

Abstract
Ethics are the moral values of human behavior and the principles which govern these values. The situation becomes challenging for a doctor when he assumes the role of researcher. The doctor-researcher has to serve both the roles and at times the zeal of an investigator has the potential to cloud the morality of the physician inside. It is very important to realize that exploiting the faith of patients is an offence that tantamount to a crime. Medical science is one discipline where the advancement of knowledge is hugely guided by research and mankind has benefitted from many experiments. However benefit and risk are the two faces of the same coin. Various unethical human experiments made us realize that the whims of researchers need to be reined and led to the evolution of the first guidelines for researcher, the Nuremberg code. Thereafter the Good Clinical Practice guidelines serve as the guiding doctrine of clinical research. The principles of ethics rest on the four pillars of autonomy, beneficence, justice, non-maleficence and recently two more pillars are added which includes, confidentiality and honesty. Ethics committees serve as a guardian of these principles. The multidisciplinary Ethics Committee ensures a competent review of the ethical aspects of the project proposal submitted and does it free from any bias or external influence. Ethical review of clinical trial applications follows a decentralized process in India, and requires Ethics Committee approval for each trial site. All Ethics committees have to be registered with Drug Controller General of India (DCGI) without which they cannot approve any clinical trial protocol and has come into effect from 25th February 2013.
KEY WORDS: Clinical research, Ethics evolution, Ethics committee, Registration of IEC
What was known?
Doctors are guided and are familiar with ‘Hippocrates oath’ or the code of medical ethics’ laid down by Medical Council of India (MCI) which mainly highlights the ‘duties and responsibilities of physician, duties of physicians to their patients/in consultation/to each other/public or paramedical profession, unethical act/misconduct related to medical practice and the punitive action if the above are breached.
Introduction
“Ethics is not definable, is not implementable, because it is not conscious; it involves not only our thinking, but also our feeling”
-Valdemar W. Setzer
Ethics are the moral values of human behavior and the principles which govern these values. Every profession is bound by code of ethics (Greek word Ethos meaning Custom or Character) and the essence of medicine as a moral community dates back to Hippocratic Oath. This oath was a guide for the physician on professional ethics and mandates that he/she would prescribe only beneficial treatments, refrain from causing harm or hurt to his/her patients,[1] and would place the interests of their patients above their own interests.[2] The situation becomes challenging for a doctor when he assumes the role of researcher. The doctor-researcher has to serve both the roles, and at times, the zeal of an investigator has the potential to cloud the morality of the physician inside. History has in its store numerous instances when the enthusiasm for knowledge breached the principles of ethics. Thus, it was realized that code of ethics for clinical research was needed and Good Clinical Practice (GCP) guidelines for human research was framed.
Science versus Ethics Vis-à-vis Researchers versus Doctor
In clinical practice, doctors pledge to treat every individual equally, irrespective of their age, disease or disability, creed, ethnic origin, gender, nationality, political affiliation, race, sexual orientation, social standing, or any other factor;[3] but when it comes to research that pledge is blurred at times, and it is forgotten that a subject for research is also a patient who has put his/her faith in the doctor for his treatment. The statement of a first-century physician from Egypt, Celsius reflects the attitude of doctor-turned-researcher who justified experiments on condemned criminals by saying that “It is not cruel to inflict on a few criminals sufferings which may benefit multitudes of innocent people through all centuries.”[4] By making this statement, the researcher has taken for granted that they are given the liberty of being reckless because criminals can be sacrificed (such as guinea pigs in laboratory). It is very important to realize that exploiting the faith of patients is an offence that tantamount to a crime.
There is another face of research, where researchers did not hesitate in self-experimentation or experimenting on family members as subject for research. The Nobel laureate Gerhard Domagk (1895–1964) discovered prontosil sodium (a sulfonamide) and first tested it on his own 6-year-old daughter who had contracted a severe streptococcal infection from an unsterilized needle and Johann Jorg (1779–1856) swallowed 17 drugs in various doses to record their properties, are only two among many such instances.
