ~200-fold fewer cysts than WT infected mice at 5 weeks and 16 weeks post-infection, respectively. This deficit was partially rescued by re-expressing CPL during the chronic stage, but not when the protease was re-expressed in only the acute stage (Fig. 2e). Overexpression of CPL under the BAG1 chronic stage specific promoter might have contributed to incomplete rescue (Supplementary Fig. 7). Finally, WT parasites and strains re-expressing CPL in the chronic stage showed normal brain cyst morphology, whereas the CPL knockout parasite cysts or those with re-expression of CPL only in the acute stage were abnormally granulated and phase dark (Fig. 2f). Collectively these findings are consistent with a vital role for CPL in persistent neural infection of a mammalian host.
To identify a potential basis for loss of viability we examined cultured cysts by microscopy and noticed that bradyzoites genetically (PΔcpl) or chemically (LHVS) deficient in CPL activity showed large, dark cytoplasmic inclusions that were absent from WT or complemented parasites (Fig. 3a,b). Stage specific CPL re-expression during the persistent stage, but not the acute stage, eliminated the inclusions. These findings suggest the aberrant structures result from lack of CPL expression during the persistent stage, and are not a legacy of CPL deficiency during acute stage infection.
Figure 3. CPL deficient bradyzoites develop Atg8-positive autophagosomes associated with the VAC.
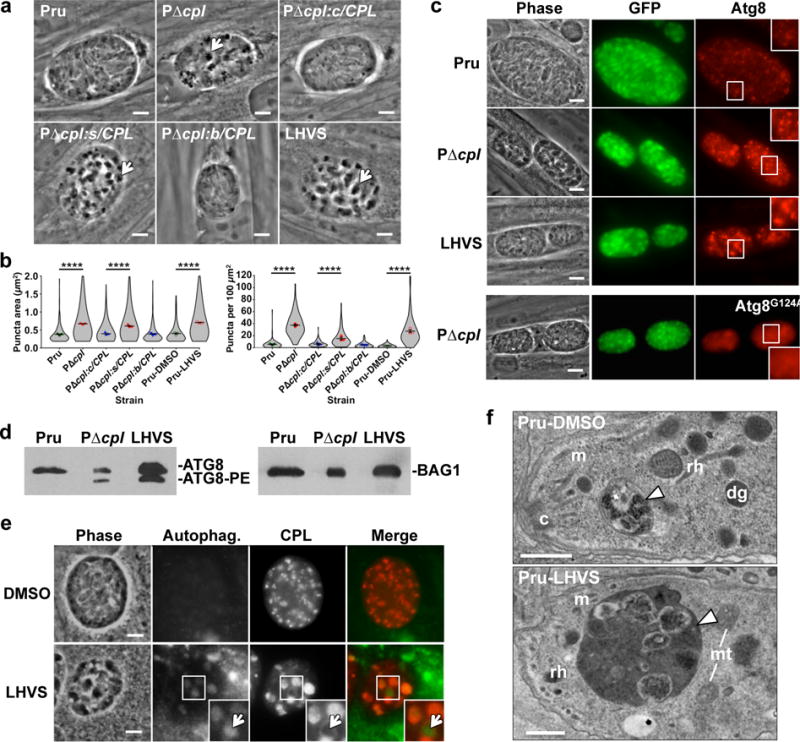
a, Phase contrast microscopy of in vitro encysted bradyzoites differentiated for 7 days. Arrows indicate cytosolic inclusions in CPL deficient strains. Scale bar, 5 μm. b, Quantification of dark inclusion size (area of individual puncta, left panel) and number (per 100 μm2 of cyst area, right panel) shown as violin plots. Data are mean ± SEM of 3 biological replicates. The following number of total cysts and puncta, respectively, were quantified: Pru 74, 409; PΔcpl 102, 3812; PΔcpl:c/CPL 120, 743; PΔcpl:s/CPL 86, 1324; PΔcpl:b/CPL 101, 464; Pru-DMSO 75, 237; and Pru-LHVS 110, 3024. Shape indicates distribution of the pooled data. ****, p<0.0001 Mann Whitney test. c, Fluorescence microscopy of autophagosomes in bradyzoites expressing tdTomato-Atg8. Bradyzoites were differentiated for 7 days, fixed, and viewed for bradyzoite specific expression of GFP and tdTomato-Atg8. Examples in the top 3 rows are of parasites expressing WT tdTomato-Atg8, whereas the bottom row shows parasites expressing a tdTomato-Atg8 bearing a glycine to alanine mutation rendering it refractory to lipidation. Scale bar, 10 μm. The same scale applies to the fluorescence images, which were captured and processed identically. d, Western blots of bradyzoite lysates probed with α-Atg8 or α-BAG1. e, Fluorescence microscopy of autophagosomes in Pru bradyzoites treated with solvent (DMSO) or 1 μM LHVS. Bradyzoites were differentiated for 7 days, treated for 4 days, stained with cytoID (green), fixed, and stained with α-CPL (red). An arrow indicates an example of CPL at the periphery of an autophagosome shown as an inset of the boxed area. Scale bar, 5 μm. The same scale applies to the fluorescence images. f, Transmission electron microscopy of bradyzoites differentiated for 1 week. Abbreviations used: c, conoid; m, microneme; rh, rhoptry; dg, dense granule; mt, mitochondrion. Scale bars, 500 nm.
