

Abstract
Reactivation of mutant p53 in human tumor cells should induce cell death by apoptosis and thus eliminate the tumor. Several small molecules that reactivate mutant p53 have been identified. Here we show that STIMA‐1, a low molecular weight compound with some structural similarities to the previously identified molecule CP‐31398, can stimulate mutant p53 DNA binding in vitro and induce expression of p53 target proteins and trigger apoptosis in mutant p53‐expressing human tumor cells. Human diploid fibroblasts are significantly more resistant to STIMA‐1 than mutant or wild type p53‐carrying tumor cells. STIMA‐1 may provide new insights into possible mechanisms of mutant p53 reactivation and thus facilitate the development of novel anticancer drugs that target mutant p53‐carrying tumors.
Keywords: Mutant p53 reactivation, STIMA-1, CP-31398, Apoptosis, Cancer therapy
1. Introduction
The tumor suppressor p53 is expressed at low levels in most cells and tissues under normal conditions. Cellular stress such as DNA damage, oncogene activation, hypoxia and telomere erosion induces p53 protein levels, leading to an array of biological responses, including cell cycle arrest and apoptosis (Vogelstein et al., 2000). p53 exerts its function mainly through transcriptional regulation of specific target genes. Transactivation of p21, Gadd45, and 14‐3‐3sigma triggers p53‐dependent cell cycle arrest, whereas transactivation of Bax, Fas, Noxa and PUMA elicits p53‐dependent apoptosis (El‐Deiry, 1998; Ryan et al., 2001). p53 is frequently mutated in human tumors, indicating a strong selection for inactivation of the p53 tumor suppressor pathway during tumor evolution (Hainaut and Hollstein, 2000; Olivier et al., 2002).
The high frequency of p53 mutations in human tumors, the high levels of mutant p53 expression in tumors, and the fact that the mutant p53‐harbouring tumors often show increased resistance to conventional chemotherapy (Greenblatt et al., 1994; Campling and El‐Deiry, 2003) makes p53 an attractive target for novel cancer therapy. Several studies have demonstrated ways to restore normal function to mutant p53 (Bykov et al., 2003). A C‐terminal peptide derived from p53 itself can restore normal function to mutant p53 and induce cell death (Selivanova et al., 1997). CDB3, a rationally designed peptide derived from the p53‐binding protein ASPP (Samuels‐Lev et al., 2001) binds to the p53 core domain and stabilizes it (Friedler et al., 2002). The small molecule CP‐31398 was shown to preserve wild type conformation of p53 upon thermal denaturation, rescue its transactivation capacity, and restore its anti‐tumor activity in vivo (Foster et al., 1999). CP‐31398 also affects wild type p53 (Luu et al., 2002; Takimoto et al., 2002; Wang et al., 2003). However, structural studies did not reveal any binding to p53 (Rippin et al., 2002). PRIMA‐1 (Bykov et al., 2002, 2002) and MIRA‐1 (Bykov et al., 2005a) were identified in a cellular screening of a chemical library from the National Cancer Institute. Both compounds induce mutant p53‐dependent apoptosis and restore native conformation, DNA binding, and transcriptional transactivation to mutant p53. PRIMA‐1 inhibits tumor growth in vivo (Bykov et al., 2002b). The molecular mechanisms for reactivation of mutant p53 by PRIMA‐1 and MIRA‐1 remain unclear. However, MIRA‐1 can potentially react covalently with thiol groups in proteins. Since p53 is known to be subject to redox regulation (Seo et al., 2002), this suggests that modifications of cysteines in the p53 core domain may play a role in mutant p53 rescue.
Despite the successful identification of several mutant p53‐targeting small molecules, further screening for new molecular scaffolds using diverse strategies is important to provide a better understanding of the mechanisms of mutant p53 reactivation. Also, the already identified lead molecules may not be suitable for clinical use due to non‐specific toxicity or undesirable pharmacodynamic properties.
Compounds structurally related to CP‐31398 were shown to possess activity against tumor cells even before the mutant p53‐targeting properties of CP‐31398 were demonstrated (Jiang et al., 1990). We synthesized and tested a number of 2‐styrylquinazolin‐4‐(3H)‐one‐related derivatives (Witt and Bergman, 2000, 2001) for mutant p53‐dependent growth suppression and identified one, STIMA‐1 (2‐vinylquinazolin‐4‐(3H)‐one) as the most potent molecule. We show here that both CP‐31398 and STIMA‐1 (SH group Targeting and Induction of Massive Apoptosis) have similar chemical activity as so called traditional Michael acceptors and that this activity is related to the observed mutant p53‐dependent growth suppression. We found that STIMA‐1 is more potent than CP‐31398 in suppressing growth of mutant p53‐expressing tumor cells. Our results provide a better understanding of possible molecular mechanisms behind reactivation of mutant p53 and will hopefully aid the design of novel and more efficient mutant p53‐targeting compounds.
2. Results
2.1. Mutant p53‐dependent growth suppression by STIMA‐1
The structural scaffold of 2‐vinylquinazolin‐4(3H)‐one has been known since 1910 (Bogert et al., 1911). Some of its derivatives have biological activity against cancer cells (Jiang et al., 1990). We noted that these compounds have some resemblance to the newly characterized mutant p53‐reactivating compound CP‐31398 (Figure 1A) (Foster et al., 1999). To investigate their anti‐tumor activity and possible effect on mutant p53, we synthesized a series of 2‐styrylquinazolin‐4(3H)‐one‐related derivatives (Witt and Bergman, 2000, 2001) and screened them for mutant p53‐dependent inhibition of cell growth in Saos‐2 and Saos‐2‐His273 cells using a WST‐1 proliferation assay. Out of 26 molecules, one compound, STIMA‐1 (Figure 1A), was identified as having significant mutant p53‐dependent growth inhibitory effect. Treatment of H1299‐His175 lung carcinoma or Saos‐2‐His273 osteosarcoma cells with 2μM STIMA‐1 for 96h and subsequent assessment of cell growth with the WST‐1 assay revealed a growth suppression of 41±3.3 for Saos‐2‐His273 cells and 50.5±0.6% for H1299‐His175 cells, while their p53 null counterparts Saos‐2 and H1299 only showed 18.5±0.6 and 2.4±2.4% growth inhibition, respectively (Figure 1B).
Figure 1.