Medical science is one discipline where the advancement of knowledge is hugely guided by research and humankind has benefitted from many experiments. However, benefit and risk are the two faces of the same coin. If there is no loss there is no gain; but the risk/loss is assumed by individuals/participants of research, and benefit/gain is reaped by a population who did not have to bear that risk. The role of ethical guideline is to establish the balance between benefit and risk and to ensure all the participants gets fair treatment that he/she expects from his/her treating physician.
Ethics is pluralistic. There can be disagreement among individuals about what is right and what is wrong, and even when they agree, the reasons can be different.
Despite the differences, the fundamental ethical principles is in harmony with the basic human rights proclaimed in the “United Nations Universal Declaration of Human Rights,” which upholds right to life, freedom from discrimination, torture and cruel inhuman or degrading treatment, freedom of opinion and expression, equal access to public services in one's country, and to medical care.[2]
Nazi experimentations during World War II are the horrendous examples of atrocious acts where thousands of war prisoners were subjected to inhuman torture in the name of research and benefit to science. This was the time when humankind realized that the whims and fancies of researchers need to be reined and led to the evolution of the first guidelines for researcher, the Nuremberg code.[5]
Evolution of Ethics Guidelines
After the World War II, trial was conducted on 23 Nazi doctors and scientists at Nuremberg for the murder of concentration camp inmates who were used as research subjects. Among the 23, 15 were convicted. Seven were condemned to death by hanging, 8 received prison sentences from 10 years to life. The trial brought to light the tortures that were conducted, and in 1947, the judgment culminated in the formulation of codes to guide research on humans, famously known as Nuremberg Code. The code highlighted on the need for informed consent, prior animal work, qualified scientists, risk justification by anticipated benefits, avoidance of physical and mental suffering, death, or disabling injury.[5]
Some researchers, however, ignored the code and continued to exploit the faith of the patients. In Willowbrook Hepatitis Study (1956), children were deliberately infected with mild form of hepatitis, and consent was obtained from parents without informing about the hazards and giving the opportunity of school admission on participation in the study.[6] In 1963, Jewish Chronic Disease Study was conducted where cancer cells were inoculated in senile subject without proper explanation on risk. Both these studies used a vulnerable group of patients who could not take independent decision.[7] The situation was highlighted in an article published in 1966 which described 22 such examples of research where there were controversies regarding the ethics, and all were conducted by reputable researchers and published in major journals (Beecher article).[8]
In view of the emerging situation, World Medical Association (WMA) General Assembly (Helsinki, Finland, 1964) developed a set of guidelines to safeguard the rights and well-being of subjects participating in clinical research. This is referred to as the Declaration of Helsinki and is revised from time to time, the last amendment in 64th WMA general assembly in Brazil, 2013. The declaration of WMA binds the physician with the words, “The health of my patient will be my first consideration,” and the International Code of Medical Ethics declares that, “A physician shall act in the patient's best interest when providing medical care.” The declaration specifically defines that the duty of the physician who are involved in medical research is to promote and safeguard the health, well-being, and rights of patients.[9]
In a very shocking turn of events, unethical research conducted by the United States Public Health Service (Tuskegee Syphilis Study) surfaced in 1972. The study was initiated in 1932 to study the natural course of syphilis on African-American participants and even after the development of penicillin, the trial participants were denied of the treatment and when it came to light it has already claimed 28 lives and led to permanent disability in 100 subjects; along with 40 wives being infected resulting in 19 cases of congenital syphilis. Not only denial but also the study misinformed the subjects and claimed spinal tapas special treatment.[10] The sheer misconduct led the US government to set up ‘International Ethical Guidelines for Biomedical Research Involving Human Subjects’ that submitted the report (Belmont report) in 1979, which stressed on three basic ethical principles: autonomy, beneficence, and justice.[11]
In the following years, various countries drafted their own guidelines of GCP, and in India, the Indian Council of Medical Research (ICMR)first released a policy statement on ethical considerations involved in research on human subjects in 1980. ICMR revised the guidelines in 2000 in the face of controversies and introduced “Ethical Guidelines for Biomedical Research on Human Subjects” and latest amendments was done in 2006.[12] In India, the ethics guidelines are given the legal status by way of Schedule Y Drugs and Cosmetics Rules, 1945 (rules 122A, 122B, 122D, 122DA, 122DAA, and 122E).[13]
With the advent of multicentric studies involving different countries, having a uniform GCP was a felt-need. For this purpose, the International Conference on Harmonisation-GCP was developed in 1996 in consideration of the current GCPs of the European Union, Japan, and the United States, as well as those of Australia, Canada, the Nordic countries, and the World Health Organization[14] [Figure 1].