We reasoned that the dark inclusions were due to accumulation of undigested material within the VAC, and that some of the material could be derived from autophagy. Autophagy is a process whereby cytoplasmic contents including organelles are membrane encapsulated and degraded within lysosomes11. Toxoplasma and related apicomplexans including malaria parasites express a limited repertoire of autophagy (ATG) proteins12. These parasites also unconventionally require certain ATG proteins for maintenance of the apicoplast13, 14, a remnant non-photosynthetic chloroplast. Autophagosome-like structures have been observed under nutrient limiting conditions15, 16, but the extent to which the parasite encounters such conditions during infection remains unknown. To define the relationship between the intracellular inclusions and autophagosomes, we ectopically expressed a copy of Toxoplasma Atg8 fused to the fluorescent protein tdTomato in bradyzoites. WT cysts showed tdTomato-Atg8 fluorescent puncta, suggestive of baseline autophagy during normal parasite encystation (Fig. 3b). However, ablation of CPL or treatment with LHVS augmented tdTomato-Atg8 fluorescence (Fig. 3b), which corresponded to the dark inclusions but not the apicoplast (Supplementary Fig. 8). Mutation of the lipidation site on Atg8 (Atg8G124A) in PΔcpl parasites caused diffuse cytosolic fluorescence, indicating that the inclusions are decorated with lipid anchored, active form of Atg8 (Fig. 3c). This finding was confirmed by western blotting for Atg8, which showed accumulation of the lipidated Atg8-phosphatidyl serine (Atg8-PE) form in PΔcpl and Pru-LHVS bradyzoites, suggesting an arrest of autophagic flux (Fig. 3d). Taken together, these imply that autophagosomes develop in WT cysts, but become much more pronounced in the absence of CPL activity. Cysts treated with LHVS developed autophagosomes containing CPL, indicating a defect in degradation of autophaged material (Fig. 3e). In some cases CPL occupied a rim around the autophagosome, suggesting recent fusion of the autophagosome and VAC (Fig. 3e). Transmission electron microscopy revealed electron dense inclusions in LHVS treated Pru bradyzoites that appeared to contain undigested material including putative organellar remnants (Fig. 3f).
Similar to Pru strain, autophagosome accumulation in ME49 bradyzoites was strictly dependent on the absence of CPL expression (Supplementary Fig. 9). ME49 CPL deficient bradyzoites also displayed remnants of organelles including ER within autophagosomes (Fig. 4a), which showed a time dependent increase in size following differentiation (Fig. 4b). WT ME49 bradyzoites presented similar VAC intraluminal structures that were more prominent in day 7 bradyzoites, suggesting active turnover of autophagic structures after differentiation. Whereas WT ME49 cysts appeared healthy on both day 4 and day 7, some day 7 MΔcpl cysts contained degenerate bradyzoites with compromised integrity (Supplementary Fig. 10), consistent with a critical function for CPL in bradyzoite viability.
Figure 4. CPL deficient bradyzoites develop undigested autophagosomes containing organellar remnants.