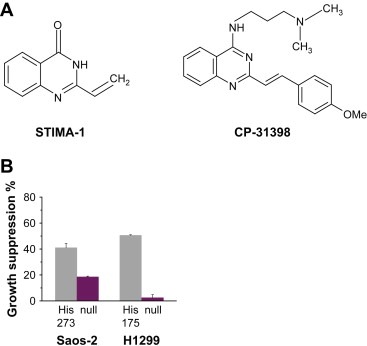
(A) Chemical structure of STIMA‐1 and CP‐31398. (B) STIMA‐1‐induces mutant p53‐dependent growth suppression in Saos‐2‐His273 and H1299‐His175 cells. Cell growth was assessed using the WST‐1 proliferation agent. Data represent mean±standard error (n=3).
2.2. Chemical reactivity of STIMA‐1 and CP‐31398
To determine the ability of STIMA‐1 and CP‐31398 to form adducts with cysteine, 2μM STIMA‐1 and 1mM CP‐31398 were dissolved in PBS and incubated with equimolar amount of N‐acetylcysteine (NAC) at 37°C for 24h. NAC is a cysteine analog that has an acetyl group instead of an amino group, leaving one SH group as the only target for alkylation. Samples were analyzed by reverse phase HPLC. Figure 2A shows that incubation of both STIMA‐1 and CP‐31398 with NAC resulted in the formation of new peaks on HPLC. Around 97% of STIMA‐1 and 20% of CP‐31398 reacted with NAC as judged by the HPLC absorbance profile. Pretreatment of H1299‐His175 cells with 5mM NAC completely blocked growth suppression induced by STIMA‐1 (Figure 2B). A similar effect was observed using the parental cells H1299 (data not shown). The effect of CP‐31398 in H1299‐His175 was only partially inhibited by pretreatment with NAC (Figure 2B).
Figure 2.
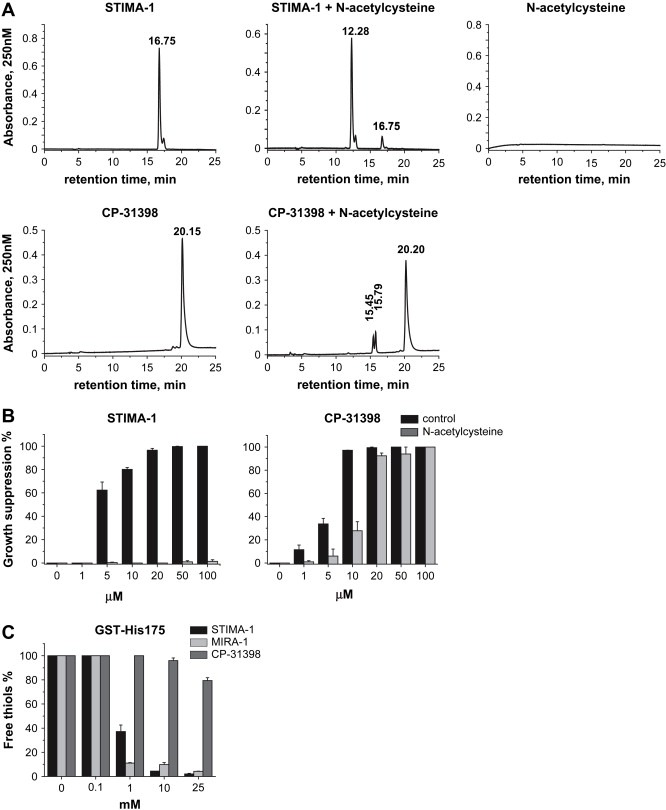
Chemical and biological reactivity of STIMA‐1 and CP‐31398. (A) STIMA‐1 (upper panel) and CP‐31398 (lower panel) form adducts with N‐acetylcysteine (NAC) as shown by HPLC analysis. (B) STIMA‐1‐induced growth suppression in H1299‐His175 cells is completely blocked by NAC according to the WST‐1 assay. The effect of CP‐31398 was only partially blocked by NAC. (C) STIMA‐1, MIRA‐1 and CP‐31398 block thiol groups in recombinant GST‐His175 mutant p53.
We also examined the effect of treatment with STIMA‐1, MIRA‐1 and CP‐31398 on the number of free thiol groups in recombinant GST‐His175 mutant p53. As shown in Figure 2C, treatment with all compounds reduced the fraction of free thiol groups in the protein. However, STIMA‐1 and MIRA‐1 were much more efficient than CP‐31398 in this regard. STIMA‐1 and MIRA‐1 treatment resulted in 2±0.6 and 4.3±0.2% free thiols, respectively, while 79±2.4% of free thiol groups were left unmodified in GST‐His175 mutant p53 upon treatment with 25mM of CP‐31398.
To investigate the ability of STIMA‐1 to bind to free thiol groups in living cells, we performed a competition assay between STIMA‐1 and a FITC‐maleimide conjugate in H1299‐His175 cells. Figure 3 shows that treatment with STIMA‐1 at 10 or 20μM caused a marked reduction in the FITC‐maleimide staining, thus confirming that STIMA‐1 blocks thiol groups also in living cells. This effect could be blocked by preincubation with NAC.
Figure 3.
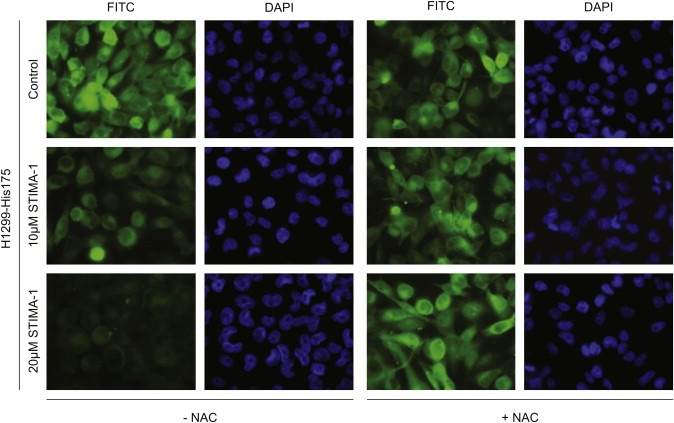
Competition for thiol groups in H1299‐His175 cells. Treatment with 10 and 20μM STIMA‐1 caused reduced staining of the FITC‐maleimide conjugate in the cells as compared to control treatment. Pretreatment with NAC inhibited this competition and did not lead to a reduction of the staining after STIMA‐1 treatment.
