

Abstract
AZD0530, an orally available Src inhibitor, demonstrated potent antimigratory and anti‐invasive effects in vitro, and inhibited metastasis in a murine model of bladder cancer. Antiproliferative activity of AZD0530 in vitro varied between cell lines (IC50 0.2 –>10μM). AZD0530 inhibited tumor growth in 4/10 xenograft models tested and dynamically inhibited in vivo phosphorylation of Src substrates paxillin and FAK in both growth‐inhibition‐resistant and ‐sensitive xenografts. The activity of AZD0530 in NBT‐II bladder cancer cells in vitro was consistent with inhibition of cell migration and stabilization of cell–cell adhesion. These data suggest a dominant anti‐invasive pharmacology for AZD0530 that may limit tumor progression in a range of cancers. AZD0530 is currently in Phase II clinical trials.
Keywords: AZD0530, Src, Preclinical, Invasion, Biomarkers, Saracatinib
1. Introduction
The constitutively active protein tyrosine kinase v‐Src was identified as the transforming element of the oncogenic Rous sarcoma retrovirus more than 30 years ago. Subsequently, the oncogenic potential of the normal cellular homolog c‐Src has been revealed (Martin, 2001). Src is the best‐understood member of a family of nine membrane‐associated, non‐receptor tyrosine kinases that also includes c‐Yes and Fyn (reviewed in Thomas and Brugge, 1997). Src transduces signals that control processes involved in cell proliferation, adhesion, and motility. Most cell types express low levels of Src; platelets, osteoclasts, and neural cells are the only normal cell types known to express high levels of Src (Thomas and Brugge, 1997). Src activity is highly regulated and activation requires dephosphorylation of regulatory tyrosine 527, autophosphorylation of tyrosine 416 within the catalytic domain, and changes in intramolecular interactions.
Deregulated elevated Src kinase activity has been reported in a wide range of human tumor types (Frame, 2002; Summy and Gallick, 2003). Clinical evidence supports a link between de‐regulation of Src activity and tumor progression and metastasis (Talamonti et al., 1993). Through its interaction with the integrin‐binding non‐receptor tyrosine kinase focal adhesion kinase (FAK), activated Src mediates the phosphorylation and recruitment of cytoskeletal proteins such as paxillin to regulate focal adhesion complexes and cell motility (reviewed in Playford and Schaller (2004)). It has been demonstrated that elevated Src activity disrupts the E‐cadherin epithelial cell–cell adhesion system (Avizienyte et al., 2002; Irby and Yeatman, 2002) and this can be restored by Src inhibition (Nam et al., 2002). In a murine model of bladder cancer metastasis, Src activity was shown to be not required for the growth of the primary xenograft, but was a critical determinant of metastasis, since the number of mice bearing metastases was dramatically reduced by the expression of a dominant‐negative mutant of Src (SrcK−) or of Csk, the natural cellular negative regulator of Src (Boyer et al., 2002). These findings support a hypothesis that a major consequence of aberrant Src signaling in tumor cells is to regulate cell adhesion and facilitate motility, promoting invasive and metastatic phenotypes.
Studies on fibroblasts engineered to express constitutively active c‐Src (thus mimicking the viral oncogene Src product), or on fibroblasts deficient in Csk, have clearly demonstrated that c‐Src under these conditions can provide a mitogenic and transforming drive (Cartwright et al., 1987; Oneyama et al., 2008; Kmiecik and Shalloway, 1987). However, there is only very limited evidence supporting mutated, constitutively active Src in human epithelial cancers (Irby et al., 1999), where de‐regulation appears predominantly driven through post‐translational modifications and intracellular interactions between Src and various activated transmembrane receptors, cytoskeletal proteins, and cytoplasmic phosphatases. Src activated under these cellular conditions in epithelial tumor cells does not appear to be linked strongly to a mitogenic phenotype (in vitro at least), with accumulating evidence suggesting that the predominant consequence of deregulated Src in tumor cells is to help facilitate a change towards a more aggressive, migratory, and invasive cellular phenotype without necessarily impacting on cellular proliferation (Boyer et al., 2002; Jones et al., 2002; Vultur et al., 2008).
The involvement of Src in tumor progression and metastasis has generated considerable interest in Src as a therapeutic anticancer target. We have reported previously the development of AZD0530, a potent, orally available Src inhibitor (Hennequin et al., 2006). AZD0530 significantly suppressed the motile and invasive nature of endocrine‐resistant breast cancer cells in vitro, effects which were increased by combination with the epidermal growth factor receptor (EGFR) inhibitor, gefitinib (Hiscox et al., 2006). Moreover, AZD0530 was able to restore tamoxifen sensitivity in tamoxifen‐resistant breast cancer cells (Chu et al., 2007).
Here, we report the preclinical pharmacology of AZD0530 with respect to proliferation, migration, and invasion in vitro, and tumor growth, metastasis, and effects on FAK and paxillin phosphorylation in vivo. We observed inconsistent inhibition of tumor cell proliferation in vitro and in vivo at therapeutically achievable concentrations of AZD0530. However, in all tumor cell types examined, inhibition of Src kinase activity was associated with reduced phosphorylation of cellular Src substrates and inhibition of cell migration. The data support the hypothesis that a key role for Src kinase in tumor progression is the emergence of an invasive tumor phenotype.
2. Results
2.1. In vitro studies
2.1.1. Inhibition of isolated protein kinase activity
AZD0530 potently inhibited Src and the other Src tyrosine kinase family members investigated (c‐Yes, Fyn, Lyn, Blk, Fgr, and Lck), with high selectivity observed against a panel of other protein kinases involved in signal transduction (Table 1), including Csk, the intracellular negative regulator of Src activation. The only other notable activities observed were versus Abl (in common with other ATP‐competitive Src inhibitors (Golas et al., 2003; Lombardo et al., 2004)) and versus activating mutant forms of the EGFR (L858R and L861Q).
Table 1.