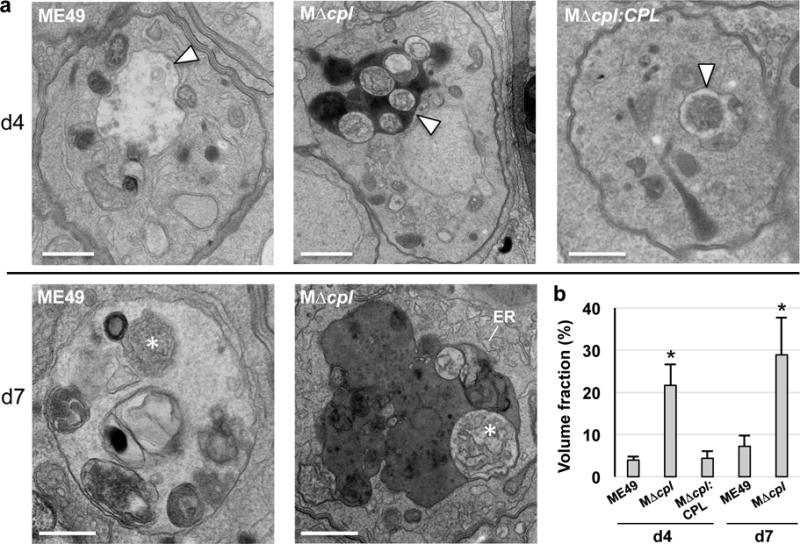
a, Electron microscopy of intracellular ME49, MΔcpl and MΔcpl:CPL comparing the morphology of VAC (arrowheads) at day 4 showing electron density for this compartment in MΔcpl bradyzoites. High magnification views at day 7 reveal cytosolic material (asterisks) resembling piecemeal of the cytoplasm (ME49) and ER (MΔcpl), suggestive of autophagy activities occurring in the VAC with delayed degradation in the mutant reflected by electron dense luminal content. Scale bars, 500 nm. b. Volume fraction of VAC from representative electron micrographs of parasitophorous vacuoles (PV) from each group (29 and 25 PV for ME49 at d4 and d7, respectively; 30 and 32 PV of MΔcpl at d4 and d7, respectively; and 25 PV for MΔcpl:CPL at d4) showing significant increase of this compartment in parasites lacking CPL compared to control strains. Volume fraction equals the area of the organelle divided by the area of the cell multiplied by 100. Bars indicate mean ± SD. *p<0.005 unpaired student’s t-test.
In summary, we show that VAC proteolytic function is required for Toxoplasma persistence in culture and in infected mice. We also find that blocking CPL activity causes accumulation of material including organellar remnants in the VAC prior to cyst death, indicating a key role for turnover of autophagosomes during Toxoplasma persistence. Whereas previous studies of autophagy in T. gondii depended upon induction via drug treatment or starvation conditions15, 16, our work suggests an intrinsic role for autophagy in bradyzoites without specific treatment. Autophagy following encystation could be required to renew organelles for the chronic stage or it might reflect a stringent response if the cyst wall physically limits resource acquisition from the host. Although the cyst wall is permeable to LHVS, this inhibitor does not cross the blood brain barrier17. Priority will be given to identifying CNS penetrant inhibitors of CPL to potentially abate Toxoplasma persistence.
METHODS
Host cells and parasite cultures
Human foreskin fibroblasts (HFF) were not specifically authenticated, but the cultures were derived from low pass cryostocs (ATCC CRL-1634) every 3 months and maintained in Dulbecco’s modified Eagle medium (DMEM Gibco) containing 10% fetal bovine serum (D10 media). T. gondii strains PruΔku80, ME49 and their mutants were propagated in vitro by serial passage on monolayers of HFF18. In vitro tachyzoite-to-bradyzoite conversion was induced by exposing parasite cultures to pH 8.2, as described previously19. Morpholine urea-leucyl-homophenyl-vinyl sulfone phenyl (LHVS) treatment of parasites was performed by daily replacement of media containing 1 μM LHVS diluted from a 1 mM stock in DMSO. HFF cells and parasite strains were routinely tested for mycoplasma contamination.
Generation of transgenic T. gondii strains
PruΔku80LUC (herein termed Pru) and PruΔku80LUCΔcpl (herein PΔcpl) strains used to generate additional transformants in this study are based on PruΔku808 and have been described previously10. CPL complement strains PΔcpl:c/CPL and PΔcpl:c/CPL* (catalytically inactive C31A mutant) where generated using vectors described in Dou et al.6 modified by replacing the chloramphenicol acetyltransferase (CAT) cDNA with that encoding bleomycin resistance (BLE). To target the complementation plasmids to the Ku80 locus by single cross-over homologous recombination, we cloned into BamHI and SacI restriction sites 3′ to BLE a 1664 bp PCR fragment encompassing the nucleotide sequence 2753961-2752298 of ME49 chromosome XI (primers ku80 Fw1/ku80 Rv1). These vectors were linearized with NdeI in the middle of the Ku80 fragment and integrated in the PruΔcpl strain, as shown Supplementary Fig. 1b. Stage specific complement strains PΔcpl:s/CPL and PΔcpl:b/CPL were created by replacing the CPL promoter in the CPL complement vector with that of SAG1 (sequence from -1185 to -1) or BAG1 (sequence from -1232 to -1) for expression exclusively in tachyzoites or bradyzoites, respectively20. SAG1/CPL and BAG1/CPL vectors were linearized with XbaI or BclI, respectively for non-disruptive single crossover integration into the SAG1 or BAG1 locus of PruΔcpl, as shown in Supplementary Fig. 1c, d.