2.3. Analysis of cell death by FACS
To confirm the growth suppression results obtained in the WST‐1 proliferation assay, we examined the ability of STIMA‐1 to induce cell death by FACS analysis. Saos‐2‐His273 cells were treated with 15μM STIMA‐1 for 96h in the presence or absence of doxycycline, which shuts off mutant p53 expression from the transfected construct. The Saos‐2‐His273 cells showed 96.6% cells with sub‐G1 DNA content in the absence of doxycycline (mutant p53 expressed) while treatment of the same cells in the presence of doxycycline (mutant p53 expression turned off) induced 46% sub‐G1 cells (Figure 4A). Results from treatment of Saos‐2‐His273 cells for 96h with increasing concentrations of STIMA‐1 in the presence or absence of doxycycline are shown in Figure 4B. This demonstrates that STIMA‐1 induces cell death in a mutant p53‐dependent manner in Saos‐2‐His273 cells. Doxycycline treatment by itself did not affect the sensitivity of the cells in the range of concentrations used in these experiments (data not shown).
Figure 4.
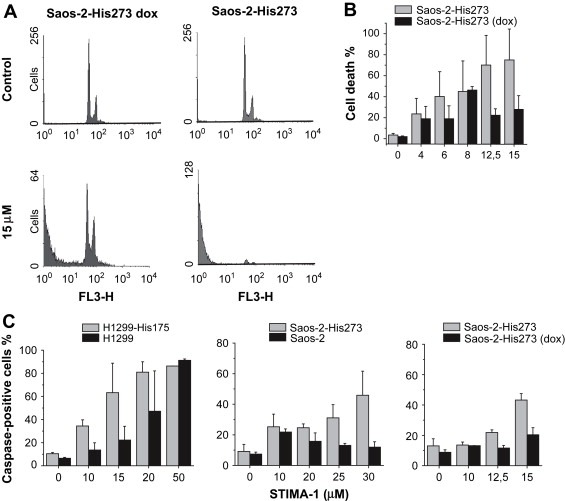
STIMA‐1 induces cell death by apoptosis. (A) FACS analysis of Saos‐2‐His273 cells before (upper panel) and after (lower panel) treatment with 15μM STIMA‐1. Doxycycline treatment shuts off expression of mutant p53 in Saos‐2‐His273 cells. (B) Quantification of the sub‐G1 cell fraction after STIMA‐1 treatment. (C) Quantification of caspase activation assays in H1299‐His175 and H1299 cells (left panel), in Saos‐2‐His273 and Saos‐2 cells (middle panel) and in Saos‐2‐His273 and Saos‐2‐His273 (+dox) cells (right panel). Data represent mean±standard error (n=2 or 3).
To test whether STIMA‐1 treatment induces caspase activity we performed a caspase activation assay (CaspaTag). After 48h of treatment with 15μM STIMA‐1, we observed 63.3±25.5% active caspase‐positive H1299‐His175 cells whereas only 22.3±11.9% of the H1299 cells became active caspase‐positive. However, treatment with 50μM of STIMA‐1 resulted in caspase activation independently of mutant p53‐expression (Figure 4C). Similarly, treatment of Saos‐2‐His273 cells with 25μM STIMA‐1 for 48h induced 31.1±8.6% active caspase‐positive cells, but only 13.1±1.3% active caspase‐positive Saos‐2 cells (Figure 4C). Thus, treatment with STIMA‐1 also triggers mutant p53‐dependent caspase activation (Figure 4C).
To confirm a mutant p53‐dependent effect we also used doxycycline to downregulate p53 expression and tested the effect of STIMA‐1. At 15μM of STIMA‐1, Saos‐2‐His273 cells showed 43.2% active caspase‐positive cells compared to 20.3% active caspase‐positive cells in doxycycline‐treated cells (Figure 4C).
2.4. Comparison of STIMA‐1 and cisplatin
To further investigate the properties of STIMA‐1, we compared its effects on tumor cells with those of cisplatin, an anticancer drug that preferentially targets tumor cells expressing wild type p53 (Borresen‐Dale, 2003). We tested both compounds in the WST‐1 proliferation assay and calculated corresponding IC50 values. An IC50 ratio was obtained for pairs of cell lines (p53 null versus mutant p53, or p53 null versus wild type p53) (Figure 5A). A high IC50 ratio between a p53 null line and its derivative line expressing exogenous mutant p53 indicates a strong preference for the mutant p53‐expressing cells. Treatment with 4μM STIMA‐1 resulted in a ratio of 3.5 for doxycycline‐treated Saos‐2‐His273 (mutant p53 off) vs. Saos‐2‐His273 cells and a 2.3 ratio for p53 null H1299 vs. H1299‐His175 cells, confirming that the mutant p53‐expressing cell lines are significantly more sensitive to STIMA‐1. In contrast, we obtained a reversed IC50 ratio of −1.3 for p53 null HCT116 cells vs. HCT116 p53+/+ cells, consistent with a weaker effect of STIMA‐1 on the wild type p53‐carrying cells compared to the p53 null cells, although the difference was not statistically significant. Treatment with 1μg/ml cisplatin had the opposite effect. The p53 null Saos‐2‐His273 (+doxycycline) and H1299 cells were more sensitive to cisplatin treatment than their mutant p53‐expressing counterparts, as shown by their reversed ratios of −2.1 and −1.6, respectively. The IC50 ratio of 1.6 between the p53 null and wild type p53‐carrying HCT116 lines demonstrates that the HCT116 p53+/+ cells are more susceptible to cisplatin‐induced cytotoxicity than the p53 null HCT116 cells. This observation is also supported by our colony formation assays (data not shown).
Figure 5.
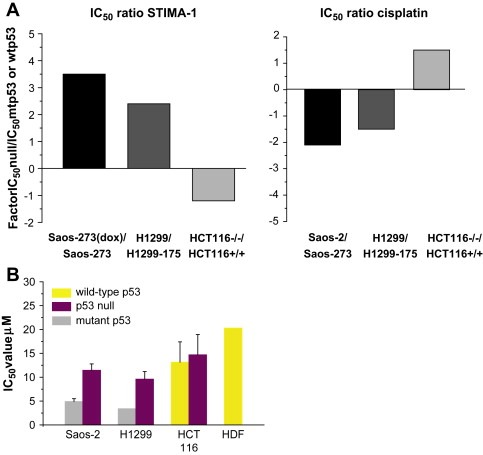
The effect of STIMA‐1 and cisplatin in cells with different p53 status. (A) Diagram showing the ratio between the IC50 values for the mutant p53‐expressing cells/wild type p53 expressing cells and the IC50 values for corresponding p53 null cells. While the Saos‐2‐His273 cells and H1299‐His175 cells are more sensitive to STIMA‐1 treatment, indicating growth suppression in a mutant p53 dependent manner, the wild type p53‐carrying HCT116 cells are more sensitive to cisplatin treatment. (B) IC50 values as determined by the WST‐1 proliferation assay. The mutant p53‐expressing H1299‐His175 and Saos‐2‐His273 cells are more sensitive to STIMA‐1 treatment than their p53 null counterparts and HCT116 p53+/+ and HCT116 p53−/− cells. Human diploid fibroblasts showed the lowest sensitivity to STIMA‐1 treatment. Data represent mean±standard error (n=2).