Inhibitory activity of AZD0530 on isolated tyrosine kinases. IC50 values are the mean of at least three measurements.
| Kinase | Definition | Mean IC50, nM |
|---|---|---|
| c‐Src | Ubiquitous Src family member | 2.7 |
| Lck | Immune cell‐restricted Src family member | <4 |
| c‐Yes | Ubiquitous Src family member | 4 |
| EGFR L861Q | Activating mutation of epidermal growth factor receptor | 4 |
| Lyn | Immune cell‐restricted Src family member | 5 |
| EGFR L858R | Activating mutation of epidermal growth factor receptor | 5 |
| Fyn | Ubiquitous Src family member | 10 |
| Fgr | Immune cell‐restricted Src family member | 10 |
| Blk | Immune cell‐restricted Src family member | 11 |
| v‐Abl | Viral Abelson tyrosine kinase | 30 |
| EGFR | Epidermal growth factor receptor tyrosine kinase | 66 |
| c‐kit | Stem cell factor receptor tyrosine kinase | 200 |
| EphA2 | Ephrin receptor tyrosine kinase | 236 |
| Csk | c‐terminal Src kinase (negative regulator of Src) | >1000 |
| PDGFRβ | Platelet‐derived growth factor receptor tyrosine kinase | >5000 |
| PDGFRα | Platelet‐derived growth factor receptor tyrosine kinase | 10,000 |
| CDK2 | Cyclin‐dependent kinase 2 | 10,000 |
| Flt‐4 (VEGFR3) | Fms‐like tyrosine kinase 4 | >10,000 |
| FGFR (FGFR1) | Fibroblast growth factor receptor tyrosine kinase | >10,000 |
| AUR‐3 | Aurora kinase‐3 | >10,000 |
| MEK | Mitogen‐activated protein kinase | 14,000 |
| KDR (VEGFR2) | Kinase insert domain‐containing receptor | 21,000 |
| Flt‐1 (VEGFR1) | Fms‐like tyrosine kinase 1 | >100,000 |
AZD0530 (10μM) was also tested for inhibitory activity versus a panel of serine/threonine kinases using a filter capture assay with 32P and showed <20% inhibition of activity (n=23).
2.1.2. Inhibition of cell proliferation
AZD0530 potently inhibited the in vitro proliferation of Src3T3 mouse fibroblasts and demonstrated variable antiproliferative activity in a range of human cancer cell lines containing endogenous Src (Table 2). Sub micromolar growth inhibition of five of the human cancer cell lines tested with AZD0530 (tumor types: colon, prostate, lung, and leukemia) was observed with IC50 values of 0.2–0.7μM. In 3‐day MTS cell proliferation assays (Promega G3580), AZD0530 inhibited in vitro proliferation of the Bcr–Abl‐driven human leukemia cell line K562 with an IC50 of 0.22μM.
Table 2.
Inhibitory activity of AZD0530 on cell line proliferation. IC50 values are the mean of at least three measurements.
| Cell line | Origin | Source | Culture conditions | AZD0530 IC50 (μM) |
|---|---|---|---|---|
| Src Y530F NIH 3T3 | Mouse embryo | Sydonia Rayter, Sugen, Inc. | DMEM+10% FCS+1% l‐glutamine | 0.08 |
| Calu‐6 | Human NSCLC | ATCC HTB‐56 | RPMI 1640+10% FCS | >10 |
| A549 | Human NSCLC | ATCC CCL‐185 | DMEM+10% FCS | 14 |
| LoVo | Human colon | ATCC CCL‐229 | DMEM+10% FCS+1% l‐glutamine | 0.2 |
| SW 403 | Human colon | ECACC 87071008 | L‐15+10% FCS | 0.6 |
| HT29 | Human colon | ECACC 85061109 | EMEM+5% FCS+1% NEAA | 6 |
| HCT 116 | Human colon | ATCC CCL‐247 | McCoy's 5a+10% FCS | >10 |
| PC‐3 | Human prostate | ATCC CRL‐1435 | RPMI+10% FCS+10% M1 | 0.7 |
| K562a | Human Ph+ leukemia | Paterson Laboratories, Christie Hospital, Manchester, UK | RPMI 1640+10% FCS | 0.2 |
| SKOV‐3 | Human ovary | ATCC HTB‐77 | Ham's F‐12+10% FCS | >10 |
| BT474C | Human breast | Vall d'Hebron University Hospital, Barcelona, Spain | DMEM+10% FCS+1% l‐glutamine | 1.6 |
| MDA‐MB‐231 | Human breast | ATCC HTB‐26 | DMEM+10% FCS+1% l‐glutamine | 3 |
| MCF‐7 | Human breast | Dr W. Heuse, Cancer Research UK, London, UK | DMEM+10% FCS+1% l‐glutamine | >10 |
| PC‐9 a | Human NSCLC | Professor K. Nishio, National Cancer Center Hospital, Tokyo, Japan | RPMI 1640+10% FCS | 0.23 |
ECACC, European Collection of Cell Cultures; NEAA, non‐essential amino acids; NSCLC, non‐small‐cell lung cancer.
3‐day MTS assay.
2.1.3. Inhibition of cell migration
In the microdroplet migration assay, AZD0530 reduced the migration of human lung cancer A549 cells in a concentration‐dependent manner (IC50 0.14μM; Figure 1A). AZD0530 at concentrations of 0.01–0.5μM demonstrated a clear dose‐dependent antimigratory effect compared with untreated controls in monolayer scratch assays of human breast cancer MDA‐MB‐231 cells (Figure 1B). AZD0530 also dose dependently inhibited the EGF‐ and collagen‐stimulated migration of NBT‐II bladder cancer cells (Figure 1C). Moreover, at a single concentration of 0.25μM, AZD0530 consistently inhibited migration in monolayer scratch assays of bladder cancer cell lines (31%, 37%, and 78% inhibition versus untreated controls in T24, SCaBER, and 1A6 cell lines, respectively).
Figure 1.