Figure 1.
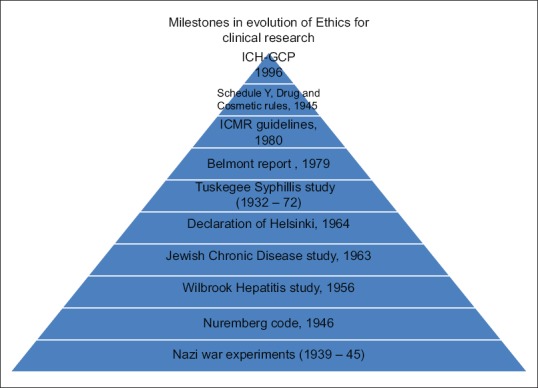
Milestones in the evolution of ethics for clinical research
Principles of Ethics in Research Involving Human Subject
The principles of ethics rest on the four pillars of autonomy, beneficence, justice, nonmaleficence,[15] and recently, two more pillars are added which includes confidentiality and honesty.
Autonomy
Autonomy is the respect for the patient's “right to self-governance, choice for care, and the right to accept or refuse treatment.”[16,17]
The principle of autonomy states that the patient has the right to make his or her own choice as to what procedure he or she aspires to have. Thus, the patient's right to an informed consent must be respected. The patient must be given the right information as what to expect, the risks involved and the alternative options available.
Beneficence
The principle of beneficence requires the practitioner to act in the patient's “best interest.” It is important for the practitioner to assess the risks versus the benefits of the procedure and maximize benefits, minimize harms. The motivation of the patient for having the procedure and how it will affect quality of life should be gauged by the physician. The physician should be specialized in the procedure and should be able to handle risks and side effects that might occur.
Justice
This principle seeks “fair treatment.” Exploitation of the patient for the sole purpose of recruitment for the study and completion of research should be refrained. The practitioner should be respectful to the patients’ wishes, understand the depth of the problem, and educate the patient about the expectation from the procedure. Risk and benefits must be equally shared by all trial participants.
Nonmaleficence
The principle of nonmaleficence requires the practitioner to “do no harm” to the patient. The practitioner should discuss of the possible side effects and complications of the trial procedure before including a person in the trial. At this point, the practitioner may suggest alternative procedures and treatments that may be more beneficial for the patient.
Confidentiality
It is essential to maintain confidentiality of all participating study patients, security of study data, photographs, biological samples, audio-visual records, etc.
Honesty
Investigator should be truthful to the study participants regarding the trial protocol, risk-benefits; and to coinvestigators, sponsors, ethics committee, and regulatory agencies regarding the adherence to trial protocol and outcome.
Indian Perspective
India in recent times has become an important country for clinical trials of international pharmaceutical companies because of abundance of patients, heterogeneous genetic population, availability of trained human resource (both doctors and support staff) and last not the least, low expenditure. A report shows that since 2004 the number of new trials has increased at 31% Compound Annual Growth Rate, and the clinical trials market has grown at 30% (almost double of the global average).[17] However, concerns were raised about ethical implications of globalization of clinical trials to developing countries[18] which are compounded by the adverse media coverage in India.
Major limitations detected at site inspection visits by regulatory authority in India include data credibility, inadequate and inaccurate records, failure to follow investigational plan, failure to notify Institutional Ethics Committee (IEC) of changes, and failure to submit progress reports. There are also concerns over areas of subject protection, namely, consent, IEC approval, reporting of adverse drug reactions (ADRs).[19]
Keeping in mind the changing situations, the Central Drugs Standard Control Organization (CDSCO) has amended the existing Schedule Y with the major thrust areas being functioning of IEC, informed consent process, ADR reporting, compensation in case of ADR.[13] Ethics committee may be considered as the “eyes and earsof CDSCO” and they are the guardian of ethics in clinical research conducted in the institute. The term, institutional review board, and independent ethics committee are used interchangeably at times. The role of ethics committee has become paramount important following the maloccurrence of events resulting from breach in ethical standard in clinical research.[19] CDSCO presently has implemented the rule for registering the ethics committee, and only those registered under CDSCO can approve the conduct of clinical trials. Thus, it has become imperative that researchers posted in those institutes having “ethics committee registered under CDSCO” can only carry out clinical trials. Thus, knowledge about ethics committee and its functioning is not only administration's prerogative but also important from researcher's view point.