ME49Δcpl (herein MΔcpl) was created using a CRISPR/Cas9 double cut strategy described by Long et al.21 whereby the first gRNA sequence (gRNA1) was targeted close to the CPL initiation codon and the second gRNA (gRNA2) near the stop codon, as shown in Supplementary Fig. 7a. Co-transfection ME49 with this sgRNA CRISPR/Cas9 plasmid (40 μg) and a PCR amplicon consisting of BLE flanked by CPL-homology arms (~3000 bp) (10 μg) successfully resulted in cpl deletion. MΔcpl:c/CPL, MΔcpl:s/CPL and MΔcpl:b/CPL complement strains were generated by random integration of the same vectors used for the Pru strains except that BLE was replaced with a dihydrofolate reductase (DHFR) cDNA conferring resistance to pyrimethamine.
Pru strains expressing tdTomato-ATG8 or a tdTomato-ATG8G124A lipidation mutant were generated by modifying the pTub-tdTomato-TgATG8-CAT plasmid described in Leveque et al.13 via replacement of CAT with BLE and cloning in a larger tubulin promoter fragment encompassing -2730 to -1 relative to the tubulin start codon. Plasmids were linearized with NarI at -1318 for single crossover integration into the tubulin locus. The tdTomato-ATG8 expression cassette was also introduced into ME49 and MΔcpl strains by random integration and chloramphenicol selection.
T. gondii transfections were performed as described previously in Lunghi et al.22. Correct integration was identified by PCR analysis of single clones using Phire tissue direct PCR Master kit (Thermo Fisher Scientific).
Immunoblotting and immunofluorescence
Immunoblotting and immunofluorescence were performed as described previously23, 24. For immunoblotting, mouse α-CPL9 and rabbit α-actin25 antibodies were diluted 1:5000 and 1:10000, respectively. For immunofluorescence, rabbit α-CPL and mouse α-CPB6 were diluted 1:500 and 1:250, respectively. Autophagosomes were stained using a cytoID® Autophagy Detection Kit 2.0 (ENZO) according to the manufacturer’s instructions.
Analysis of VAC size
Syringe released tachyzoites were allowed to invade HFF cells for 2 h at 37°C followed washing with PBS. Newly invaded parasites were again syringe released and affixed to slides coated with Cell-Tak™ (Corning), fixed with 4% paraformaldehyde, and stained with mouse α-CPB to identify the VAC. Images were acquired by focusing on the VAC signal in the maximum number of parasites within a field of view and captured on a Zeiss Axiovert Observer Z1 inverted fluorescence microscope equipped with a 100X and an AxioCAM MRm camera and processed using Zeiss Axiovision 4.3 software. For Pru, PΔcpl, PΔcpl:c/CPL, and PΔcpl:c/CPL strains VAC size was measured with Zeiss Axiovision 4.3 software by defining the area of α-CPB immunofluorescence. VAC measurements were only performed on VACs that were within focus. Equal parameters for the capture and enumeration of images were consistently applied to all samples. We cannot rule out that fixation differentially affected VAC size for the different strains assessed in the study.
Bradyzoite conversion, viability, and analysis of dark inclusions
Bradyzoite conversion was assessed by counting vacuoles. Vacuoles displaying more than 50% of parasites stained by the rabbit ααBAG1 antibodies were considered positive. HFF monolayers grown onto coverslips were infected with mechanically extruded tachyzoites for 16 h and then the media were replaced with the alkaline media to induce bradyzoite conversion. At least 200 parasite vacuoles were counted in two independent experiments for each time point.
Loss of bradyzoite viability was measured by counting GFP positive versus GFP negative cysts at different time points of conversion. Samples were blinded. Infected HFF monolayers were cultivated in alkaline media for 1, 2 or 3 weeks and then fixed in 4% paraformaldehyde for 20 min and at least 100 cysts were assessed for GFP positivity under fluorescence microscope.