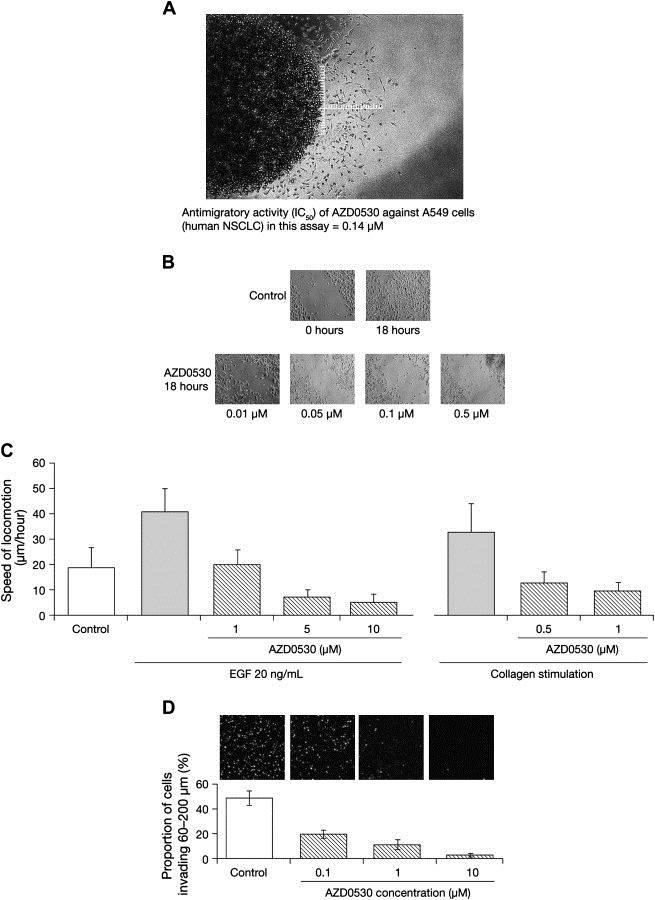
AZD0530 antimigratory and anti‐invasive activity. (A) An illustration of the microdroplet migration assay. The image shows human NSCLC A549 cells migrating out from an agarose microdroplet after 72h of culture, and indicates how cell migration is measured. (B) Representative time‐lapse photography images (magnification ×200) in which AZD0530 shows dose‐dependent antimigratory activity compared with untreated controls in human breast cancer cells (MDA‐MB‐231) after 18h treatment in a monolayer scratch assay. (C) AZD0530 exhibited dose dependent inhibition of NBT‐II cell motility. EGF (20ng/mL)‐ and collagen‐induced cell motility assessed by videomicroscopy in vitro (mean+SEM of at least three experiments). (D) In vitro invasion of HT1080 cells through a 3D fibrillar collagen matrix. Exposure of HT1080 cells to AZD0530 resulted in a dose‐dependent reduction in the proportion of cells invading beyond a depth of 60μm into a 3D collagen type I matrix. Bars represent mean±SEM of three experiments. Representative images (magnification ×200) of invaded, Hoechst‐labeled cells at a depth of 60μm into the 3D collagen matrix are shown.
2.1.4. Inhibition of cell invasion
AZD0530 treatment significantly impaired the invasion of HT1080 cells through a 3‐dimensional (3D) collagen matrix (Figure 1D). In contrast to other 3D invasion assays, our assay (see Methods) monitors the proportion of total cells within a 3D matrix invading beyond a specified depth (60μm), thereby distinguishing repressed cellular invasion from cell loss that may be due to impaired adhesion, proliferation, or excessive cell death. This method confirms that AZD0530 exposure produces a significant and dose‐dependent inhibition of the invasive properties of HT1080 cells.
2.1.5. Inhibition of cell scattering
AZD0530 1μM completely inhibited EGF‐induced cell scattering in NBT‐II bladder cancer cells, restoring cell–cell adhesion and the localization of desmoplakin to desmosomal junctions (Figure 2A).
Figure 2.

AZD0530 effects on cell scattering and paxillin phosphorylation. (A) AZD0530 inhibited EGF (20ng/mL)‐mediated loss of desmoplakin immunoreactivity from the surface of NBT‐II cells in monolayer culture. (B) AZD0530 inhibited tyrosine phosphorylation of paxillin Y118 in NBT‐II cells in vitro. Relative phosphorylation levels were quantified by densitometry. (C) Effects of AZD0530 on the intracellular distribution of phosphorylated paxillin in NBT‐II cells in vitro.
2.1.6. Inhibition of paxillin phosphorylation
AZD0530 dose dependently inhibited phosphorylation of the Src substrate paxillin in NBT‐II bladder cancer cells (Figure 2B). Moreover, AZD0530 treatment (1μM) induced a relocalization of paxillin from the cell membrane into the cell cytoplasm (Figure 2C) further supporting a role for Src in the early development of cell migratory behavior.
2.1.7. Effects on the EGFR
Given the moderate inhibitory effect observed for AZD0530 against the EGFR in the isolated kinase assay (IC50 66nM; Table 1), further studies were performed to investigate AZD0530 effects on the EGFR. In a head‐to‐head assay measuring EGFR phosphorylation in KB cells, AZD0530 exhibited an IC50 of 1.25μM compared with 11nM for gefitinib, an EGFR tyrosine kinase inhibitor. In isolated kinase assays, AZD0530 exhibited IC50s of 5 and 4nM respectively, for EGFR point‐mutant isoforms L858R (Millipore 14‐626) and L861Q (Millipore 14‐627). Effects on the EGFR L858R isoform were further investigated in PC‐9 cells, a non‐small‐cell lung cancer (NSCLC) cell line harboring this EGFR mutation. AZD0530 inhibited growth of PC‐9 cells with an IC50 of 0.23μM in 3‐day MTS cell proliferation assays (Promega G3580).
2.1.8. Enzyme kinetics
Pre‐incubation of Src kinase with 100nM AZD0530 (in the presence of 160μM ATP) resulted in >95% inhibition of Src activity. A subsequent 10‐fold dilution to 10nM AZD0530 in the presence of a high concentration of ATP (1.60mM) led to a regain of Src activity, with only 30% inhibition, suggesting that inhibition was reversible. A similar degree of inhibition was observed for the same final concentrations of AZD0530 and ATP without the dilution step.
The reversibility studies implied that AZD0530 was competitive with ATP, as has been reported for other anilinoquinazolines (Bridges, 2001). ATP‐competitive inhibition was confirmed in experiments where both the concentration of ATP and that of AZD0530 were varied at fixed concentrations of the substrate, Src II peptide. The estimated inhibition constant, K is was 1.5 and the 95% confidence interval (CI) was 1.2, 1.9nM. This is lower than the IC50 of 2.7nM reported in Table 1 because it relates to a situation in the absence of competing ATP. AZD0530 was shown to follow pure noncompetitive kinetics when the concentration of Src II peptide was varied at 250μM ATP, with an apparent K i′ of 10nM (95% CI 8.4, 12.0; Figure 3A and B), which corresponds to a K i of 1.1nM (95% CI 0.9, 1.3) in the absence of ATP. Many kinases have structural differences between their active and inactive forms (Huse and Kuriyan, 2002). Inactivated Src adopts a closed conformation and AZD0530 binds approximately 10‐fold more weakly to inactive Src (K d 11nM; 95% CI 9.3, 14).
Figure 3.