2.5. Effect of STIMA‐1 on normal cells and tumor cells with different p53 status
Further, we tested the effect of STIMA‐1 on normal human diploid fibroblasts (HDF). The cells were treated with 0–100μM of STIMA‐1 for 96h and analyzed for growth suppression by the WST‐1 assay. The IC50 value for STIMA‐1 in human diploid fibroblasts was 20.3μM (Figure 5B). For comparison, we calculated IC50 values for human tumor cell lines. The IC50 values for the H1299‐His175 and Saos‐2‐His273 cells were 3.4μM and 4.9μM, respectively, which is significantly lower than the IC50 values for their p53 null counterparts Saos‐2 (11.4μM) and H1299 (9.6μM). Furthermore, the IC50 values for the wild type p53‐carrying HCT116 p53+/+ cells and the isogenic HCT116 p53−/− cells were 13.2μM and 14.7μM, respectively (Figure 5B). Thus, human tumor cells expressing exogenous His175 or His273 mutant p53 show a significantly higher sensitivity to STIMA‐1 as compared to p53 null or wild type p53‐carrying human tumor cells and normal human diploid fibroblasts.
2.6. STIMA‐1 induces expression of p53 target genes
In order to test the ability of STIMA‐1 to rescue the transcriptional transactivation function to mutant p53, we examined if STIMA‐1 was able to induce p53 target genes at the protein level. First, we looked at the p53 expression levels in the different cells and sublines. Doxycyline treatment shut off p53 expression in both H1299‐His175 and Saos‐2‐His273 cells. Expression of p53 was only observed in untreated H1299‐His175 and Saos‐2‐His273 (Figure 6A). The level of p53 was unchanged upon STIMA‐1 treatment (data not shown). Treatment of H1299‐His175 cells with STIMA‐1 resulted in induction of the p53 targets p21 and PUMA at 8h after treatment, as well as Bax after 24h of treatment, while no changes were observed in parental H1299 cells (Figures 6B–D). Thus, STIMA‐1 treatment induced expression of all three p53 target genes in a mutant p53‐dependent manner. However, we did not detect any upregulation of p21 or PUMA in SW480 colon carcinoma cells expressing endogenous mutant p53 upon treatment with 20μM STIMA‐1 for 8, 16 and 24h (data not shown). The wild type p53‐carrying HCT116 cells showed a weak upregulation PUMA in response to STIMA‐1 treatment (data not shown).
Figure 6.
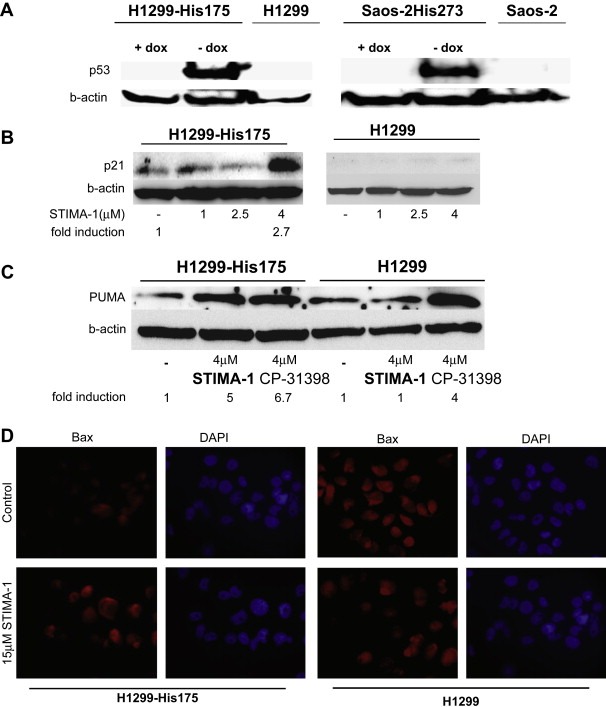
Expression of p53 and p53 target genes. (A) Western blot analysis of p53 in doxycycline‐treated H1299‐His175 and Saos‐2‐His273 cells. (B–D) Western blot analysis of the expression of p21 (B) and PUMA (C), and analysis of Bax expression (D) by immunofluorescence staining in STIMA‐1‐treated H1299 and H1299‐His175 cells. (C) PUMA protein expression is also induced by treatment with CP‐31398.
2.7. STIMA‐1 enhances DNA binding of mutant p53
To elucidate whether STIMA‐1 can restore DNA binding to mutant p53, we tested H1299‐His175 cells in the TransAM DNA binding assay. The wild type p53‐expressing MCF7 cells treated with H2O2 were used as positive control (100%; Figure 7). As expected, untreated p53 null H1299 cells showed almost no DNA binding (3.6±2.5%). The H1299‐His175 had a DNA binding activity of 32.9±16%. Treatment with 8μM STIMA‐1 for 6h resulted in a 3‐fold increase in p53 DNA binding as compared to untreated H1299‐His175 cells. STIMA‐1‐treated H1299 cells did not show any increase in DNA binding (Figure 7). Competition with a DNA oligonucleotide containing a p53 binding site inhibited the observed DNA binding of p53 from MCF7 cells, but did not significantly reduce the DNA binding of STIMA‐1‐treated His175 mutant p53 in H1299 cells (data not shown). This indicates that mutant p53 DNA binding induced by STIMA‐1 is qualitatively different from wild type p53 DNA binding.
Figure 7.
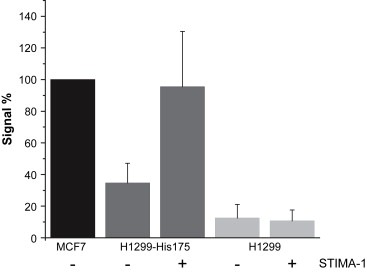
Effect of STIMA‐1 on p53 DNA binding as assessed using the TransAM assay. (A) The diagram shows p53 DNA binding activity in MCF7 cells carrying wild type p53 (positive control) and STIMA‐1‐treated H1299‐His175 and untreated parental p53 null H1299 cells. Treatment with 8μM STIMA‐1 enhances p53 DNA binding in H1299‐His175 cells to levels comparable to those in MCF7 cells.