Viability of bradyzoites was assessed by combining plaque assay and qPCR analysis of genome number. In vitro cysts were generated by maintaining infected HFF in alkaline media in six-well plates. After selected times, infected monolayers were washed 3 times with HBSS and cysts contained in one well were mechanically extruded from the HFFs by syringing several times through 26G needles. Released cysts were centrifuged at 1500g for 10 min at RT and resuspended in 500 μl of HBSS. Then, 500 μl of pre-warmed 2X pepsin solution (0.026% Pepsin in 170 mM NaCl and 60 mM HCl, final concentration) was added and the sample was incubated for 30 min at 37°C. Reactions were stopped by adding 94 mM Na2CO3, removing the supernatant after centrifugation at 1500g for 10 min at RT and resuspending pepsin treated parasites in 1 ml of DMEM without serum. After calculating number of parasites per ml, 100 or 500 parasites per well were added to six-well plates containing confluent monolayers of HFFs in D10 media. Parasites were grown undisturbed for 12 days and plaque assays were performed by ethanol fixation for 2 min, staining with 0.2% crystal violet for 5 min and followed by several washes with double-distilled H2O. Images of the wells were scanned, and plaque number and size were analyzed with ImageJ. Seven hundreds μl of the initial 1 ml of pepsin treated parasites were used for gDNA purification using the DNeasy Blood & Tissue Kit (Qiagen). Genomic DNA was eluted in a final volume of 100 μl and 8 μl was analyzed by qPCR using the tubulin primers TUB2.RT.F and TUB2.RT.R to determine the number of parasite genomes per μl. qPCR analyses were performed using Brilliant II SYBR green QPCR Master Mix (Agilent) and the Stratagene Mx3000PQ-PCR machine.
To quantify dark inclusions in bradyzoites, images of in vitro differentiated cysts were taken using a Zeiss Axio inverted microscope equipped with an Axiocam MRM CCD camera using a 100X oil objective. ImageJ was used to identify cysts by Max Entropy thresholding on the GFP channel followed by selection of objects between 130–1900 square microns. Particles (puncta) under the GFP mask were further analyzed by automatic local thresholding on the phase image using the Phansalkar method with the following parameters: radius = 5 μm; k-value = 0.5; r-value = 0.5. Dark inclusions (puncta) were measured from the resulting binary mask by particle analysis according to the following: size = 0.18–2.00 μm; circularity = 0.50–1.00. These parameters were chosen based on accuracy in a head-to-head comparison against manual counting. A total of 10,013 puncta were analyzed from 668 cysts.
Assessment of Atg8 lipidation
Parasites grown in HFF cells were converted to bradyzoites in alkaline medium for 5 days and treated with 1 μM LHVS in conversion medium for an additional 2 days. Bradyzoites were isolated by scraping and syringing infected monolayers several times through a 26G needle and subsequent pepsin treatment for 30 min at 37°C to release bradyzoites and selectively kill tachyzoites. Parasite pellets were solubilized in SDS sample buffer and lysates corresponding to 3×106 parasites per lane were separated in 15% SDS-PAGE gels containing 6 M urea, transferred to PVDF membranes and incubated with affinity purified rabbit α-TgATG8 (diluted 1:500; kindly provided by William Sullivan Jr). Blots were then incubated with an anti-rabbit HRP antibody and immunoreactive bands were visualized with the West Pico Chemiluminescent Substrate (Thermo Scientific) and exposure to film. Blots were then stripped and reprobed with rabbit α-BAG1 (1:5000)26 as a loading control and to assess bradyzoite conversion.
Electron microscopy of in vitro cysts
HFF monolayers grown in six-well plates were infected with 5×104 tachyzoites in D10 media. After 16 h D10 media were replaced with alkaline media (pH 8.2) to induce bradyzoite conversion. In vitro cysts were processed for electron microscopy (EM) analysis either 4- or 7-days post bradyzoite conversion. EM Preparation of samples was carried out by washing the infected monolayers with cold PBS 3 times followed by fixation with 2.5% glutaraldehyde (EMS, catalog number 16210) in 0.1 M sodium buffer (pH 7.4) for 1 hour at RT. After fixation, infected cells were gently scraped to detach large sheets, centrifuged at 3000 rpm for 10 min and washed 3 times with 0.1 M cacodylate (pH 7.4) buffer and stored in the same buffer at 4°C until processed for EM as described27 before examination with a Philips CM120 Electron Microscope (Eindhoven, the Netherlands) under 80-kV. Quantitative morphometric analysis of VAC from 25 to 32 representative electron micrographs from each group (ME49, MΔcpl and MΔcpl:CPL) we performed at low magnification to ensure the entire parasite fit into the field of view and could be analyzed as described28 by using the standard formula for randomly orientated cell and structures.