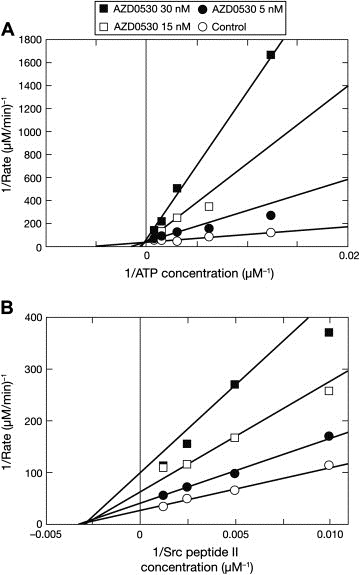
AZD0530 enzyme kinetics. (A) Competitive kinetics when the concentrations of AZD0530 and ATP are varied in the presence of 1mM Src II peptide, with best‐fit values of Kis 1.5nM, maximum velocity (Vmax) 0.03μM/min, and ATP KM 190μM. (B) Pure noncompetitive kinetics when the concentrations of AZD0530 and Src II peptide are varied in the presence of 1.6mM ATP, with best‐fit values of apparent Ki′ 10nM; Vmax 0.04μM/min, and Src peptide II KM 330μM.
2.2. In vivo studies
2.2.1. Pharmacokinetics
The AZD0530 plasma concentration 6h after oral dosing increased proportionally to the dose (Figure 4A).
Figure 4.
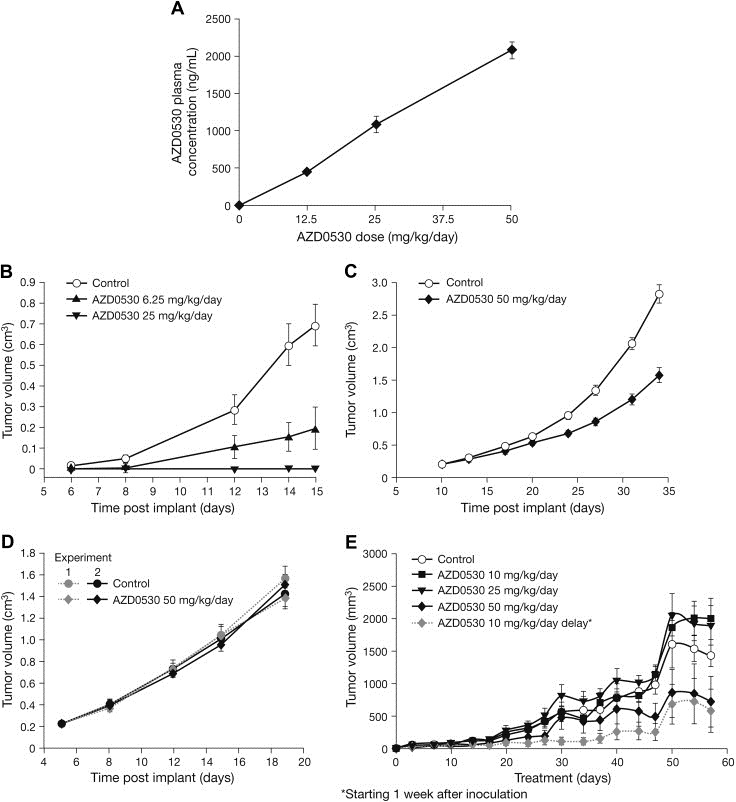
AZD0530 pharmacokinetics and effects on xenograft tumor growth. (A) AZD0530 plasma concentration (ng/mL) 6h post‐terminal dose in female nude mice bearing LoVo tumors (n=7 per group). (B–D) Effect of once‐daily AZD0530 on tumor growth in female nude mice (n=7 per group) bearing (B) Src3T3 allografts, (C) human NSCLC (Calu‐6) xenografts, and (D) human colon cancer (LoVo) xenografts. (E) Effects of AZD0530 at 10 (n=14), 25 (n=14) or 50mg/kg/day po (n=7) on the same day as cell inoculation or 1 week after inoculation on the growth of NBT‐II tumor cells grown as sc xenografts in nude mice. Mean±SEM.
2.2.2. Src3T3 allografts and xenografts
AZD0530 treatment potently inhibited the proliferation of subcutaneously transplanted Src3T3 fibroblasts in mice (Figure 4B) and rats (Hennequin et al., 2006) in a dose‐dependent manner. In both models, significant inhibition of tumor growth was seen at doses ≥6mg/kg/day (60% inhibition in mice [P<0.01] and 98% inhibition in rats [P<0.001] versus animals treated with vehicle) and, at the maximum doses investigated, complete tumor growth inhibition was observed (100% inhibition at 25mg/kg/day in mice and 10mg/kg/day in rats) (Hennequin et al., 2006).
2.2.3. Human tumor xenografts
Once‐daily oral dosing of AZD0530 resulted in a moderate growth delay in 4/10 xenograft models tested (Table 3). In the remainder of the xenograft panel, AZD0530 treatment failed to inhibit primary tumor growth. Representative xenograft growth delay curves for an AZD0530 growth‐inhibition‐sensitive (Calu‐6) and an AZD0530 growth‐inhibition‐insensitive model (LoVo) are shown in Figure 4C and D, respectively. AZD0530 did not affect any aspect of tumor vascularity in xenograft models as assessed by blood vessel number or CD31 positive vessels (data not shown).
Table 3.
Effect of once‐daily AZD0530 50mg/kg on human tumor cell xenograft growth in female nude mice.
| Cell line | Origin | Total number of cells inoculated | Average tumor volume before randomization (cm3) | Number of mice randomized to each group (treatment/control) | Duration of dosing (days) | Mean inhibition of tumor volume relative to control (%) |
|---|---|---|---|---|---|---|
| MDA‐MB‐231 | Breast ER− | 5×106 | 0.062 | 9/9 | 27 | 40 |
| AsPc‐1 | Pancreas | 1×107 | 0.3 | 10/10 | 15 | 42 |
| Calu‐6 | Lung | 1×106 | 0.2 | 7/8 | 28 | 40–50a |
| BT474C | Breast ER+ | 1×107 | 0.4 | 12/15 | 21 | 23–34b |
| ZR‐75‐1 | Breast ER+ | 1×107 | 0.5 | 10/10 | 15 | None |
| Colo205 | Colon | 3×106 | 0.3 | 9/9 | 18 | None |
| KB | Oral carcinoma | 5×106 | 0.3 | 8/8 | 10 | None |
| HT29 | Colon | 1×107 | NA | 10/10 | 36–91 | None |
| LoVo | Colon | 1×107 | 0.3 | 7/8 | 14 | None |
| HPAC | Pancreas | 1×107 | 0.3 | 15/15 | 21 | None |
EMEM, Earle's minimal essential medium; NA, not applicable, treatment started on day of inoculation.
