

Abstract
Consenting to participate in a clinical research study after being properly and correctly informed upholds the basic ethical principle of “autonomy” in human research. The informed consent is a process by which the physician sensitizes the patient about the nature, procedures, risks benefits, treatment schedules, etc of the study in a language that is non-technical and understandable by the study participant. Informed consent document (ICD) has got two parts: the ‘Subject Information Sheet’ and the ‘Informed Consent Form’ (ICF); and they have to be approved by the Institutional Ethics Committee (IEC) before administration. Consent should be obtained without any coercion. In case of a situation where a participant is not able to give informed consent (e.g. unconscious, minor or those suffering from severe mental illness or disability) or is illiterate, it has be obtained from a legally acceptable representative (LAR). If the participant or LAR is unable to read/write, then an impartial witness should be present during the entire informed consent process and must append his/her signatures to the consent form. For children < 7 years, verbal consent is essential and for mature minors (age group 7 to 18 years) informed assent should be obtained.
KEY WORDS: Informed consent form, informed consent document assent, LAR, vulnerable population
What was known?
Doctors are aware of taking consent of patient during intervention or procedures wherein the focus is on the description of the risks/complications that are involved with the procedure, and obtaining the signature of the patient and relative who give their consent.
Introduction
The informed consent process upholds the basic ethical principle of “autonomy” in human research. In any clinical trial, a freely given, informed written consent has to be obtained from each research participant to comply with the requirements of Good Clinical Practices (GCP),[1] the Indian Council of Medical Research (ICMR) guidelines,[2] and Schedule Y.[3]
Informed Consent Process
The informed consent is a process by which the physician sensitizes the patient about the nature of the study in a language that is nontechnical and understandable by the study participant. Informed consent document (ICD) has got two parts: the “subject information sheet” and the “informed consent form” (ICF), and they have to be approved by the Institutional Ethics Committee (IEC) before administration. The format of ICF should be in accordance with the Appendix V of Schedule Y (Annexure III).[4] The content of the document should be brief, lucid, nontechnical, and simple enough to be understood by the participant. Consent should be obtained without any coercion.
Informed consent process is of paramount importance to safeguard the autonomy of a person with regard to his willingness to participate in a trial. It is especially relevant in the present day scenario when the mistrust among the study participants is growing, and according a recent report in 26% of Indian patients, mistrust is the barrier for study participation.[5] The proper consent process can rebuild this trust and can bridge the rapport between the physician and study participants. The issue has come into attention with the Supreme Court order on October 21, 2013,[6] with respect to five clinical trials, where informed consent, which is the most important aspect of GCP, was not properly obtained.
Role of a Legally Acceptable Representative and Impartial Witness
In case of a situation where a participant is not able to give informed consent (e.g., unconscious, minor, or those suffering from severe mental illness or disability), it has be obtained from a legally acceptable representative (LAR). A LAR is an individual or a legal body authorized under applicable law to consent, on behalf of a prospective participant, to the individuals’ participation in the clinical trial. If the participant or LAR is unable to read/write, then an impartial witness should be present during the entire informed consent process and must append his/her signatures to the consent form. An impartial witness is a person who is independent of the trial and cannot be unduly influenced by the people involved with the trial and who attends the informed consent process if the participant or the participant's LAR cannot read and who reads the ICF and any other written information supplied to the participant. Usually, the patient party of the subsequent patient is taken as impartial witness. Staff nurse or technician is usually not regarded as impartial witness as they can be unduly influenced by the investigator. In case of a matured minor (as defined by the ICMR guidelines as an individual <18 years but ≥7 years), informed assent needs to be taken apart from parental/LAR consent as explained later.[2]
Practical tip: The next educated patient or his/her relative in the OPD can act as the best LAR.
Flowcharts describing the process of obtaining ICF a literate participant, illiterate participant, and when both participant and LAR are illiterate are shown in Figures 1-3, respectively.
Figure 1.

Flowcharts describing the process of obtaining informed consent from a literate participant
Figure 3.