3. Discussion
The fact that a large fraction of human tumors carry p53 mutation and that those tumors in general show poor response to currently used anticancer therapy (Levine, 1997) has stimulated attempts to design novel anticancer drugs that target mutant p53. Chemical library screening and other approaches have allowed the identification of several other low molecular weight compounds that target mutant p53 and/or preferentially induce cell death in mutant p53‐expressing tumor cells, including CP‐31398 (Foster et al., 1999), ellipticine (Shi et al., 1998), amifostine, WR1065 (North et al., 2002) and glycerol (Ohnishi et al., 1999). We have screened the Diversity set from the National Cancer Institute and identified two low molecular weight compounds, PRIMA‐1 (Bykov et al., 2002b) and MIRA‐1 (Bykov et al., 2005a), that induce mutant p53‐dependent apoptosis through restoration of the tumor suppressor function to mutant p53. Nevertheless, further studies aimed at the identification of novel mutant p53‐targeting compounds are imperative, since only a small fraction of identified lead substances are likely to reach the clinic. In addition, analysis of structural scaffolds of different mutant p53‐targeting molecules may provide clues as to the molecular mechanisms of mutant p53 reactivation.
The compound 2‐styrylquinazolin‐4(3H)‐one has been known since 1910 (Bogert et al., 1911) and some of its derivatives have shown activity against cancer cells (Jiang et al., 1990). These chemicals show some resemblance to the recently characterized mutant p53‐reactivating molecule CP‐31398 (Foster et al., 1999). To explore the anti‐tumor effect of this class of compounds, we synthesized a series of 2‐styrylquinazolin‐4(3H)‐one‐related derivatives (Witt and Bergman, 2000, 2001) and tested them for mutant p53‐dependent inhibition of cell growth. This allowed identification of the compound STIMA‐1. We found that STIMA‐1 targets human tumor cells expressing two hot spot mutant p53 proteins belonging to the two main classes, conformational mutants (His175) and DNA contact mutants (His273). This was assessed by the WST‐1 proliferation assay, FACS analysis of caspase activation and Western blot analysis of p53 target protein expression. STIMA‐1 appears at least as potent and selective for mutant p53‐expressing cells as PRIMA‐1 (Bykov et al., 2002b) and MIRA‐1 (Bykov et al., 2005a). Comparison of IC50 values for tumor cell lines and human diploid fibroblasts confirmed that STIMA‐1 preferentially targets cells expressing mutant p53, while human diploid fibroblasts are more resistant. Like PRIMA‐1 and MIRA‐1, STIMA‐1 triggers activation of caspases, indicating that it induces cell death through apoptosis. Western blot analysis showed that STIMA‐1 induces Bax, PUMA and p21 in mutant p53‐expressing cells, while no induction of p53 target genes was observed in the corresponding p53 null cells. In contrast, CP‐31398 induces PUMA in p53 null H1299 cells, and others have observed upregulation of p21 in HCT116 p53−/− cells (Luu et al., 2002) and p53‐independent cell death in glioma cells (Wischhusen et al., 2003). Furthermore, gene array profiles indicate a p53‐independent effect of CP‐31398 (Takimoto et al., 2002). These results suggest that CP‐31398 has biochemical functions other than its ability to reactivate and stabilize p53. STIMA‐1 treatment stimulates DNA binding of mutant p53 in H1299‐His175 cells (Figure 6). However, our competition experiments indicated that STIMA‐1‐induced mutant p53 DNA binding is different from wild type p53 DNA binding. Further analysis is required to clarify the nature of the interaction between mutant p53 and DNA in STIMA‐1‐treated cells.
The ability of STIMA‐1 to preferentially kill mutant p53‐carrying tumor cells was clearly evident in our comparison of STIMA‐1 and cisplatin, a compound that forms various adducts with DNA (Kelland and Farrell, 2000) and is widely used in clinical cancer therapy (Dorr and Von Hoff, 1994). STIMA‐1 and cisplatin showed opposite patterns of activity in the examined tumor cell lines. Cisplatin inhibited growth of tumor cell lines in wild type p53‐dependent manner and mutant p53‐expression rendered cells resistant to cisplatin in agreement with previously published results (Matas et al., 2001; Bykov et al., 2005b). In contrast, STIMA‐1 triggered mutant p53‐dependent growth suppression and apoptosis in tumor cells. This demonstrates that the mechanism of action of STIMA‐1 is entirely different from that of cisplatin and emphasizes its potential for development of mutant p53‐targeting anticancer drugs.
CP‐31398, on the other hand, was suggested to act as a DNA‐damaging agent (Rippin et al., 2002). However, wild type p53 stabilization caused by CP‐31398 rather results from reduced ubiquination than phosphorylation of p53 at Ser‐15 or Ser‐20 (Wang et al., 2003). Phosphorylation at these two sites is often related to DNA damage. Our experiments demonstrated that CP‐31398 as well as STIMA‐1 and MIRA‐1 react with N‐acetylcysteine and alkylate thiols in p53. The reactivity of CP‐31398 was substantially lower than that of STIMA‐1 and MIRA‐1.
The exact molecular mechanism of mutant p53 reactivation by CP‐31398, MIRA‐1 and STIMA‐1 is not known. However, the shared ability of all three molecules to alkylate thiol groups provides an interesting clue. It is known that the redox status of p53 is important for its tumor suppressor function (Hainaut and Milner, 1993). Oxidation and reduction of p53 will affect thiol groups on the p53 surface. Oxidation of these cysteines is likely to result in the formation of inter‐ and intramolecular disulfide bridges resulting in the loss of active conformation and the formation of large inactive aggregates (Hainaut and Milner, 1993; Sun et al., 2003). Interestingly, the thiol‐reducing amifostine derivative WR1065 has been shown to restore wild type conformation to Met272 mutant p53 (North et al., 2002). It is noteworthy that although STIMA‐1 and MIRA‐1 do not share any overall structural similarity, both compounds contain reactive double bonds that enable them to participate in the reaction of nucleophilic addition or so called Michael addition. In other words, these substances are good Michael acceptors. A similar reactive double bond coupled to an electron‐withdrawing group that is characteristic of Michael acceptors (Smith and March, 2001) is also present in the CP‐31398 scaffold. However, at least two factors mitigate the activity of the carbon‐carbon double bond in CP‐31398. First, it is less polarized than that in STIMA‐1 and MIRA‐1, and second, the reactive site is less accessible for nucleophilic substrates due to the presence of a bulky side‐chain containing amino groups as well as the aryl ring. This is in full agreement with our data showing that CP‐31398 has significantly less chemical reactivity with N‐acetylcysteine and is less efficient in blocking free thiols in p53 compared to STIMA‐1 and MIRA‐1. Thus, all three molecules could potentially alkylate nucleophils in cells and form adducts preferentially with thiol and amino groups in proteins. The presence of the reactive double bond in MIRA‐1 is critical for its mutant p53‐dependent effect (Bykov et al., 2005a). This chemical property is likely to be responsible for at least some of the biological activities of CP‐31398 and STIMA‐1. It is conceivable that alkylation of one or several of the 10 cysteine residues in the p53 core domain is responsible for the mutant p53‐dependent effect of these compounds, possibly through stabilizing the native fold of the p53 core domain. The extent of thiol alkylation has been shown to affect p53 DNA binding; low levels of thiol alkylation were found to stimulate p53 DNA binding (Bykov et al., 2005a; Bhanoori et al., 2003) whereas high levels were suppressive (Bykov et al., 2005a; Rainwater et al., 1995). Low levels of thiol alkylation may reduce protein aggregation by blocking the ability of cysteines to form disulfide bridges upon oxidation. Moreover, alkylation of thiols by small molecules may affect protein conformation.