Range of means observed across eight separate studies.
Range of means observed across two separate studies.
2.2.4. NBT‐II bladder cancer metastasis model
In nude mice bearing subcutaneous xenografts of NBT‐II bladder cancer cells, once‐daily AZD0530 treatment at doses of 10, 25, and 50mg/kg/day resulted in a small, non significant delay in xenograft growth at the highest dose only (50mg/kg/day; Figure 4E). In contrast, all three doses of AZD0530 resulted in a reduction in the number of mice from which tumor colonies could be grown from mesenteric lymph node extracts (Figure 5A). In an additional experiment, this antimetastatic effect of AZD0530 did not appear to be affected by a 7‐day delay between cell inoculation and drug administration. One out of seven mice developed metastases in a group that started treatment with AZD0530 50mg/kg/day 7 days after tumor cell inoculation, and 1/7 developed metastases in a group that started treatment with the same dose immediately after cell inoculation.
Figure 5.
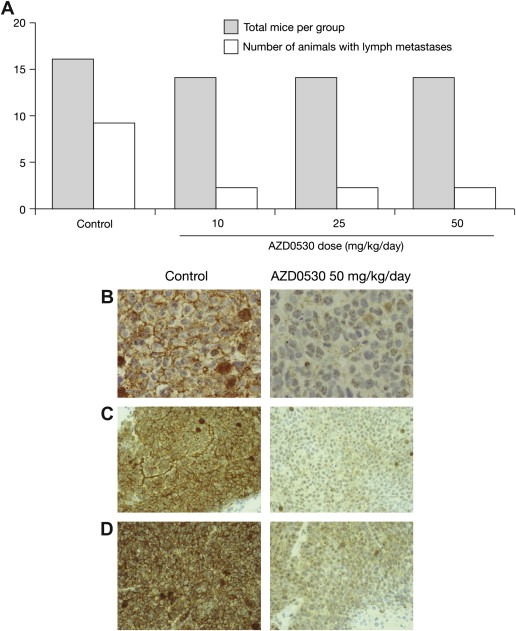
AZD0530 effects on metastasis and phosphorylation of Src substrates FAK and paxillin. (A) Effects of increasing doses of AZD0530 on metastasis to lymph nodes after injection of NBT‐II cells into nude mice. (B–D) Immunohistochemical analysis after 28 (Calu‐6) or 14 (LoVo) days once‐daily oral treatment with vehicle alone or AZD0530 50mg/kg. AZD0530 inhibited phosphorylation of paxillin (pY31) in (B) human Calu‐6, and (C) LoVo xenografts grown subcutaneously in nude mice. (D) AZD0530 inhibited phosphorylation of FAK (pY861) in LoVo human xenografts grown subcutaneously in nude mice. Images are at magnifications of (B) ×40 and (C and D) ×20.
2.2.5. Phosphorylation of Src kinase substrates FAK and paxillin
Levels of phosphorylated FAK (pY861) and paxillin (pY31), assessed by immunohistochemistry using phosphospecific antibodies, were reduced markedly in tumors from animals bearing both growth‐inhibition‐sensitive and growth‐inhibition‐insensitive xenografts after 14–28 days' treatment with AZD0530 50mg/kg/day (Figure 5B–D). Antibodies detecting non‐phospho epitopes of FAK or paxillin showed no effect on the expression of either protein (data not shown).
3. Discussion
The discoveries of the first oncogenic kinase, v‐Src, and its cellular homolog, Src, have led to significant progress in our understanding of the biochemistry of both normal and malignant cells (Martin, 2001). Substantial evidence implicates deregulated Src in the development of solid tumors, and Src inhibition is a potential therapeutic strategy in many common cancers (Frame, 2002; Summy and Gallick, 2003). In addition, Src activity is essential for osteoclast bone resorptive activity, suggesting that Src inhibitors could have an additional benefit in the management of malignant bone disease (Metcalf et al., 2002).
The present studies investigated the preclinical properties of the novel Src inhibitor AZD0530. Isolated enzyme assays demonstrated that AZD0530 is a potent, reversible, Src inhibitor that has high selectivity when compared with a range of protein tyrosine kinases involved in signal transduction. Inactivation of Src by AZD0530 is achieved by competitive binding of AZD0530 to an ATP site (Hennequin et al., 2006; Noble et al., 2004), and is consistent with the crystal structure of a complex of AZD0530 with inactivated Src. AZD0530 affinity for activated Src is 10‐fold higher than for inactivated Src. This observation could explain the inconsistent abrogation of Src autophosphorylation (pY419) observed in Western blots after treatment with AZD0530 in preclinical models (data not shown). Subsequent studies focused on downstream substrates of Src (FAK and paxillin) and confirmed these as more consistent biomarkers of Src tyrosine kinase and AZD0530 pharmacodynamic activity.
AZD0530 caused a dramatic inhibition of human non‐small‐cell lung (A549) and human breast (MDA‐MB‐231) cancer cell migration in vitro at concentrations well below the antiproliferative IC50 values in these two cell lines. In migration scratch assays using a range of human tumor cell lines, consistent antimigratory activity was seen using a single concentration of 250nM AZD0530. Furthermore, in NBT‐II bladder cancer cells, inhibition of cell migration in response to treatment with AZD0530 was accompanied by reduced phosphorylation of the Src substrate paxillin, which has previously been shown to be critically required for cell migration (Boyer et al., 1997; Valles et al., 2004). These data are consistent with the well‐established role of Src in cell migration, and further support the association of Src activity with an invasive tumor cell phenotype. Inhibition of Src activity also inhibited the invasion of HT1080 cells into matrigel or collagen in a 3D invasion assay. Although the invasion of HT1080 cells into 3D fibrillar collagen gels was inhibited by diverse protease inhibitors, a compensatory mechanism, the so‐called mesenchymal to amoeboid transition, required treatment with an inhibitor cocktail targeting multiple extracellular proteases including, matrix metalloproteinases, and serine proteases to be fully effective (Wolf et al., 2003; Friedl and Wolf, 2003). Such tumor plasticity permits continued invasion through 3D collagen in the presence of these inhibitors and may in part explain the failure of MMP inhibitors to impede tumor progression in clinical trials. In contrast, the data obtained with AZD0530 indicate that in HT1080 cells, which are known to express both MMP and serine proteases, inhibition of Src kinase circumvents such compensatory responses, suggesting that inhibitors of Src kinase target an integral component or multiple components of the invasive process, and may present significant utility as an anti‐invasive therapy in vivo.