Ethics Committee
Ethical review of clinical trial applications follows a decentralized process in India and requires ethics committee approval for each trial site. The ethics committees are based at clinical or academic institutions and hospitals. Ethics committee is an independent body that plays the pivotal role in ensuring that a trial is conducted in accordance with GCP guidelines and to safeguard the safety and well-being of subjects participating in a clinical trial. Ethics committee ensures a competent review of all the ethical aspects of the project proposal submitted and does it free from any bias or external influence. In institutions where a scientific review board is not present, the ethics committee assumes the additional responsibility of reviewing the scientific rationality of the research proposal submitted.
An ethics committee should be constituted with at least seven members and appoints from among its members a chairperson (from outside the institution) and a member secretary (generally from the parent institution to conduct committee business). To represent differing viewpoints, the members should be a mix of medical/nonmedical and scientific/nonscientific persons, including the lay public. The composition should be as follows:
Chairperson
One to two basic medical scientists (preferably one pharmacologist)
One to two clinicians from various institutions
One legal expert or retired judge
One social scientist/representative of nongovernmental voluntary agency
One philosopher/ethicist/theologian
One lay person from the community
Member secretary.
If the institution specializes in certain areas of research, it is desirable to include a member from specific patient groups (e.g., HIV AIDS, genetic disorders, etc.) in the ethics committee as much as possible. If required, subject experts could be invited to offer their views, but would not have any voting rights. There should be appropriate gender and age representation on the ethics committee.
According to Indian GCP, ICMR guidelines, and Schedule Y, the ethics committee review should be conducted through formal meetings and should not resort to decisions through a circulation of proposals or E-mails. The committee should meet at regular intervals and should not keep a decision pending for >3–6 months, which should be defined in the standard operating procedure (SOP). Proper record keeping of all meetings, decisions should be done. The ethics committee is not only entrusted with reviewing proposals but also reviewing ongoing trials by reviewing the periodic study progress reports furnished by the investigators, and/or monitoring and internal audit reports furnished by the sponsor, and/or by self visiting the study sites. In the case of clinical trial-related injury or death, the ethics committee should also review and make recommendations for compensation to be paid by the sponsor within a certain time frame.
A clinical trial should be initiated at an investigator site only and only after obtaining written approval of the respective IES. Any amendment to the approved trial documents requires a fresh ethics committee approval. The minimum clinical trial documents that should be reviewed by the committee are as follows:
Protocol (for scientific rationale)
Informed consent document (for the safety and welfare of research participants)
Informed consent document's vernacular translation
Investigator brochure for information regarding clinical and nonclinical data of the investigational product
Study advertisement for participant recruitment or any other written information to the patient
Grants, payments, insurance documents.
Added responsibility of ethics committee
In academic trials (e.g., postgraduate thesis/investigator- initiated trials in academic institutions), the ethics committee of the institute decides whether the protocol is to be sent to the regulatory body (DCGI) for approval before initiation of the trial. In the event of not receiving any reply from the office of the DCGI by 30 days, the trial can be initiated, but also the record of the communication must be retained by the ethics committee.