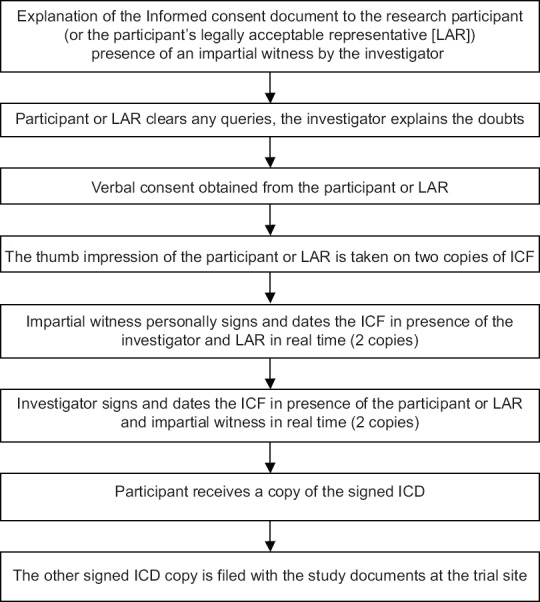
Flowcharts describing the process of obtaining informed consent when both participant and legally acceptable representative are illiterate
Figure 2.
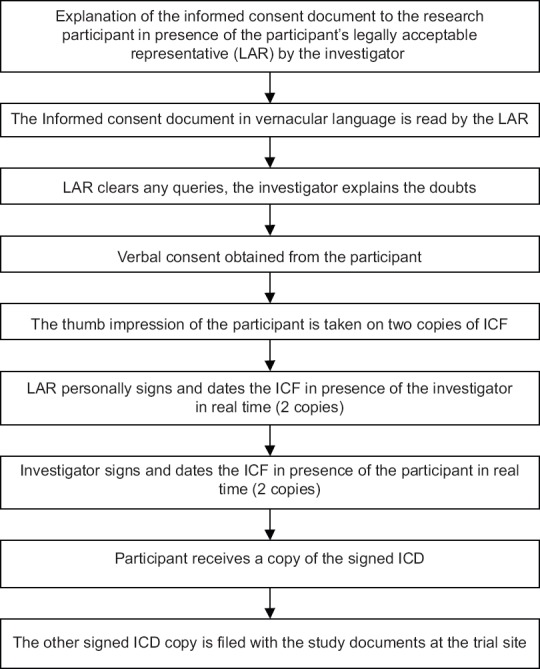
Flowcharts describing the process of obtaining informed consent from an illiterate participant
Audiovisual Recording of Informed Consent Process
In spite of having a nicely drafted ICD, it was observed that the elements of informed consent were not followed in letter and spirit. In view of the recent scenario, CDSCO has emphasized that the Rule 122DAC (which highlights that clinical trials be conducted following GCP guidelines) be strictly followed. Accordingly, the office of DCGI has made it mandatory that the audiovisual (AV) recording of process (in effect from November 2013) of providing information to the participant and his/her understanding on such consent has to be done with proper maintenance of confidentiality.[7] However, in the fifth amendment of the Drug and Cosmetic Rules, it was stated that the investigator should obtain AV recording of the informed consent process for vulnerable participants in clinical trials of new chemical entity or new molecular entity. In cases where clinical trials are conducted on anti-human immunodeficiency virus (HIV) and antileprosy drugs, the investigator should only obtain an audio recording of the informed consent process.
To identify the participant/LAR/impartial witness, his/her photo ID needs to be documented. The video camera for the AV recording needs to be of adequate enough to capture the facial details of study participant, LAR/impartial witness (if any), and investigator/authorized person present during the consent process. The place where the AV recording is performed should be conducive to recording of disturbance-free audio and video of the consent process. It is important that, during the process of AV recording, unrelated persons/patients at the hospital are not included in this study. AV recording of informed consent process and other related documents should be preserved safely after the completion/termination of the study for at least a period of 5 years if it is not permanently.[8]
Practical tips:
The AV or audio record is a confidential document that has to be archived like all other trial-related documents
Any changes in the ICF after the trial commencement have to be re-approved by the IEC before it is administered
The ICF should be written in English and in the vernacular language that the trial participant understands
Every ICF should be signed by the participant and the investigator and dated at real time. A copy of the signed ICF is retained by the investigator and another copy is given to the participant
The investigator or a person knowledgeable about the trial and designated by the investigator should obtain informed consent.