AZD0530 demonstrated in vitro and in vivo antiproliferative activity against Src3T3 fibroblasts that overexpress a constitutively active form of Src. The effect of AZD0530 on the growth of human cancer cell lines and tumor xenografts expressing endogenous Src was more variable and generally modest in comparison with the Src3T3 model. The transforming, mitogenic properties of both v‐Src and constitutively active c‐Src in fibroblasts suggest that high endogenous Src activity could provide a mitogenic drive in human epithelial cancer cells. However, published data do not support a direct association of Src kinase activity with cell proliferation, and suggest that low, rather than high, levels of activated Src may be a determinant of growth sensitivity to Src inhibitors (Emaduddin et al., 2008; Jones et al., 2002; Rosen et al., 1986). Our data further highlight the lack of correlation between the inhibition of proliferation in vitro and inhibition of xenograft growth in vivo. For example, treatment of Calu‐6 (NSCLC) cells with AZD0530 in vitro resulted in minimal antiproliferative activity (IC50>10μM, Table 2) whereas the growth of Calu‐6 xenografts in vivo was inhibited by ∼50% (although no dose–response relationship was observed). In the case of the colon cancer cell lines HT29 and LoVo, which have relatively high and low Src kinase activity, respectively (Bolen et al., 1987; Emaduddin et al., 2008), sensitivity to AZD0530 in vitro segregated with low Src activity. Although no inhibition of in vivo tumor growth was observed for either of these cell lines, Src kinase activity was clearly inhibited as evidenced by the reduced phosphorylation of Src kinase substrates FAK and paxillin (Figure 5). Taken together these data suggest that both cellular context (in vitro versus in vivo setting) and cell‐line‐specific Src kinase activity and signaling may determine response to Src kinase inhibitors, rather than direct inhibition of a Src mitogenic phenotype.
The growth delay observed in vivo was not attributable to an anti‐angiogenic effect, as blood vessel density staining was not affected in the AZD0530‐treated animals (data not shown). Possible explanations for the anti‐growth activity observed are (a) an indirect effect on growth by AZD0530 through inhibition of local migration and invasion of the tumor cells, or (b) an as yet unknown underlying molecular phenotype, possibly influenced by Src kinase dependent aspects of the tumor and host microenvironment that contribute to local regulation of tumor growth. In the three human colon tumor xenograft models investigated, AZD0530 had no effect on tumor growth, but immunohistochemistry revealed that AZD0530 was biologically active, as shown by the inhibition of FAK and paxillin phosphorylation on tyrosine residues known to be direct targets for Src kinase. Although Src kinase activity has been suggested to support angiogenic behavior in endothelial cells (Eliceiri et al., 1999; Lesslie and Gallick, 2005) we were unable to demonstrate any effect of Src kinase inhibition on tumor vasculature in xenograft tumors shown to be responsive to vascular endothelial growth factor targeted agents (Calu‐6, data not shown). Taken together, these data support the hypothesis that Src is acting primarily to promote invasive tumor progression in the majority of solid tumor types. These data are consistent with previously reported data for dasatinib, a Src/Abl inhibitor, which inhibits integrin‐mediated motility and adhesion in colorectal cancer cell lines (Serrels et al., 2006), and migration and invasion of sarcoma cell lines (Shor et al., 2007).
In isolated kinase assays, AZD0530 inhibited the EGFR with approximately 20‐fold lower potency than its effects on Src. Functional assays in cell culture showed only modest effects on EGFR phosphorylation. However, AZD0530 exhibited more potent effects on the EGFR activating point mutants L858R and L861Q. These results are consistent with recent findings that AZD0530 inhibited proliferation and migration more potently in NSCLC cell lines harboring mutant EGFR isoforms than in NSCLC cell lines harboring wild‐type EGFR (Helfrich et al., 2006). Mutations in the EGFR that modulate sensitivity of cancers to EGFR inhibitors can also modulate sensitivity to Src inhibition (Fu et al., 2008; Song et al., 2006), raising the possibility of additional utility of AZD0530 in cancers harboring mutant EGFR.
AZD0530 exhibited moderate activity against Abl kinase in the isolated kinase assay. Affinity for Abl appears to be a common characteristic of the small‐molecule ATP‐competitive Src inhibitors, although this effect is smaller with AZD0530 than with dasatinib and bosutinib (Golas et al., 2003; Lombardo et al., 2004). Moreover, AZD0530 exhibited moderate effects (IC50 0.22μM) in a functional assay of Abl inhibition using human leukemia cells that are growth driven through Bcr–Abl activity. In contrast, dasatinib and bosutinib demonstrated IC50 values of 1nM and 20nM, respectively, for growth inhibition in the same cells (Manley et al., 2005). AZD0530's activity at Abl is not currently considered sufficient to provide a strong rationale for its clinical development in Bcr–Abl‐driven leukemias.
Further investigation of AZD0530 in other tumor models is warranted to define its biochemical and anticancer activity. In the current study, AZD0530 was tested for tumor growth inhibitory activity in the xenograft models available at the time, and this did not provide a complete overlap with the panel of cell lines tested for AZD0530 antiproliferative effects in vitro. However, it is likely that tumor models that assess the antiproliferative activity of a test compound against a cancer cell line or a primary tumor subcutaneous xenograft will have limited value in assessing the activity of an anti‐invasive agent. Such agents may provide benefit in terms of inhibiting local invasive growth or metastatic spread of an established cancer, rather than reducing primary tumor growth per se. Indeed, Src inhibition has recently been shown to significantly reduce metastasis in xenograft models of pancreatic, bladder, and colorectal cancer (Green et al., 2005; Phillips et al., 2007; Trevino et al., 2006).