Competition assay with LHVS
Brains containing ME49 cysts were harvested from infected mice and homogenized in 1 ml of 1X PBS by sequential syringing through 19G and 23G. Homogenized brain material were then incubated for 30 min with either 200 nM BODIPY-LHVS (BO-LHVS) alone or a 1:1 or 1:10 mixture of BO-LHVS and LHVS. After three washes with 1X PBS, brain material was fixed with 4% paraformaldehyde and stained with rabbit anti-TgCPL antibodies at a 1:200 dilution followed by Alexa 594-conjugated goat anti-rabbit antibodies. Stained brain material was then allowed to adhere on slides coated with Cell-tak adhesive (Corning). Images were captured with a Zeiss Axiovert Observer Z1 inverted fluorescence microscope and an AxioCAM MRm camera and processed using Zeiss Axiovision 4.3 software.
Mouse studies
Seven-week-old female C57BL/6 mice (Charles River) were randomly assigned to groups and infected intraperitoneally (i.p.) with 105, 106 or 107 tachyzoites of WT or transgenic PruΔku80 strains (type II genotype). Group sizes and number of experiments are as follows: Pru 105, 25 mice total, 4 experiments; Pru 106, 20 mice total, 3 experiments; Pru 107, 5 mice total, 1 experiment; PΔcpl 105, 10 mice total, 1 experiment; PΔcpl 106, 5 mice/group, 1 experiment; PΔcpl 107, 20 mice total, 3 experiments. Mice were humanely euthanized 4 weeks post-infection. Assessing statistical differences in cyst burden between mice infected with Pru and PruΔcpl was complicated by the absence of cyst data from the mice that did not survive the infection. Since mice that die from the infection often have higher burden and they become moribund and must be euthanized before the 4 weeks when cysts are counted, enumeration of cysts from those that survive probably underestimates differences. Accordingly, statistically analysis was provided. WT or genetically manipulated ME49 strains were injected with 500 tachyzoites i.p. in 7-week-old female CBA/J mice (Jackson Laboratory). Data were pooled from two experiments with 13 mice per group in experiment 1 and 7 mice per group in experiment 2. Mice infected with ME49 strains were euthanized humanely at 5 or 16 weeks post infection. The mouse brains were placed in 1 ml sterile PBS and individually minced with scissors, vortexed, and homogenized by three or four passages through a 22-G needle and syringe. Two 100 μl brain samples (200 μl total) from mice infected with PruΔku80 strains were placed on a glass microscope slide and then mounted on a coverslip. Cysts were enumerated without knowledge of the sample’s treatment group under an inverted fluorescence phase-contrast microscope by exploiting fluorescence emitted by GFP expressed under the control of the bradyzoite specific LDH2 promoter3. Three 10-μl samples (30 μl total) of brain homogenates from mice infected with ME49 strains were instead analyzed by phase-contrast microscopy to enumerate cysts. Sample sizes were chosen based on previous studies. Mice were randomly assigned to groups and samples were blinded for enumeration of cysts. Animal studies described herein adhere to a protocol was approved by the Committee on the Use and Care of Animals of the University of Michigan.
Bioluminescence imaging
Seven-week old female albino C57B/6 mice (4 per strain, based on investigator experience) were IP injected with 106 Pru or 4×106 PruΔcpl parasites, which constituted equal inoculation of infectious parasites based on plaque assays. Bioluminescence was assessed as described10. Briefly, images were taken at 1 at 2, 4, 5, 6, 7 and 8 days post-infection using a Xenogen IVIS200 system 10 min after IP injected with 200 μl of 40 mg/ml D-luciferin in PBS. Mice were anesthetized with inhaled isoflurane during imaging. The intensities of bioluminescence signals were quantified using LiveImage software coupled with the Xenogen IVIS200. All 4 mice in both groups were alive for imaging on day 6 and 3 mice in both groups were imaged on day 7. One Pru infected mouse and with 3 PΔcpl infected mice were imaged on day 8 when the experiment was terminated.
Quantitative PCR
Brains of infected and uninfected control mice were dissected, weighed, and immediately stored at -20°C. Tissue was later thawed, and 25 mg of tissue per sample was homogenized with a pestle in 1X PBS and then passaging 4-5 times through a 22-gauge needle. Genomic DNA was extracted using the Qiagen DNeasy Blood and Tissue Kit. To create a standard curve, known numbers of cultured tachyzoites were spiked into 25 mg of homogenized brain tissue from age matched uninfected mice at concentrations of 10−1, 100, 101, 102, 103, 104, and 105 tachyzoites per mg of brain tissue. Fifty nanograms of DNA from each sample were used to run quantitative real-time PCR using primers against a Toxoplasma specific 529 bp repeat element29 (Tox-9 Forward, 5′ AGGAGAGATATCAGGACTGTAG, Tox-11 Reverse 5′ GCGTCGTCTCGTCTAGATCG). Treated and control sample CT values were compared against the standard curve to extrapolate the number of Toxoplasma genomes per brain. All samples were run in biological and technical triplicate. Samples with a CT value at or above 40 were considered to have undetectable amplification, and were set to zero Toxoplasma genomes/brain.