Studies of the ability of STIMA‐1 to inhibit tumor growth in vivo have been hampered by difficulties in dissolving the compound in PBS for in vivo doses of 1 and 10mg/kg. Therefore mice were treated by i.p. injections of STIMA‐1 in suspension. We did not observe any effect on the growth of H1299‐His175 human tumor xenografts, nor any toxicity (data not shown). It is possible that the poor solubility of STIMA‐1 prevented its distribution and activity in vivo. Incorporation of charged groups into the molecular scaffold of STIMA‐1 and/or improved pharmacological formulation for in vivo studies may overcome these problems.
In conclusion, we have identified STIMA‐1, a low molecular weight compound that selectively targets mutant p53‐carrying tumor cells. The identification and characterization of novel structural scaffolds with mutant p53‐targeting capacity such as STIMA‐1 may help understanding the molecular mechanisms of mutant p53 rescue and thus facilitate the design of more potent and selective mutant p53‐targeting anticancer drugs.
4. Materials and methods
4.1. Cells and cell culture
The human Saos‐2 osteosarcoma and H1299 lung adenocarcinoma cell lines are p53 null. The sublines Saos‐2‐His273 and H1299‐His175 carry tetracycline‐regulated mutant p53 constructs (Tet‐Off). To downregulate mutant p53 expression, Saos‐2‐His273 cells were treated with doxycycline for 1week (5μg/ml) and H1299‐His175 for 2days (2μg/ml) and another week with 0.5μg/ml. The human colon carcinoma cell line HCT116 carries wild type p53 (p53+/+) and the isogenic HCT116 (p53−/−) cells are p53 null. Cells were grown at 37°C with 5% CO2 in Iscove's modified Dulbecco's medium containing 10% FBS, 2mM l‐glutamine, 40μl/ml gentamycin. Human diploid fibroblasts and SW480 colon adenocarcinoma cells (endogenous His273/Ser309 mutant p53) were grown as the other cells except that 2.5μg/ml plasmocin was used instead of gentamycin.
4.2. Synthesis of STIMA‐1
2‐Styrylquinazolin‐4(3H)‐one and related molecules (i.e. the double bond conjugated with an aryl group) have traditionally been prepared by condensation of 2‐methyl‐quinazolin‐4(3H)‐one with e.g. benzaldehyde and anis aldehyde. This methodology (Witt and Bergman, 2000) cannot be used for the preparation of the parent compound, 2‐vinylquinazolin‐4(3H)‐one. Instead, this molecule was prepared using a ring synthesis. Thus, anthranilamide was acylated with 3‐chloropropionyl chloride and the amide obtained was subsequently treated with sodium hydroxide in ethanol which resulted in the desired elimination and concomitant formation of the heterocyclic ring. The desired molecule 2‐vinylquinazolin‐4(3H)‐one, STIMA‐1, was obtained in excellent yield.
4.3. WST‐1 cell proliferation assay
Cells were placed at a density of 3000 cells per well in 100μl medium in 96‐well plates, cultured overnight and then treated with different STIMA‐1 concentrations. After 96h, WST‐1 cell proliferation reagent (Roche, Switzerland) was added and samples were incubated at 37°C for 1h and absorbance measured at 450nm.
4.4. HPLC analysis
The HPLC assay was performed on a Beckman instrument operated with System Gold and coupled to a 156 diode array detector module. The Kromasil C18 column 4.5×250mm, 5μm particle size was used. The separation was performed employing a gradient elution in which 50mM ammonium formate pH 4.6 was mixed with methanol. The elution started with 30% methanol immediately followed by a linear gradient for 20min up to 75% methanol. The flow rate was 0.7ml/min. The column temperature was kept ambient for experiments with STIMA‐1 or at +42°C for CP‐31398. The products were detected by UV diode array detector.
4.5. Free thiol group determination assay for GST‐His175 mutant p53
Recombinant GST‐His175 mutant p53 was treated with the tested substances on ELISA plates for 2h at 37°C or with DMSO and washed twice with PBS. Next the samples were incubated with Maleimide‐Biotin conjugate that alkylates all accessible free thiol groups, (Pierce, IL) for 1h at room temperature. The wells were then washed with PBS, blocked with 5% skim milk, washed and probed with Avidin‐HRP conjugate and washed again. The signal was developed with TMB HRP substrate (Pierce, IL), samples were treated with HCL and absorbance was scored at 450nm by ELISA reader. The preparation of recombinant p53 protein contains ∼1–10mM DTT and glutathione. Therefore we used STIMA‐1 concentrations exceeding these amounts of reducing agents.
4.6. Competition for thiol groups in living cells
Cells were seeded on a 16‐well chamber slide at a density of 10,000 cells per well per 200μl. The next day 100μl medium were removed and STIMA‐1 was added at 10 or 20μM. After 6h the FITC‐maleimide conjugate was added to the cells at a final concentration of 5μM. After 90min the cells were fixed with 4% formaldehyde, permeabilized with 0.2% Triton‐X, washed, mounted, sealed with nail polish, and examined by microscopy.
4.7. Flow cytometry
For DNA fragmentation assays, cells were grown on 12‐well plates at an initial density of 10,000cells/cm2 and treated with STIMA‐1 the next day. After 96h incubation cells were harvested by trypsinization, fixed with 70% ethanol, treated with RNase A (0.25mg/ml) and stained with propidium iodide (0.05mg/ml). Samples were analyzed on a FACScan flow cytometer (Becton Dickinson, CA) according to standard procedures. For caspase activation assay cells were cultured in 12‐well plates (10,000cells/cm2) overnight and treated with STIMA‐1. After 48h cells were harvested by trypsinization and labelled with FLICA reagent (CaspaTag TM Pan Fluorescein Caspase (VAD) activity kit, Intergen, UK) according to the manufacturer's instructions. Samples were analyzed on a FACScan.