In a recent report, it was revealed that approval letter of IEC's has deficiencies in various aspects, including composition, quorum, and review of insurance and clinical trial agreement. This highlights the gaps in education and training of IEC members.[20] With reports of IEC malfunction pouring in the media,[21] CDSCO has taken stern steps in streamlining the IEC functioning. The Schedule Y is amended by inserting a rule 122DD which specifies the detail procedures for the registration of ethics committee.[22]
Registration of Ethics Committee
As per rule 122DD, all ethics committees have to be registered with Drug Controller General of India (DCGI) without which they cannot approve any clinical trial protocol and has come into effect from February 25, 2013.[22] For the purpose of registration, application has to be sent by the ethics committee to CDSCO as per the requirement specified in Appendix VIII of Schedule Y [Annexure I] along with a checklist available from CDSCO website.[23] The information that is required to be submitted by the applicant for registration of the ethics committee are:
Name of the ethics committee
Authority under which the ethics committee has been constituted, membership requirements, the term of reference, conditions of appointment, and the quorum required
The procedure for resignation, replacement, or removal of members
Address of the office of the ethics committee
Name, address, qualification, organizational title, telephone number, fax number, E-mail, mailing profile of the chairman
Name, address, qualification, organizational title, telephone number, fax number, E-mail, mailing profile of the members of the ethics committee. The information should also include member's specialty (primary, scientific, nonscientific), members affiliation with institution, and patient group representation if any
Details of supporting staff
Details of the type of clinical research reviewed by the existing committee (e.g., pharmaceuticals, devices, epidemiological, retrospective, herbals, etc.), documents reviewed for any clinical trial protocol, including informed Consent documents, information in respect of number of meetings of the committee and documentation of the minutes of meetings of these committees concerning clinical trial, information regarding review of serious adverse events reported during conduct of clinical trial
The SOPs to be followed by the committee in general
The SOPs to be followed by the committee for vulnerable population
Policy regarding training for new and existing members along with the SOPs
Policy to monitor or prevent the conflict of interest along with SOPs
Details of any previous audit or inspection of the committee.
The licensing authority (CDSCO) after being satisfied with the requirements grants registration for 3 years from the date of issue after which the ethics committee has to apply for reregistration within 3 months from expiry. For reregistration, GCP training certificate of each member of the ethics committee and information on monitoring of ongoing trial has been made mandatory. By registering with the CDSCO, the IEC commits itself for safeguarding the rights, safety, and well-being of the trial subjects by:[23]
Reviewing and according its approval to a clinical trial in accordance with Schedule Y and GCP and also to carry ongoing review of the trial at appropriate intervals
In the case of any SAE, the committee would analyze and forward its opinion as per procedures specified under Appendix XII of Schedule Y
-
The committee would allow inspectors or officials authorized by the CDSCO to enter its premises to inspect any record, data, or any document related to clinical trial and also would provide adequate replies to any query raised by such inspectors or officials (in relation to the conduct of clinical trial)
Maintain adequate and accurate records after the completion or termination of the study for not <5 years from the date of completion or termination of the trial (Both in hard and soft copies).
The ethics committTo comply with the regulatory environment in India, the ethics committee is required to have records and access to the written SOPs, national and international guidelines, constitution and composition of the ethics committee the curriculum vitae of all its members, copies of all the trial documents received for review, all the correspondence between the investigator and the committee, agenda and minutes of all the ethics committee meetings, final reports of all the studies it approved.
CDSCO is strict about certain parameters while registering the ethics committee and that has be kept in mind while applying:[23]
Chairperson: The chairperson has to be outside the institute, and the principal/director of the institute cannot be chosen as the chairperson for the purpose of autonomy of the committee. The principal/director may the member secretary for operational feasibility. It also has to be kept in mind that a chairperson cannot serve the dual purpose of lay person/pharmacologist/legal expert or any other essential membership criteria of IEC laid down by Schedule Y; and separate representation of that member category has to be their in the committee. In case the chairperson is absent for a particular meeting, the committee can choose any member who are present, to function as the chairperson for that meeting; but the person has to be from outside the institute
The lay person in the committee: The idea behind inclusion of lay person is to have a person in the committee who is representative of the study population; thus having a person from the creamy layer of the society undermines the very essence of the logic. CDSCO is strict that lay should come from the society and free of any conflict-of-interest. Appointing the secretary, account officer, librarian of the institution as the lay person is unacceptable
Legal expert: A legal expert can be a practicing lawyer or a retired judge; not just anyone who has the degree of Bachelor of Legislative Law (LLB) and has never practiced law
Authority under which committee is constituted: For the purpose of autonomy, it is desired that committee members (including the member secretary) are chosen by the chairperson and not by the principal/director/any other person belonging to the institute
Conflict of interest: It should be mentioned in the SOP that all members having conflict of interest would refrain from the discussion on that particular proposal. At the end of the meeting, the members should sign the undertaking that they had no conflict of interest
Research involving vulnerable population: The SOP must clarify how the ethics committee is going to handle the research involving vulnerable population or else it may spell out that it will be decided on case-to-case basis
GCP training of members: It is mandatory that members of ethics committee are trained in GCP, and it is essential to submit their certificates of their training while applying for the registration.