Cytokine analysis
Seven-week-old female C57B/6 mice injected with 106 Pru or 4×106 PruΔcpl parasites were euthanized by CO2 fixation at 1, 2, 4, 5, 6, 7, 8, 14, and 21 days post-infection. Uninfected controls were euthanized at 1, 6, 14, and 21 DPI to be used as baseline controls. Blood was collected by cardiac puncture and serum separated by centrifugation. Levels of cytokines (IFNγ, IL-10, and TNFα) were assessed by capture ELISA. Data was analyzed and graphed in GraphPad Prism 6.
Data Availability
Authors can confirm that all relevant data are included in the paper and/or its supplementary information.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01AI120627 (VBC and MDC) and R01GM111703 (MB), and R01AI060767 (IC). ZD and AJS received fellowship support from the American Heart Association. BMH and GK were supported by National Institutes of Health training grant T32AI007528. OLM was supported by a National Institutes of Health Ruth Kirschstein National Research Service Award F31AI118274. We thank Sasha Meshinchi and Joel Whitfield at the University of Michigan for help with electron microscopy and cytokine analysis, respectively, along with the Johns Hopkins University Microscopy facility. Drs. Louis Weiss, William Sullivan, Jr., and L. David Sibley generously provided antibodies for this study.
Footnotes
Author Contributions
M.D.C., Z.D., M.L., O.L.M., G.K., M.H.H., A.J.M., B.M.H., C.E., and V.B.C. designed the experiments, M.D.C., A.J.S., A.J.M., B.M.H., and V.B.C. wrote the manuscript. W.L. performed chemical synthesis. M.D.C., Z.D., M.L., O.L.M., G.K., M.H.H., T.L.S., A.J.S., A.J.M., B.M.H., and I.C. performed the experiments. C.E., M.B., and S.B. provided advice or reagents that were essential for completion of the study. All authors discussed the findings and commented on the manuscript.
Author Information
References
- 1.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 2.Sutterland AL, et al. Beyond the association Toxoplasma gondii in schizophrenia, bipolar disorder, and addiction: systematic review and meta-analysis. Acta Psychiatr Scand. 2015;132:161–179. doi: 10.1111/acps.12423. [DOI] [PubMed] [Google Scholar]
- 3.Bharti AR, et al. Latent Toxoplasma Infection and Higher Toxoplasma gondii Immunoglobulin G Levels Are Associated With Worse Neurocognitive Functioning in HIV-Infected Adults. Clin Infect Dis. 2016 doi: 10.1093/cid/ciw655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parussini F, Coppens I, Shah PP, Diamond SL, Carruthers VB. Cathepsin L occupies a vacuolar compartment and is a protein maturase within the endo/exocytic system of Toxoplasma gondii. Mol Microbiol. 2010;76:1340–1357. doi: 10.1111/j.1365-2958.2010.07181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miranda K, et al. Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole. Mol Microbiol. 2010;76:1358–1375. doi: 10.1111/j.1365-2958.2010.07165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dou Z, Coppens I, Carruthers VB. Non-canonical Maturation of Two Papain-family Proteases in Toxoplasma gondii. J Biol Chem. 2013;288:3523–3534. doi: 10.1074/jbc.M112.443697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pittman KJ, Aliota MT, Knoll LJ. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics. 2014;15:806-2164–15-806. doi: 10.1186/1471-2164-15-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox BA, et al. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot Cell. 2011;10:1193–1206. doi: 10.1128/EC.00297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson ET, et al. Toxoplasma gondii cathepsin L is the primary target of the invasion-inhibitory compound morpholinurea-leucyl-homophenyl-vinyl sulfone phenyl. J Biol Chem. 2009;284:26839–26850. doi: 10.1074/jbc.M109.003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dou Z, McGovern O, Di Cristina M, Carruthers V. Toxoplasma gondii ingests and digests host cytosolic proteins mBio. 2014;5(4):e01188–14. doi: 10.1128/mBio.01188-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besteiro S. Which roles for autophagy in Toxoplasma gondii and related apicomplexan parasites? Mol Biochem Parasitol. 2012;184:1–8. doi: 10.1016/j.molbiopara.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Leveque MF, et al. Autophagy-Related Protein ATG8 Has a Noncanonical Function for Apicoplast Inheritance in Toxoplasma gondii. MBio. 2015;6:e01446–15. doi: 10.1128/mBio.01446-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomlins AM, et al. Plasmodium falciparum ATG8 implicated in both autophagy and apicoplast formation. Autophagy. 2013;9:1540–1552. doi: 10.4161/auto.25832. [DOI] [PubMed] [Google Scholar]