The data presented here support a mechanistic role of endogenous Src in promoting an invasive cell phenotype through the regulation of cell adhesion, migration, and invasion. AZD0530 inhibited cancer cell proliferation in vitro and in vivo in a constitutively active Src fibroblast tumor model, and in some human tumor cell lines. The lack of consistent antiproliferative activity and the underlying mechanism of such diverse responses require further investigation in additional tumor models. Effects on models of invasion were more clear cut: AZD0530 inhibited invasion and migration of cancer cells in vitro, and inhibited phosphorylation of proteins controlling migration in vitro and in vivo. In an in vivo model of bladder cancer metastasis, AZD0530 inhibited the number of lymph node metastases, detected through colony forming assays, thus recapitulating the effects seen with dominant‐negative Src and Csk constructs in earlier studies in this model (Boyer et al., 2002). We and others have recently demonstrated activity of AZD0530 in inhibiting metastasis in pancreatic cancer models (Green et al., 2005) and in an orthotopic model of human colorectal carcinoma (Phillips et al., 2007). Together with the results presented here, these data suggest that AZD0530 may provide clinical benefit by preventing or delaying tumor progression through inhibition of tumor cell migration and invasion. AZD0530 is currently in Phase II clinical trials.
4. Experimental procedures
4.1. Isolated protein kinase assay
Inhibition of tyrosine kinase activity was examined using an enzyme‐linked immunosorbent assay (ELISA) with recombinant catalytic domains of a panel of receptor and non‐receptor tyrosine kinases (in some cases only part of the catalytic domain was used). This method has been described previously (Plé et al., 2004). AZD0530 dose ranges varied depending on the activity versus the particular kinase tested, but were typically 0.001–10μM.
Specificity assays against a panel of serine/threonine kinases were performed using a filter capture assay with 32P. Briefly, multidrop 384 plates containing 0.5μL AZD0530 or controls (dimethyl sulfoxide [DMSO] alone or pH 3.0 buffer controls) were incubated with 15μL of enzyme plus peptide/protein substrate for 5min before the reaction was initiated by the addition of 10μL of 20mM Mg.ATP. For all enzymes the final concentration was approximated to the Michaelis constant (K m). Assays were carried out for 30min at room temperature before termination by the addition of 5μL orthophosphoric acid. After mixing, the well contents were harvested onto a P81 Unifilter plate, using orthophosphoric acid as the wash buffer. Microcal Origin software (vs. 3.78, Microcal Software, Inc., Northampton, MA, USA) was used to interpolate IC50 values by nonlinear regression.
4.2. Cell lines
The cell lines used were mouse NIH 3T3 fibroblasts engineered to overexpress a constitutively active form of human Src (Src3T3) with a point mutation at the negative regulatory tyrosine site in the c‐terminal domain (Y530F) (Plé et al., 2004) and human cancer cell lines containing endogenous Src (Table 2). Src3T3 fibroblasts form colonies in soft agar and grow subcutaneously in immunocompromised athymic rats and mice in vivo, while the wild‐type parental 3T3 cells do not. Src3T3 cells will grow in medium containing as little as 0.5% fetal calf serum (FCS; used in the assay conditions), whereas wild‐type non‐Src‐transfected 3T3 cells will not grow in these low serum conditions. AZD0530 functional activity against Abl kinase was assessed in an in vitro proliferation assay using K562 human leukemia cells known to be growth driven through Bcr–Abl activity (Lozzio and Lozzio, 1975). NBT‐II rat bladder cancer cells were sourced and cultured as described previously (Boyer et al., 2002).
4.3. Cell proliferation assay
Cell proliferation was assessed using a colorimetric 5‐bromo‐2′‐deoxyuridine (BrdU) Cell Proliferation ELISA kit (Roche Diagnostics GmbH), as described previously (Plé et al., 2004). Briefly, cells were plated onto 96‐well plates (1.5×104cells/well), the following day 0.039–20μM AZD0530 in DMSO (at a final concentration of 0.5%) was added and the cells were incubated for 24h. The cells were pulse labeled with BrdU for 2h and fixed. Cellular DNA was then denatured with the provided solution and incubated with antiBrdU peroxidase for 90min. Following three washes with phosphate‐buffered saline, tetramethylbenzidine substrate solution was added and the plates were incubated on a plate shaker for 10–30min until the positive control absorbance at 690nm was approximately 1.5 absorbance units.
4.4. EGFR phosphorylation assay
KB (nasopharyngeal carcinoma) cells were seeded at 5000cells/well in 96‐well plates and cultured for 72h in Rosewell Park Memorial Institute (RPMI) 1640 media with 10% FCS, followed by 24h' incubation with serum‐free RPMI 1640. Cells were treated with compound for 90min at concentrations ranging from 0 to 10μM. Cells were incubated with 15ng/mL EGF ligand (concentration required to increase receptor phosphorylation to 90% of maximum) for 5 min prior to lysis. Levels of phosphorylated EGFR were measured using the human phospho‐EGFR Duoset ELISA kit (R&D Systems, DYC1095).
4.5. Microdroplet migration (chemokinesis) assay
The microdroplet migration assay assesses the ability of test compounds to inhibit the random motility (chemokinesis) of human epithelial A549 lung cancer cells (American Type Culture Collection [ATCC] CCL 185), which were routinely cultured in Dulbecco's modified Eagle's medium (DMEM)+10% FCS. The assay method has been described in full previously (Plé et al., 2004). Briefly, A549 cells (2×107/mL) were suspended in warm DMEM (37°C) containing 0.3% agarose. The suspension was pipetted into 96‐well plates (2μL/well) and chilled briefly on ice to allow the agarose microdroplet to gel. When set, 90μL of chilled RPMI 1640 medium (Gibco Invitrogen Cell Culture, Paisley, UK) was added to each well, followed by 10μL AZD0530 (0.02–5μM). The plates were incubated for 72h at 37°C to allow migration to occur. Cell migration was measured by taking several equidistant measurements from around the agar droplet to the migrating front of the motile cells. An illustration of the microdroplet migration assay is shown in Figure 1A, indicating how cell migration was measured.
4.6. Monolayer scratch assay
Human MDA‐MB‐231 breast cancer cells and T24, SCaBER, and 1A6 bladder cancer cells were grown as monolayers in adapted six‐well tissue culture plates. Confluent monolayers were gently scraped with a sterile pipette tip to form a scratch and AZD0530 (0.01–0.5μM) was added to the wells in medium (phenol red‐free DMEM [Gibco Invitrogen Cell Culture] supplemented with 10% charcoal/dextran‐treated FCS and 1% l‐glutamine). Cell movement back into the area of the scratch was recorded by time‐lapse photography over 18h.
