

1.
Recent developments in whole‐genome Single Nucleotide Polymorphism (SNP) analysis have spurred a number of Genome‐Wide Association Studies (GWAS) in breast, prostate, and up‐coming testicular and bowel cancer (Easton et al., 2007; Eeles et al., 2008). Until now the genetic variants that have been revealed by these studies are responsible for comparatively small increases in relative risk of disease and it is unlikely that highly predictive genetic variants will emerge from subsequent studies. This may be seen by those arguing that a change in strategy is called for as evidence of the weakness of these studies. But is this a fair inference? Hardly any higher relative risk and more penetrant alleles should have been expected from these studies in the first place; had these alleles existed they would have been found through the extensive linkage studies of the past 20 years in family studies of sufficient size (Oldenburg et al., 2007; Balmain et al., 2003). This holds true even if one takes into consideration the multistage design of GWAS studies that is aimed at “amplifying” the hereditary effect by using individuals with a highly familial component of the disease as a starting point. Experiments such as the whole‐genome association analyses funded by Cancer Research UK (Easton et al., 2007; Eeles et al., 2008) constitute a brave and important exploratory step. It is the kind of experiment one has little chance of knowing whether it is worth doing before it is actually done. The experiments that have already been made represent an unprecedented effort invaluable for the scientific community given the amount of money and the effort made by the projects' principle investigators to bring together so many research groups in order to collect sufficient number of patients for such large multistage studies.
The accumulation of a critical mass of data is of crucial importance. Large amounts of data have been generated already, the money is well spent, and it is of huge importance that this data is available to the scientific community so that it can be analysed in various novel ways. There are two alternative ways in which we can proceed from here: we can either (a) increase the power of these studies by including more and more samples in both the case and the control groups or (b) we can elaborate on the data analysis concentrating on clinical and molecular subtypes for each of the diseases studied. The enlarged GWAS studies are bound to confirm the finding(s) of the original studies, however, it is doubtful whether they will reveal novel susceptibility loci, particularly if the data are analysed in the same way as before. The main focus of data analysis in GWAS studies so far has been on reproducibility of findings using classical hazard ratio analysis, which means that markers with extremely low p‐values have been favoured. This requirement for certainty when studying the effects of 300,000 and 500,000 putative factors for susceptibility (corresponding to the size of the SNP arrays used) has caused investigators to report only on susceptibility alleles of universal impact for all individuals, rather than high penetrance alleles in a discreet subgroup of patients, which remain insignificant in the overall pool. Using this type of statistical analysis on GWAS studies can be compared to attempting to describe quantum mechanics using tools from classical Newtonian physics. Therefore we strongly advocate proceeding in the alternative direction: first identifying discreet molecular and clinical subtypes for each disease and subsequently elaborating on the data analysis including probability models, pattern recognition and Bayesian neuronal networking models. Our personal experience is that there is a common scepticism towards these approaches rooted on a “what you do not see cannot be there” attitude, and that it may be rather difficult to publish work that employs these approaches.