- 15.Besteiro S, Brooks CF, Striepen B, Dubremetz JF. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS Pathog. 2011;7:e1002416. doi: 10.1371/journal.ppat.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh D, Walton JL, Roepe PD, Sinai AP. Autophagy is a cell death mechanism in Toxoplasma gondii. Cell Microbiol. 2012;14:589–607. doi: 10.1111/j.1462-5822.2011.01745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barclay J, et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. 2007;130:225–234. doi: 10.1016/j.pain.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 18.Di Cristina M, et al. Transformed Toxoplasma gondii tachyzoites expressing the circumsporozoite protein of Plasmodium knowlesi elicit a specific immune response in rhesus monkeys. Infect Immun. 1999;67:1677–1682. doi: 10.1128/iai.67.4.1677-1682.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galizi R, et al. Evidence of tRNA cleavage in apicomplexan parasites: Half-tRNAs as new potential regulatory molecules of Toxoplasma gondii and Plasmodium berghei. Mol Biochem Parasitol. 2013;188:99–108. doi: 10.1016/j.molbiopara.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Di Cristina M, et al. Temporal and spatial distribution of Toxoplasma gondii differentiation into Bradyzoites and tissue cyst formation in vivo. Infect Immun. 2008;76:3491–3501. doi: 10.1128/IAI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long S, Wang Q, Sibley LD. Analysis of Noncanonical Calcium-Dependent Protein Kinases in Toxoplasma gondii by Targeted Gene Deletion Using CRISPR/Cas9. Infect Immun. 2016;84:1262–1273. doi: 10.1128/IAI.01173-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lunghi M, Galizi R, Magini A, Carruthers VB, Di Cristina M. Expression of the glycolytic enzymes enolase and lactate dehydrogenase during the early phase of Toxoplasma differentiation is regulated by an intron retention mechanism. Mol Microbiol. 2015;96:1159–1175. doi: 10.1111/mmi.12999. [DOI] [PubMed] [Google Scholar]
- 23.Di Cristina M, Spaccapelo R, Soldati D, Bistoni F, Crisanti A. Two conserved amino acid motifs mediate protein targeting to the micronemes of the apicomplexan parasite Toxoplasma gondii. Mol Cell Biol. 2000;20:7332–7341. doi: 10.1128/mcb.20.19.7332-7341.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spano F, et al. The SAG5 locus of Toxoplasma gondii encodes three novel proteins belonging to the SAG1 family of surface antigens. Int J Parasitol. 2002;32:121–131. doi: 10.1016/s0020-7519(01)00349-6. [DOI] [PubMed] [Google Scholar]
- 25.Dobrowolski JM, Sibley LD. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 1996;84:933–939. doi: 10.1016/s0092-8674(00)81071-5. [DOI] [PubMed] [Google Scholar]
- 26.Zhang YW, et al. Disruption of the Toxoplasma gondii bradyzoite-specific gene BAG1 decreases in vivo cyst formation. Mol Microbiol. 1999;31:691–701. doi: 10.1046/j.1365-2958.1999.01210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coppens I, Joiner KA. Host but not parasite cholesterol controls Toxoplasma cell entry by modulating organelle discharge. Mol Biol Cell. 2003;14:3804–20. doi: 10.1091/mbc.E02-12-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Opperdoes FR, et al. Purification, morphometric analysis, and characterization of the glycosomes (microbodies) of the protozoan hemoflagellate Trypanosoma brucei. J Cell Biol. 1984;98:1178–1184. doi: 10.1083/jcb.98.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reischl U, Bretagne S, Kruger D, Ernault P, Costa JM. Comparison of two DNA targets for the diagnosis of Toxoplasmosis by real-time PCR using fluorescence resonance energy transfer hybridization probes. BMC Infect Dis. 2003;3:7. doi: 10.1186/1471-2334-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Authors can confirm that all relevant data are included in the paper and/or its supplementary information.