Exemption from Institutional Ethics Committee Review
Proposals which have less than “minimal risk” are exempt from review by the IEC. “Minimal risk” is the risk anticipated not greater than that encountered in routine daily life activities of general population. Examples of such proposals are research on educational practices (instructional strategies, curricula, or classroom management), research involving the collection of or study of existing data, documents, records, pathological or diagnostic specimens if these sources are publicly available and do not identify participants.
Expedited Institutional Ethics Committee Review
Proposals with no more than minimal risk can be considered for expedited review. For example, minor deviations from originally approved research during the period of approval, continuing review of approved projects, research involving clinical materials (data, documents, records, and specimens) that have been collected for nonresearch (clinical) purposes and during emergency situations (disasters, outbreaks).
With increased number of clinical research and trials taking place, the responsibility of the ethics committee has increased manifold. Improved ethics committee functioning is the need of the hour so that the research participants are protected, and ethical clinical research is done.
Tips for Facing the Ethics Committee
The ethics committee is strict regarding certain aspects of protocol, and these are the essential elements the investigator should be careful while submitting
Informed consent document: The protocol must contain the vernacular version of the patient information sheet and the informed consent form
Audio-visual recording of the informed consent process: This is relevant when the research involves new molecular entity or vulnerable population
Vulnerable population: Research involving vulnerable population is better avoided unless such subjects are specific beneficiaries of the research
Serious adverse events: The protocol must elaborate the timeline for reporting of serious adverse events to the ethics committee, regulatory body (DCGI), sponsors (if relevant) and head of institution; and also management protocol of serious adverse events
Compensation in serious adverse events: The insurance certificate for compensation and management of serious adverse events should be annexed with the protocol (relevant for sponsored trial). For academic trials, corpus fund earmarked for such purpose should be maintained and mentioned
Placebo-controlled trial: Better to avoid Placebo-controlled trial if standard care exist for the disease and active-control should be used in place of placebo
Advertisements for recruitment of trial participants: This should be better avoided
Compensation for participation in trial: Monetary compensation in excess of travel allowance and daily wage loss should be avoided
Registration of trial in clinical trial registry: This is to be done for all clinical trials and optional for observational studies
Submission of progess report: It is essential to document in the protocol the plan for submission of progress report.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
What is new?
When a doctor becomes a researcher, the duties and responsibilities are compounded and he has to serve the role of both a doctor and researcher maintaining the rules and regulations of “Good Clinical Practice (GCP)” in clinical research laid down by International Conference on Harmonisation-GCP (ICH-GCP), Government of India (Indian-GCP), Schedule Y and also are guided by the meeting resolution of World Medical Association (WMA) e.g., Helsinki declaration and is guided by the principles of autonomy, non-maleficence, justice, beneficence, confidentiality and honesty.
Acknowledgment
The authors thankfully acknowledge Prof. Debabrata Bandyopadhyay (Professor and Head, Department of Dermatology, Medical College, Kolkata) and Professor Avijit Hazra (Professor, Department of Pharmacology, IPGME&R, Kolkata) for reviewing the manuscript and providing valuable inputs.
References
- 1.Hippocratic Oath. Ethical Code. [Last accessed on 2017 Apr 13]. Available from: http://www.britannica.com/topic/Hippocratic-oath .
- 2.Williams JR, editor. Medical Ethics Manual. France: World Medical Association; 2009. [Google Scholar]
- 3.Williams JR. Declaration of Geneva. Medical Ethics Manual. France: World Medical Association; 2009. [Google Scholar]
- 4.Participants in Clinical Research. [Last accessed on 2017 Apr 13]. Available from: http://www.icmr.nic.in/bioethics/cc_biothics/presentations/sym_pune/UGs/Research%20Ethics.pdf .