Requirements
Any changes in the ICF after the trial commencement have to be re-approved by the IEC before it is administered
The ICF should be written in English and in the vernacular language that the trial participant understands
Every ICF should be signed by the participant and the investigator and dated at real time
A copy of the signed ICF is retained by the investigator and another copy is given to the participant
The investigator or a person knowledgeable about the trial and designated by the investigator should obtain informed consent.
Elements that Need to be Included in the Informed Consent Form
Schedule Y states the checklist that is required to be incorporated into the ICF. They are as follows:
-
1.
The study involves research and an explanation of its nature and purpose
Practical tip: ICD may start with a line “We invite you to take part in a research study entitled.****”
-
2.
The expected duration of the participant's participation
Practical tip: A heading in the ICD may be inserted on “Time duration of the procedures and study” which may state the following:
“If you agree to take part in this study, your involvement will last approximately ‘n’ weeks/months/years. You will be asked to return to the clinic nth times. Each clinic visit will take approximately ‘n’ minutes”
-
3.
The procedures to be followed
Practical tip: Any minor procedure (e.g. venipuncture) should also be mentioned
-
4.
Any reasonably foreseeable risks or discomforts to the participant resulting from his/her participation and whether the project involves more than minimal risk
Practical tip: A heading in the ICD may be inserted on “Discomfort and Risk” which may state the following:
“While on the study, you are at risk for the following side effects (and the side effects to be enumerated). Most of them are listed below, but they will vary from person to person”
-
5.
The investigational product may fail to achieve the intended therapeutic effect
Practical tip: It is best to insert a line “However, there is no guarantee that you will benefit from being in this research”
-
6.
In the case of a placebo-controlled trial, the placebo administered to the participant(s) shall not have any therapeutic effect
Practical tip: It is best to insert a line “half/one-third/exact percentage of the study participants are going to get dummy medicine and unlikely to achieve any benefit”
-
7.
Any benefits or prorated payment to the participant or others reasonably expected from the research; if no benefit is expected, the participant should be made aware of this
Practical tip: A heading in the ICD may be inserted on “Potential Benefit” which may state the following:
“Possible benefits to the participant: The possible benefit you may experience in this research includes treatment of your disease condition
Possible benefits to others: The results of this research may guide the future treatment of ‘xyz’ disease condition
You will be given trial medications free of cost and investigations will be borne by the hospital”
-
8.
The disclosure of specific appropriate alternative procedures or therapies available to the participant
Practical tip: A heading in the ICD may be inserted on “Alternative therapies” which may state the details with their pros and cons
-
9.
The mechanism by which confidentiality of records identifying the participant will be maintained and who will have access to the participant's medical records
Practical tip: A heading in the ICD may be inserted on “Statement of confidentiality” which may state the following:
“Your research records that are reviewed, stored, and analyzed at ‘abc’ medical college and hospital will be kept in a secured area in department of ‘xyz’. Your blood samples collected for research purposes will be labeled with your initials and will be stored in department of ‘uvx’. For research records sent to examiner, you will not be identified by name, address, or phone number. The records and blood samples may include your initials. The list that matches your name with the code number will be kept in a locked file in department of ‘ijk’
In the event of any publication or presentation resulting from the research, no personally identifiable information will be shared
We will keep your participation in this research study confidential to the extent permitted by law. However, it is possible that other people may become aware of your participation in this study. For example, the following people/groups may inspect and copy records pertaining to this research
- The investigator and his/her team
- The Institutional Ethics Review Board (a committee that reviews and approves research studies) and
- The Drug Regulatory Authority
Some of these records could contain information that personally identifies you. Reasonable efforts will be made to keep the personal information in your research record private and confidential, but absolute confidentiality cannot be guaranteed”
-
10.
Clinical trial treatment schedule(s) and the probability for random assignment to each treatment
Practical tip: A heading in the ICD may be inserted on “Treatment schedule” which may state the “the details of the procedures and follow-up” and “they will be randomly assigned to each treatment arm”
-
11a.