4.8. Colony formation assay
Cells were plated as described above. The next day cells were treated with STIMA‐1 or cisplatin for 24h, trypsinized and seeded in 25cm2 flask (120 cells per flask from initial density) and cultured for 7days. Colonies were stained with Giemsa and counted.
4.9. Western blotting
Cells were seeded on 6‐well plates at the density of 10,000cells/cm2 and treated with STIMA‐1 the following day. After 8h, cells were harvested by trypsinization, lysed and analyzed by Western blotting according to standard procedures. Equal amount of total protein extract was separated on SDS‐PAGE and transferred onto Hybond ECL nitrocellulose membranes (Amersham Pharmacia Biotech, Sweden). Anti‐MDM2 (sc‐813, Santa Cruz Biotechnology, CA), anti‐p21 (BD Biosciences, CA), anti‐PUMA (Anti‐PUMA ab9643, Abcam, UK) and anti‐β‐actin (AC‐15, Sigma, Sweden) were used. Primary antibodies were detected by anti‐mouse HRP or anti‐rabbit HRP (Amersham Pharmacia Biotech, Sweden). Protein bands were visualized using Super Signal West Femto Maximum Sensitivity Substrate (Pierce, IL).
4.10. Immunofluorescence staining
Cells (10,000) were plated in chamber slides (Nalge Nunc International, NY) and treated with STIMA‐1 the following day. After 24h, cells were fixed in 4% formaldehyde for 10min and permeabilized in 0.2% Triton‐X for 2min. Cells were then stained by first incubating with anti‐Bax (sc‐6236, Santa Cruz Biotechnology, CA) for 1h in a wet chamber, followed by PBS washes and incubation with a TexasRed‐conjugated anti‐rabbit antibody (Vector laboratories, CA) for 30min. The slides were finally washed in PBS and mounted with Vectashied hardest mounting medium with DAPI (Vector laboratories, CA). Images were captured on a Zeiss Axioplan 2 microscope connected to an AxioCam HRm Camera.
4.11. Trans AM assay
Cells (1,200,000) were plated in 10cm plates overnight and then treated with STIMA‐1. After 6h, cells were harvested by trypsinization. The cells nuclear protein content was separated from the cytoplasmic fraction by using a nuclear extract kit (Nuclear Extract Kit, Active Motif, Belgium). Protein concentration of the nuclear extract was measured by Bradford according to standard procedures. Equal amount of total nuclear protein was loaded on to a 96‐well plate coated with an immobilized oligonucleotide containing a p53 consensus binding site (TransAM, p53 Transcription Factor Assay Kit, Active Motif, Belgium). Nuclear extract of H2O2 treated MCF7 cells (provided by the manufacturer) served as a positive control. Anti‐p53 and anti‐rabbit HRP were used to quantify the amount of bound p53 protein. The HRP signal was developed by a substrate provided by the manufacturer and samples were analyzed by ELISA reader at 450nm. The experiment was performed at least twice in duplicates.
Acknowledgements
This work was supported by the Swedish Cancer Society (Cancerfonden), the Cancer Society of Stockholm (Cancerföreningen), the Ingabritt & Arne Lundberg Foundation, the Karolinska Institute, and the EU 6th Framework Program. The information in this document is provided as is and no guarantee or warranty is given that the information is fit for any particular purpose. The user thereof uses the information at its sole risk and liability. The community is not liable for any use that may be made of the information contained herein. Klas G. Wiman and Vladimir J.N. Bykov are cofounders and shareholders of APREA AB, a company that develops p53‐based cancer therapy.
Zache Nicole, Lambert Jeremy M.R., Rökaeus Nina, Shen Jinfeng, Hainaut Pierre, Bergman Jan, Wiman Klas G., Bykov Vladimir J.N., (2008), Mutant p53 targeting by the low molecular weight compound STIMA‐1, Molecular Oncology, 2, doi: 10.1016/j.molonc.2008.02.004.
References
- Bhanoori, M. , Yellaturu, C.R. , Ghosh, S.K. , Hassid, A. , Jennings, L.K. , Rao, G.N. , 2003. Thiol alkylation inhibits the mitogenic effects of platelet-derived growth factor and renders it proapoptotic via activation of STATs and p53 and induction of expression of caspase1 and p21(waf1/cip1). Oncogene 22, 117–130. [DOI] [PubMed] [Google Scholar]
- Bogert, M.T. , Beal, G.D. , 1911. Researches on quinazolones. XXVI. Synthesis of some stilbazoles, hydrazones and Schiff bases in the 4-quinazolone group. J. Am. Chem. Soc. 32, 1654–1664. [Google Scholar]
- Borresen-Dale, A.L. , 2003. TP53 and breast cancer. Hum. Mutat. 21, 292–300. [DOI] [PubMed] [Google Scholar]
- Bykov, V.J. , Issaeva, N. , Selivanova, G. , Wiman, K.G. , 2002. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis 23, 2011–2018. [DOI] [PubMed] [Google Scholar]
- Bykov, V.J. , Issaeva, N. , Shilov, A. , Hultcrantz, M. , Pugacheva, E. , Chumakov, P. , Bergman, J. , Wiman, K.G. , Selivanova, G. , 2002. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8, 282–288. [DOI] [PubMed] [Google Scholar]
- Bykov, V.J. , Selivanova, G. , Wiman, K.G. , 2003. Small molecules that reactivate mutant p53. Eur. J. Cancer 39, 1828–1834. [DOI] [PubMed] [Google Scholar]
- Bykov, V.J. , Issaeva, N. , Zache, N. , Shilov, A. , Hultcrantz, M. , Bergman, J. , Selivanova, G. , Wiman, K.G. , 2005. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J. Biol. Chem. 280, 30384–30391. [DOI] [PubMed] [Google Scholar]
- Bykov, V.J. , Zache, N. , Stridh, H. , Westman, J. , Bergman, J. , Selivanova, G. , Wiman, K.G. , 2005. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 24, 3484–3491. [DOI] [PubMed] [Google Scholar]
- Campling, B.G. , El-Deiry, W.S. , 2003. Clinical implications of p53 mutations in lung cancer. Methods Mol. Med. 75, 53–77. [DOI] [PubMed] [Google Scholar]
- Dorr, R.T. , Von Hoff, D.D. , 1994. Cancer Chemotherapy Handbook second ed. Appleton and Lange; Norwalk, CT: [Google Scholar]