A further criticism of the rationale behind GWAS studies is based on the argument that inferring an individual's cancer risk from a limited number of alleles is probably a flawed proposition. The opponents argue that given that the majority of such genes will be inherited randomly, the predisposing alleles will tend to be balanced out by those that do not predispose to the condition (i.e. ‘the “good” alleles will tend to balance out the “bad” ones’). Therefore the distribution/shape of the risk profile would tend to be so narrow as to be fairly ineffective for the great majority of people. This is an argument we disagree with. Indeed, it is unlikely that common susceptibility markers will be found for the total population or “the great majority of people”, but it is of equal health benefit to identify the susceptibility markers for different subpopulations at risk. That the “good” and the “bad” alleles would balance each other does not mean that there is no point in identifying them by well designed epidemiological studies. It is necessary to perform stratified analysis and identify markers specific for groups of individuals at risk, sharing common familial background or developing common cancer phenotype. This can be illustrated with our own studies of breast cancer. Recently, GWAS analysis of breast cancer revealed SNPs in five novel genes associated to susceptibility: TNRC9, FGFR2, MAP3K1, H19 and LSP1 (Easton et al., 2007). The results were confirmed for FGFR2 and TNRC9 in two independent studies (Hunter et al., 2007; Stacey et al., 2007). While there is little doubt that the novel susceptibility markers identified by such highly powered studies are genuine, the mechanism by which they cause the susceptibility remains unravelled. Pooling of such a large amount of cases leads inevitably to conceal the various histological and clinico‐pathological subtypes, suggesting that the observed genes are either of universal importance for breast cancer development, or are associated with a subgroup which dominates the overall pool or with any subgroup but with an association sufficiently strong to dominate the overall result. Breast cancer patients can be divided into five distinct molecular subtypes based on their expression profiles (Perou et al., 2000). The existence of these five subgroups, Luminal A, Luminal B, Basal‐like, ErbB2+, and Normal‐like, has been confirmed in independent data sets (Sorlie et al., 2003) and they are associated with different clinical outcome (Sorlie et al., 2001). If the probability to develop a given subclass of breast cancer is genetically determined we might expect to find that the newly discovered susceptibility genes (Easton et al., 2007) are differentially expressed in the various tumor subclasses, and that their transcription is regulated in cis by SNPs within them. With this in mind, we retrieved the mRNA expression data of TNRC9, FGFR2, MAP3K1, H19 and LSP1 from 112 breast tumors representing all five subclasses (Sorlie et al., 2003). Recently we reported that significantly different mRNA levels between the subtypes were found for all the five genes by ANOVA analysis (Nordgard et al., 2007).
Further understanding of the molecular mechanisms underlying the tumor subclassification and the role of these novel susceptibility genes play in developing these subtypes is necessary. One known strong factor underlying the subclassification is the estrogen receptor (ER). The relationship between the ER and its ligand, oestradiol, and the enzymes that synthesize it is not well understood. In a previous study we found that the expression of a set of nine mRNA transcripts of members of the oestradiol metabolic and signalling pathways, including CYP19 (aromatase), was significantly different in the five molecular subtypes (Kristensen et al., 2005). We have previously reported a SNP in the 3′‐untranslated area of CYP19 mRNA to be associated to higher mRNA levels and tumor size in locally advanced breast cancers (stage III/IV) (Kristensen et al., 2000). The distribution of the different alleles of this SNP is also significantly different in the five molecular subtypes (Fig. 1). Since the induction and promotion of carcinogenesis is attributed to ER receptor mediated stimulation of proliferation of pre‐malignant breast epithelial cells, the amount of produced oestrogen and its availability at the time of this stimulation is likely to be of importance for the further development of the disease, and may ultimately reflect in the tumor classes observed by gene expression profiling. This illustrates the necessity to conduct stratified SNP‐disease association studies and stratification of patients by their molecular subtypes may give much more power to the classical case control studies, and genes of no or borderline overall significance may be highly penetrant for certain subclasses, and therefore identifiable.
Figure 1.
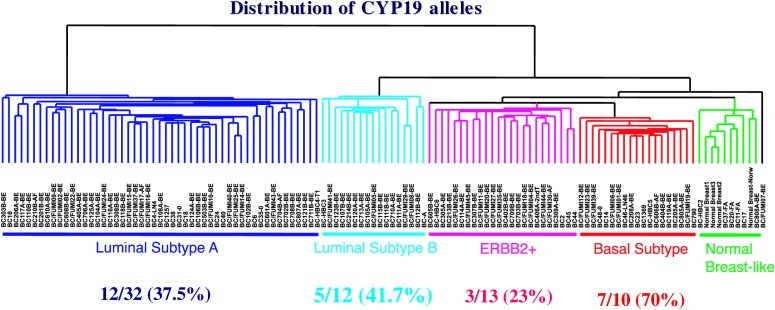
Distribution of the TT genotypes (homozygous for the variant allele) in the molecular subclasses of breast cancer defined by whole‐genome mRNA expression. TT homozygotes are significantly more frequent in patients developing basal‐like tumors (p=0.05).
In conclusion
The GWA studies should continue; simply because they have already been started, the investments have been made and stopping half‐way is counterproductive. The returns so far may seem to someone low due to inadequately conservative approaches to data analysis (which has nothing to do with the experimental design itself), and this will change with time.