The policy for compensation for participation should be stated clearly. Practical tip: A heading in the ICD on “Compensation for participation” which may state the following: “Participation in this study will be free of cost to you. Drug and clinic visits will be provided without any cost to you. You will be given Rs. 500/- as reimbursement for local conveyance as approved by IEC.”
-
11b.
The policy on compensation and/or medical treatment(s) available to the participant in the event of a trial-related injury, disability, or death
Practical tip: A heading in the ICD may be inserted on “Compensation for injury” which may state the following:
In case of injury occurring to you during the study, free medical management and cost of procedures required for management shall be given as long as required or till such time it is established that the injury is not related to the study, whichever is earlier.
In case the injury occurring to you is related to the study, you will be entitled for financial compensation, according to the Licensing Authority defined under clause (b) of Rule 21, and the financial compensation will be over and above any expenses incurred on your medical treatment. In case, there is no permanent injury, the amount of compensation shall be commensurate with the nature of non-permanent injury and the loss of your wages. In the case of your death related to the study, your nominee(s) would be entitled for financial compensation, as per order of the Licensing Authority defined under clause (b) of Rule 21, and the financial compensation will be over and above any expenses incurred on your medical treatment. The expenses on medical management and financial compensation in case of injury to you due to the study shall be bourne by the sponsor.
-
12.
Principal investigator (PI) and co-investigator(s) contact information
Practical tip: A heading in the ICD may be inserted on “Contact Information for Questions or Concerns” which states the following: you have the right to ask any questions you may have about this research. If you have questions, complaints or concerns, or believe, you may have developed an injury related to this research, contact the following contact personnel
This is to be followed by the name of and contact information of PI and Co-PI
-
13.
EC contact information
-
14.
The participant's responsibilities in participating in the trial
Practical tip: It is best to insert a line “If you choose to take part in this research, your major responsibilities will include taking the medicines as advised by your physician, coming for follow-up as suggested”
-
15.
Participation is voluntary, the participant can withdraw from the study at any time, and refusal to participate will not involve any penalty or loss of benefits to which the participant is otherwise entitled Practical tip: A heading in the ICD may be inserted on “Voluntary Participation” which states the following:
“Taking part in this research study is voluntary. If you choose to take part, you have the right to stop at any time. If you decide not to participate or if you decide to stop taking part in the research at a later date, there will be no penalty or loss of benefits to which you are otherwise entitled
If you will be participating in another clinical trial while in this research, you should discuss the procedures and/or treatments with your physician or the investigators. This precaution is intended to protect you from possible side effects from interactions of research drugs, treatments, or testing”
-
16.
Foreseeable instances under which the investigator(s) may remove the participant without his/her consent
Practical tip: It is best to insert a line “Your investigator may take you out of the research study without your permission. Some possible reasons for this are noncompliance, not appearing at follow-up”
-
17.
The consequences of a participant's decision to withdraw from the research, and procedures for orderly withdrawal by the participant
Practical tip: It is best to insert a line (vide supra in voluntary participation) “if you decide to stop taking part in the research at a later date, there will be no penalty or loss of benefits to which you are otherwise entitled”
-
18.
If any new significant findings develop which may affect the participant's willingness to continue, the participant or LAR or guardian will be informed
Practical tip: It is best to insert a line “During the course of the research, you will be provided with any significant new findings that may affect your willingness to continue participating in this research”
-
19.
The particular treatment or procedure may involve risks to the participant (or to the embryo or fetus, if the participant is or may become pregnant), which are currently unforeseeable
-
20.
Additional costs to the participant that may result from participating in the study
-
21.
If genetics and/or HIV testing is to be conducted, counseling before consent for testing must be given as per national guidelines
-
22.
If any biological sample is taken from the participant, the storage period of biological sample, related data, and options for future use will be stated to the participant which he might agree or refuse
-
23.
The participant's right to prevent the use of his/her biological sample (DNA, cell line, etc.) at any time during the conduct of the research
-
24.
The participant should be told about any publication of his medical information, including unidentifiable photographs and pedigree charts as a part of the research done
-
25.
The informed consent form should contain the name of the participant, date of birth/age, address of the participant, qualification, occupation, annual income, name age address contact number of the nominee in case of trial related death of the participant. The relation of the nominee to the participant should also be mentioned. These details are required during compensation if such requirements arise.