- 5.Nuremberg Military Tribunal. The Nuremberg Code. JAMA. 1996;276:1691. [PubMed] [Google Scholar]
- 6.Krugman S. The Willowbrook hepatitis studies revisited: Ethical aspects. Rev Infect Dis. 1986;8:157–62. doi: 10.1093/clinids/8.1.157. [DOI] [PubMed] [Google Scholar]
- 7.Sanmukhani J, Tripathi CB. Ethics in clinical research: The Indian perspective. Indian J Pharm Sci. 2011;73:125–30. doi: 10.4103/0250-474x.91564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beecher HK. Ethics and clinical research. N Engl J Med. 1966;274:1354–60. doi: 10.1056/NEJM196606162742405. [DOI] [PubMed] [Google Scholar]
- 9.WMA Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. [Last accessed on 2017 Apr 13]. Available from: http://www.wma.net/en/30publications/10policies/b3/
- 10.Thatte U. Ethical issues in clinical research. In: Gupta SK, editor. Basic Principles of Clinical Research and Methodology. 1st ed. New Delhi: Jaypee Brothers; 2007. pp. 58–73. [Google Scholar]
- 11.The Belmont Report. National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research. Washington, DC: U.S. Government Printing Office; 1979. [Google Scholar]
- 12.Ethical Guidelines for Biomedical Research on Human Subjects. New Delhi: Indian Council of Medical Research; 2006. [Last accessed on 2017 Apr 13]. Available from: http://www.icmr.nic.in/ethical_guidelines.pdf . [Google Scholar]
- 13.Schedule Y. Drug and Cosmetic Rule. Government of India. 1945. [Last accessed on 2014 Oct 13]. Available from: http://www.rgcb.res.in/wp-content/uploads/2014/07/Schedule-Y.pdf .
- 14.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6(R1) 1996 [Google Scholar]
- 15.Beauchamp TL, Childress JF. Principles of Biomedical Ethics. 5th ed. Oxford: Oxford University; 2001. [Google Scholar]
- 16.Chung KC, Pushman AG, Bellfi LT. A systematic review of ethical principles in the plastic surgery literature. Plast Reconstr Surg. 2009;124:1711–8. doi: 10.1097/PRS.0b013e3181b98a9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennelly J. Medical ethics: Four principles, two decisions, two roles and no reasons. J Prim Health Care. 2011;3:170–4. [PubMed] [Google Scholar]
- 18.Glickman SW, McHutchison JG, Peterson ED, Cairns CB, Harrington RA, Califf RM, et al. Ethical and scientific implications of the globalization of clinical research. N Engl J Med. 2009;360:816–23. doi: 10.1056/NEJMsb0803929. [DOI] [PubMed] [Google Scholar]
- 19.Bhatt A. Indian clinical trials: Paradigm shift from speed to quality? Perspect Clin Res. 2012;3:1–3. doi: 10.4103/2229-3485.92299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taur SR, Bavdekar SB, Thatte UM. Survey of ethics committee protocol approval letters: Compliance with Schedule Y/ICMR guidelines 2006. Indian J Med Ethics. 2011;8:214–6. doi: 10.20529/IJME.2011.083. [DOI] [PubMed] [Google Scholar]
- 21.Nagarajan R. Experiments with Untruth. [Last accessed on 2016 Nov 01]. Available from: http://www.articles.timesofindia.indiatimes.com/2011-07-10/specialreport/29757613_1_clinical-trials-independent-ethics-committeeindian-council .
- 22.Amendment to the Drugs & Cosmetics Rules 1945, Gazette Notification [GSR 72 (E)] 2013. [Last accessed on 2017 Apr 13]. Available from: http://cdsco.nic.in/html/G.S.R%2072(E)%20dated%2008.02.2013.pdf .
- 23.System for the Pre-Screening of the Applications for Registration of Ethics Committee. [Last accessed on 2017 Apr 13]. Available from: http://www.cdsco.nic.in/writereaddata/CHECKLIST%20FOR%20SUBMISSION%20192013.pdf .