Only genotyping more samples will provide large enough sample subsets for stratified analysis with sufficiently unified clinical and molecular profiles to lead us to the particular contribution of the “good” and “bad” alleles. Funding should be provided to encourage these stratified analyses.
The raw data should be made available to the scientific community. This would promote the development of novel methodologies for analysis of data generated by large scale SNP studies, in an analogous way to the bioinformatics boom that was triggered by the flood of data generated by mRNA expression microarray platforms.
Kristensen Vessela N., Børresen-Dale Anne-Lise, (2008), SNPs associated with molecular subtypes of breast cancer: On the usefulness of stratified Genome‐wide Association Studies (GWAS) in the identification of novel susceptibility loci, Molecular Oncology, 2, doi: 10.1016/j.molonc.2008.02.003.
References
- Balmain, A. , Gray, J. , Ponder, B. , Mar 2003. The genetics and genomics of cancer. Nat. Genet. 33, 238–244. [DOI] [PubMed] [Google Scholar]
- Easton, D.F. , Pooley, K.A. , Dunning, A.M. , Pharoah, P.D. , Thompson, D. , Ballinger, D.G. , Struewing, J.P. , Morrison, J. , Field, H. , Luben, R. , Wareham, N. , Ahmed, S. , Healey, C.S. , Bowman; SEARCH collaborators, R. , Meyer, K.B. , Haiman, C.A. , Kolonel, L.K. , Henderson, B.E. , Le Marchand, L. , Brennan, P. , Sangrajrang, S. , Gaborieau, V. , Odefrey, F. , Shen, C.Y. , Wu, P.E. , Wang, H.C. , Eccles, D. , Evans, D.G. , Peto, J. , Fletcher, O. , Johnson, N. , Seal, S. , Stratton, M.R. , Rahman, N. , Chenevix-Trench, G. , Bojesen, S.E. , Nordestgaard, B.G. , Axelsson, C.K. , Garcia-Closas, M. , Brinton, L. , Chanock, S. , Lissowska, J. , Peplonska, B. , Nevanlinna, H. , Fagerholm, R. , Eerola, H. , Kang, D. , Yoo, K.Y. , Noh, D.Y. , Ahn, S.H. , Hunter, D.J. , Hankinson, S.E. , Cox, D.G. , Hall, P. , Wedren, S. , Liu, J. , Low, Y.L. , Bogdanova, N. , Schürmann, P. , Dörk, T. , Tollenaar, R.A. , Jacobi, C.E. , Devilee, P. , Klijn, J.G. , Sigurdson, A.J. , Doody, M.M. , Alexander, B.H. , Zhang, J. , Cox, A. , Brock, I.W. , MacPherson, G. , Reed, M.W. , Couch, F.J. , Goode, E.L. , Olson, J.E. , Meijers-Heijboer, H. , van den Ouweland, A. , Uitterlinden, A. , Rivadeneira, F. , Milne, R.L. , Ribas, G. , Gonzalez-Neira, A. , Benitez, J. , Hopper, J.L. , McCredie, M. , Southey, M. , Giles, G.G. , Schroen, C. , Justenhoven, C. , Brauch, H. , Hamann, U. , Ko, Y.D. , Spurdle, A.B. , Beesley, J. , Chen, X. , kConFab; AOCS Management Group, Mannermaa, A. , Kosma, V.M. , Kataja, V. , Hartikainen, J. , Day, N.E. , Cox, D.R. , Ponder, B.A. , 2007. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447, 1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeles, R.A. , Kote-Jarai, Z. , Giles, G.G. , Olama, A.A. , Guy, M. , Jugurnauth, S.K. , Mulholland, S. , Leongamornlert, D.A. , Edwards, S.M. , Morrison, J. , Field, H.I. , Southey, M.C. , Severi, G. , Donovan, J.L. , Hamdy, F.C. , Dearnaley, D.P. , Muir, K.R. , Smith, C. , Bagnato, M. , Ardern-Jones, A.T. , Hall, A.L. , O'Brien, L.T. , Gehr-Swain, B.N. , Wilkinson, R.A. , Cox, A. , Lewis, S. , Brown, P.M. , Jhavar, S.G. , Tymrakiewicz, M. , Lophatananon, A. , Bryant, S.L. , The UK Genetic Prostate Cancer Study Collaborators, British Association of Urological Surgeons' Section of Oncology, The UK ProtecT Study Collaborators, Horwich, A. , Huddart, R.A. , Khoo, V.S. , Parker, C.C. , Woodhouse, C.J. , Thompson, A. , Christmas, T. , Ogden, C. , Fisher, C. , Jamieson, C. , Cooper, C.S. , English, D.R. , Hopper, J.L. , Neal, D.E. , Easton, D.F. , Feb 10, 2008. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hunter, D.J. , Kraft, P. , Jacobs, K.B. , Cox, D.G. , Yeager, M. , Hankinson, S.E. , Wacholder, S. , Wang, Z. , Welch, R. , Hutchinson, A. , 2007. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 39, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen, V.N. , Harada, N. , Yoshimura, N. , Haraldsen, E. , Lonning, P.E. , Erikstein, B. , Kåresen, R. , Kristensen, T. , Børresen-Dale, A.L. , 2000. Genetic variants of CYP19 (aromatase) and breast cancer risk. Oncogene 19, 1329–1333. [DOI] [PubMed] [Google Scholar]
- Kristensen, V.N. , Sørlie, T. , Geisler, J. , Langerød, A. , Yoshimura, N. , Kåresen, R. , Harada, N. , Lønning, P.E. , Børresen-Dale, A.L. , 2005. Gene expression profiling of breast cancer in relation to estrogen receptor status and estrogen-metabolizing enzymes: clinical implications. Clin. Cancer Res. 11, 878–883. [PubMed] [Google Scholar]
- Nordgard, S.H. , Johansen, F.E. , Alnæs, G.I. , Naume, B. , Børresen-Dale, A.L. , Kristensen, V.N. , 2007. Genes harbouring susceptibility SNPs are differentially expressed in the breast cancer subtypes. Breast Cancer Res. 9, 113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg, R.A. , Meijers-Heijboer, H. , Cornelisse, C.J. , Devilee, P. , Aug 2007. Genetic susceptibility for breast cancer: how many more genes to be found?. Crit. Rev. Oncol. Hematol. 63, (2) 125–149. [DOI] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de, R.M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , 2000. Molecular portraits of human breast tumours. Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.B. , van de, R.M. , Jeffrey, S.S. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U.S.A. 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Tibshirani, R. , Parker, J. , Hastie, T. , Marron, J.S. , Nobel, A. , Deng, S. , Johnsen, H. , Pesich, R. , Geisler, S. , 2003. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. U.S.A. 100, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey, S.N. , Manolescu, A. , Sulem, P. , Rafnar, T. , Gudmundsson, J. , Gudjonsson, S.A. , Masson, G. , Jakobsdottir, M. , Thorlacius, S. , Helgason, A. , Aben, K.K. , Strobbe, L.J. , Albers-Akkers, M.T. , Swinkels, D.W. , Henderson, B.E. , Kolonel, L.N. , Le Marchand, L. , Millastre, E. , Andres, R. , Godino, J. , Garcia-Prats, M.D. , Polo, E. , Tres, A. , Mouy, M. , Saemundsdottir, J. , Backman, V.M. , Gudmundsson, L. , Kristjansson, K. , Bergthorsson, J.T. , Kostic, J. , Frigge, M.L. , Geller, F. , Gudbjartsson, D. , Sigurdsson, H. , Jonsdottir, T. , Hrafnkelsson, J. , Johannsson, J. , Sveinsson, T. , Myrdal, G. , Grimsson, H.N. , Jonsson, T. , von Holst, S. , Werelius, B. , Margolin, S. , Lindblom, A. , Mayordomo, J.I. , Haiman, C.A. , Kiemeney, L.A. , Johannsson, O.T. , Gulcher, J.R. , Thorsteinsdottir, U. , Kong, A. , Stefansson, K. , 2007. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 39, 865–869. [DOI] [PubMed] [Google Scholar]