Compensation for Participation in Research
The ICF should include a statement that describes the anticipated payment to the participant for his participation as a research subject in the clinical trial. This payment is to reimburse the inconvenience and time spent, expenditure for travel, food, and loss of wages. Such payment should not be so excessive as to induce a patient to participate. The ICF also includes a statement that states that compensation and medical treatment will be available if any adverse event occurs. The Ethics Committee approves all the payments, reimbursements that will be provided to the participant. When a legal representative or guardian gives consent on behalf of an incompetent participant, no remuneration should be offered except to refund out-of-pocket expenses.
Rights of the Research Participant
The ICF addresses the basic rights of a research participant during an informed consent process. The participation in the research is entirely voluntary, and the participant may refuse to participate in the trial or withdraw his assent at any time during the ongoing study. Such refusal will not involve in any penalty, and such patient will receive his due standard care from the physician. The research participant has the right to be informed about the purpose, anticipated duration of the research study, study procedures, any potential benefits or risks, any compensation for participation or injury/treatment, and any significant new information regarding the research study. Participants have the right to privacy and confidentiality, and a statement must be provided in the ICF that recognizes this right. The investigator may assign serial numbers or initials to protect the identity, records, or data of the participants. The research participant has his right to inquire about any information about the trial and report any adverse event that might occur during the course of the trial. The contact information of the investigator and Ethics Committee are provided in the ICF to aid the participants seek out the investigator for any query or appeal in case of violation of his rights. The GCP and Declaration of Helsinki state that the participant's right to safety and protection of his health takes precedence over the objectives of biomedical research.[1]
Special Circumstances Involving Informed Consent Process
Special circumstances may arise in case of emergencies, dealing with unconscious patients, children, elderly age groups, and others. Schedule Y and ICMR guidelines mention the provision for protecting the rights for such research participants.
Medical emergencies
The life of the participant becomes of paramount importance in such situation, and treating such a research participant without his/her consent is permissible if the participant is unconscious, mentally ill, or gravely sick, and no one who is authorized to provide consent is unavailable. The LAR or guardian, if available, can give consent in such cases on behalf of the patient. Consent from the patient is taken once he regains consciousness or recovers his mental capacity to understand the research study. The ICF should address such circumstance if it is so anticipated. Such provision must be explicitly stated in the protocol.
Vulnerable population
The GCP defines “vulnerable” population as those research participants who have diminished autonomy such as those with incurable diseases, in nursing homes, in detention, unemployed or impoverished, prisoners, students, service personnel, in emergency rooms, homeless persons, nomads, refugees, and any ethnic or racial minority groups.[1] Such individuals should not be included unless the research is essential to promote the health of the population represented, and this research cannot instead be performed on other participants. The mode of consent for these participants must be carefully considered and approved by the Ethics Committee.
Geriatric population
Schedule Y states that geriatric population can be included in Phase II and III trials when the disease intended to be treated is typically a disease of aging, or the new drug is likely to differ in the safety and efficacy response of geriatric patients compared to nongeriatric population.[3]
Children/minors
According to the ICMR guidelines,[2] minor refers to an individual <18 years. The Schedule Y[3] states that children will not be involved in research that could be carried out equally in adults, and the research purpose is to obtain knowledge relevant to health-care needs of children. Pediatric participants are dependent on their legal representatives or guardians to provide consent on their behalf. However, all pediatric patients should be informed to the fullest extent possible about the study in nontechnical, simple understandable language. If the child refuses to participate, his refusal should be respected even if the parent or guardian consents. This brings us to the concept of “informed assent” in which mature minors (age group 7–18 years) consent is verbally taken after they have read and understood a specially designed assent form. Such assent form should be more simplistic than the standard ICF so that the child can understand the contents. The parent/LAR will sign and the investigator will sign indicating that informed assent has been taken. Thus, for mature minors, two sets of consent forms are required, one the ICF for the guardian and the other is the informed assent form.