- El-Deiry, W.S. , 1998. Regulation of p53 downstream genes. Semin. Cancer Biol. 8, 345–357. [DOI] [PubMed] [Google Scholar]
- Foster, B.A. , Coffey, H.A. , Morin, M.J. , Rastinejad, F. , 1999. Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510. [DOI] [PubMed] [Google Scholar]
- Friedler, A. , Hansson, L.O. , Veprintsev, D.B. , Freund, S.M. , Rippin, T.M. , Nikolova, P.V. , Proctor, M.R. , Rudiger, S. , Fersht, A.R. , 2002. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. U.S.A. 99, 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt, M.S. , Bennett, W.P. , Hollstein, M. , Harris, C.C. , 1994. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54, 4855–4878. [PubMed] [Google Scholar]
- Hainaut, P. , Hollstein, M. , 2000. p53 and human cancer: the first ten thousand mutations. Adv. Cancer Res. 77, 81–137. [DOI] [PubMed] [Google Scholar]
- Hainaut, P. , Milner, J. , 1993. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res. 53, 4469–4473. [PubMed] [Google Scholar]
- Jiang, J.B. , Hesson, D.P. , Dusak, B.A. , Dexter, D.L. , Kang, G.J. , Hamel, E. , 1990. Synthesis and biological evaluation of 2-styrylquinazolin-4(3H)-ones, a new class of antimitotic anticancer agents which inhibit tubulin polymerization. J. Med. Chem. 33, 1721–1728. [DOI] [PubMed] [Google Scholar]
- Kelland, L.R. , Farrell, N.P. , 2000. Platinum-based Drugs in Cancer Therapy Humana Press; Totowa, NJ: [Google Scholar]
- Levine, A.J. , 1997. p53, the cellular gatekeeper for growth and division. Cell 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Luu, Y. , Bush, J. , Cheung, K.J. , Li, G. , 2002. The p53 stabilizing compound CP-31398 induces apoptosis by activating the intrinsic Bax/mitochondrial/caspase-9 pathway. Exp. Cell Res. 276, 214–222. [DOI] [PubMed] [Google Scholar]
- Matas, D. , Sigal, A. , Stambolsky, P. , Milyavsky, M. , Weisz, L. , Schwartz, D. , Goldfinger, N. , Rotter, V. , 2001. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. Embo J. 20, 4163–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North, S. , Pluquet, O. , Maurici, D. , El-Ghissassi, F. , Hainaut, P. , 2002. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53(V272M)) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol. Carcinog. 33, 181–188. [DOI] [PubMed] [Google Scholar]
- Ohnishi, T. , Matsumoto, H. , Wang, X. , Takahashi, A. , Tamamoto, T. , Ohnishi, K. , 1999. Restoration by glycerol of p53-dependent apoptosis in cells bearing the mutant p53 gene. Int. J. Radiat. Biol. 75, 1095–1098. [DOI] [PubMed] [Google Scholar]
- Olivier, M. , Eeles, R. , Hollstein, M. , Khan, M.A. , Harris, C.C. , Hainaut, P. , 2002. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat. 19, 607–614. [DOI] [PubMed] [Google Scholar]
- Rainwater, R. , Parks, D. , Anderson, M.E. , Tegtmeyer, P. , Mann, K. , 1995. Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 15, 3892–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippin, T.M. , Bykov, V.J. , Freund, S.M. , Selivanova, G. , Wiman, K.G. , Fersht, A.R. , 2002. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119–2129. [DOI] [PubMed] [Google Scholar]
- Ryan, K.M. , Phillips, A.C. , Vousden, K.H. , 2001. Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol. 13, 332–337. [DOI] [PubMed] [Google Scholar]
- Samuels-Lev, Y. , O'Connor, D.J. , Bergamaschi, D. , Trigiante, G. , Hsieh, J.K. , Zhong, S. , Campargue, I. , Naumovski, L. , Crook, T. , Lu, X. , 2001. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 8, 781–794. [DOI] [PubMed] [Google Scholar]
- Selivanova, G. , Iotsova, V. , Okan, I. , Fritsche, M. , Strom, M. , Groner, B. , Grafstrom, R.C. , Wiman, K.G. , 1997. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 3, 632–638. [DOI] [PubMed] [Google Scholar]
- Seo, Y.R. , Kelley, M.R. , Smith, M.L. , 2002. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc. Natl. Acad. Sci. U.S.A. 99, 14548–14553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, L.M. , Fan, Y. , Myers, T.G. , O'Connor, P.M. , Paull, K.D. , Friend, S.H. , Weinstein, J.N. , 1998. Mining the NCI anticancer drug discovery databases: genetic function approximation for the QSAR study of anticancer ellipticine analogues. J. Chem. Inf. Comput. Sci. 38, 189–199. [DOI] [PubMed] [Google Scholar]
- Smith, M.A. , March, J. , 2001. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure John Wiley & Sons; New York: [Google Scholar]
- Sun, X.Z. , Vinci, C. , Makmura, L. , Han, S. , Tran, D. , Nguyen, J. , Hamann, M. , Grazziani, S. , Sheppard, S. , Gutova, M. , Zhou, F. , Thomas, J. , Momand, J. , 2003. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxid. Redox Signal 5, 655–665. [DOI] [PubMed] [Google Scholar]
- Takimoto, R. , Wang, W. , Dicker, D.T. , Rastinejad, F. , Lyssikatos, J. , El-Deiry, W.S. , 2002. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol. Ther. 1, 47–55. [DOI] [PubMed] [Google Scholar]
- Vogelstein, B. , Lane, D. , Levine, A.J. , 2000. Surfing the p53 network. Nature 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Takimoto, R. , Rastinejad, F. , El-Deiry, W.S. , 2003. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol. Cell. Biol. 23, 2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischhusen, J. , Naumann, U. , Ohgaki, H. , Rastinejad, F. , Weller, M. , 2003. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 22, 8233–8245. [DOI] [PubMed] [Google Scholar]
- Witt, A. , Bergman, J. , 2000. Synthesis and reactions of some 2-Vinyl-3H-quinazolin-4-ones. Tetrahedron 56, 7245–7253. [Google Scholar]
- Witt, A. , Bergman, J. , 2001. Total syntheses of the benzodiazepine alkaloids circumdatin F and circumdatin C. J. Org. Chem. 66, 2784–2788. [DOI] [PubMed] [Google Scholar]