Pregnant and nursing mothers
Pregnant or nursing women should only be included if the drug in trial is intended to be used by pregnant or nursing mothers or fetuses, and data on the nonpregnant women are unsuitable. Such research should carry less than minimal risk to the developing fetus and such participants should not be debarred unnecessarily from benefit from investigations, drugs, vaccines, or other agents that promise therapeutic or preventive benefits during pregnancy or lactation.
Mentally impaired subjects
Since such patients are not capable of consenting on their own, the proxy consent is obtained from the legal guardian or representative.
The informed consent process is thus much beyond just a sheet of paper on which the participant signs but is also the beginning of a good researcher and participant bonding. A well-taken consent makes the participant confident about the new arena of clinical research that he is faced with, makes him continue through all the follow-ups of the study, and adheres to study intervention regimen.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
What is new?
In clinical research, the crux of ethics lies on the Informed consent process which ensures the most important principle, autonomy. The informed consent document is a robust document which has got a section on patient information sheet detailing the research procedure, its benefits, risks, responsibilities of participants, compensation, confidentiality, alternative options and most importantly voluntary participation. The process of obtaining consent in literate, illiterate, children are also important and has to be ensured that the participant in not forced/coaxed to agree. Understanding that there may be breech in obtaining the informed consent, audio-visual recording of the process is also warranted in trials involving new molecular entities in vulnerable population.
Acknowledgment
The authors thankfully acknowledge Prof. Debabrata Bandyopadhyay (Professor and Head, Department of Dermatology, Medical College, Kolkata) and Professor Avijit Hazra (Professor, Department of Pharmacology, IPGME and R, Kolkata) for reviewing the manuscript and providing valuable inputs.
References
- 1.Good Clinical Practices for Clinical Research in India (IN-GCPs). Central Drugs Standard Control Organization, Ministry of Health and Family Welfare, Government of India; 10 May. 2003. [Last accessed on 2017 May 13]. Available from: http://www.cdsco.nic.in/
- 2.Ethical Guidelines for Biomedical Research on Human Participants (ICMR Guidelines). Indian Council of Medical Research, Department of Health, Ministry of Health and Family Welfare, Government of India; October. 2006. [Last accessed on 2017 Mar 02]. Available from: http://www.icmr.nic.in/ethical_guidelines.pdf .
- 3.Drugs and Cosmetics (IInd Amendment) Rules, 2005-Notification: Schedule Y-Requirements and Guidelines for Permission to Import and/or Manufacture of New Drugs for Sale or to Undertake Clinical Trials (Amended Version) (Schedule Y. Central Drugs Standard Control Organization, Ministry of Health and Family Welfare, Government of India; 20 January. 2005. [Last accessed on 2017 Jan 02]. Available from: http://rgcb.res.in/uploads/2014/07/Schedule-Y.pdf .
- 4.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6(R1) 1996. [Last accesed on 2017 Mar 06]. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Concept_papers/Q10_Concept_Paper.pdf .
- 5.Shah JY, Phadtare A, Rajgor D, Vaghasia M, Pradhan S, Zelko H, et al. What leads Indians to participate in clinical trials? A meta-analysis of qualitative studies. PLoS One. 2010;5:e10730. doi: 10.1371/journal.pone.0010730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.W.P.(C) No. 33/2012 of Swasthya Adhikar Mancha, Indore & Anr. vs. Ministry of Health and Family Welfare & Ors with W.P.(C) No. 779/2012. [Last accessed on 2016 Dec 09]. Available from: http://www.cdsco.nic.in/writereaddata/minutes_of_the%2013th%20_Apex_committee_meeting%2015%2004%2014(1).pdf .
- 7.F No. GCT/20/SC/Clin./2013. Dated 19th November 2013. DCGI. DGHS. Ministry of Health and Family Welfare. Govt. of India. 2013. [Last accessed on 2017 May 05]. Available form: http://www.cdsco.nic.in/writereaddata/Guidance_for_AV%20Recording_09.January.14.pdf .
- 8. [Last accessed on 2017 Apr 13];Guidelines on Audio-Visual Recording of Informed Consent Process in Clinical Trial. Directorate General of Health Services Ministry of Health & Family Welfare Govt. of India 9th January. 2014 Available from: http://www.cdsco.nic.in/writereaddata/Guidance_for_AV%20Recording_09.January.14.pdf . [Google Scholar]