4.7. NBT‐II cell adhesion and migration assays
The effects of AZD0530 on EGF‐ and collagen‐induced motility of NBT‐II cells were assessed by videomicroscopy as described previously (Valles et al., 2004). The effect of AZD0530 on EGF‐induced cell dispersion was evaluated by immunofluorescence with anti‐desmoplakin antibodies to monitor the EGF‐induced loss of desmosomes from the cell periphery, as reported before (Boyer et al., 1997).
4.8. Assessment of paxillin phosphorylation
The effects of AZD0530 on paxillin phosphorylation was assessed in NBT‐II cells that had been stimulated with 100ng/mL EGF for 15min in the presence of increasing amounts of AZD0530. Paxillin tyrosine phosphorylation was revealed by immunoblotting with polyclonal antibodies against phospho Y118‐paxillin and total paxillin.
4.9. 3D invasion assay
Fibrillar collagen type I gels (1mg/mL final concentration) were prepared by neutralizing a solution of collagen type I (Vitrogen 100, Cohesion Corp.) with 1/10 volume 10× DMEM concentrate, diluted to a final concentration of 1× with distilled H2O, to which 1/11 volume 0.1N NaOH was added. Fibrillar collagen gels (80μL) were set within the upper chamber of a 24‐well transwell insert, above an 8μm pore‐size polycarbonate filter (Costar, Corning Inc.) at 37°C for 18h prior to cell seeding. Human HT1080 fibrosarcoma cells (ATCC CCL 121; DMEM [Gibco Invitrogen Cell Culture], supplemented with 0.2% FCS and 2mM l‐glutamine) were seeded on top of a collagen gel in an upper transwell chamber (1×104cells per transwell). DMEM supplemented with 10% FCS and 2mM l‐glutamine (750μL total volume) was placed in the lower chamber to provide a chemotactic gradient. AZD0530 (0.1, 1, and 10μM) or vehicle (0.1% DMSO) was added to the cells prior to seeding and also to media within the lower chamber. Transwells were incubated at 37°C for 72h and then incubated with 10μM Hoechst 33342 (Molecular Probes Europe) in serum‐free DMEM for 30min at 37°C. Invading cells were visualized by confocal microscopic analysis using a Bio‐Rad Radiance 2000 multiphoton confocal illumination unit attached to a Nikon Eclipse inverted microscope. Quantification of cell invasion was performed with modification to the protocol as described previously (Carragher et al., 2006). Briefly, using a 20× objective, optical sections were scanned at 20μm intervals from the collagen gel surface. Image‐Pro analysis software (Media Cybernetics) was applied to identify the number of positive pixels (Hoechst stain above a threshold) and segment into individual nuclei per optical section. The accumulated sum of the positive nuclei present in the optical sections between 60 and 200μm below the top of collagen gels was expressed as a percentage of the total number of positive nuclei on top and within the collagen gel. This value, therefore, represents the proportion of total cells within each sample invading beyond a depth of 60μm and up to 200μm.
4.10. Enzyme kinetics
Investigation of the reversibility and the mechanism of AZD0530 inhibition was conducted using a full‐length activated human Src (phosphorylated at tyrosine 416, Upstate Biotech, Dundee, UK) in a continuous, coupled assay adapted from Jenkins (1991). ATP and peptide substrate (Src II peptide; Merck Bioscience, Darmstadt, Germany) concentrations were varied in turn (ATP 40–1280μM; Src II peptide 100–800μM), in conjunction with AZD0530 (0–30nM), at saturating concentrations of the non‐varied substrate (ATP 1.6mM; Src II peptide 1.0mM). The binding affinity of AZD0530 for inactivated Src (phosphorylated at tyrosine 527, not tyrosine 416) was measured using a BIAcore inhibition‐in‐solution assay (Karlsson et al., 2000). The assay followed competition binding between AZD0530 and an immobilized ureidoquinazoline (Sullivan et al., 2005) for binding to Src. Data analysis was performed by unweighted nonlinear regression using GraFit, version 5 (Erithacus Software Ltd, Horley, UK) and an F‐test (Mannervik, 1982) was used to identify the most suitable equation.
4.11. Xenograft studies
Female athymic mice (nu/nu: Alpk; Alderley Park) and rats (RH‐rnu/rnu; Harlan, France) were housed and maintained as previously described (Wilkinson et al., 2007). All animal studies were conducted in accordance with the UK Home Office Animal (Scientific Procedures) Act 1986 and French Ministries of Agriculture and Research (Directive N86/609 CEE du 24 Nov. 1986). Src3T3 and human tumor lines (as indicated in Table 3) were inoculated subcutaneously in the left flank of animals. Tumor growth was monitored by bi‐dimensional caliper measurements twice weekly. The tumor volume was calculated by the following formula: (length×width)×√(length×width)×(π/6) and supported by excision and weighing of tumors at the end of the studies. Dosing started when the average tumor volume reached 0.2–0.5cm3 (except MDA‐MB‐231 and HT29; see Table 3). Animals were treated once daily by oral gavage with either vehicle alone or AZD0530 6.25–50mg/kg for 10–91 days (Table 3). Tumor growth inhibition was calculated as described previously (Wedge et al., 2005). For pharmacokinetic and pharmacodynamic analysis animals were humanely sacrificed and samples (plasma and tumor) were collected. Tumor samples were homogenized with 5 volumes of water and extracted with chloroform. Plasma and tumor samples were analyzed for AZD0530 concentration using high‐performance liquid chromatography with tandem mass spectrometric detection after solid‐phase extraction.
4.12. Metastasis studies
Female nude mice (nu/nu Swiss strain) aged 7 weeks old were injected subcutaneously with 5×106 NBT‐II cells. Treatment with AZD0530 at 10 (n=14), 25 (n=14) or 50mg/kg/day po (n=7) was initiated on the same day as cell inoculation. In an additional cohort of seven mice, treatment with AZD0530 50mg/kg/day po was initiated one week after inoculation. Treatment with AZD0530 continued for 2 months. Control mice received vehicle alone. Tumor growth was assessed weekly by measuring two perpendicular diameters with a caliper. Tumor volume (V) was calculated as follows: V=a 2×b/2 where a=tumor width (mm) and b=tumor length (mm). When the tumors reached a volume of 1500–2000mm3, the mice were sacrificed and lymph nodes harvested. The presence of macroscopic metastases in the lymph nodes, liver, and lungs was assessed at autopsy. To assess the presence of micrometastases, fragments of lymph nodes or other organs (liver, lungs) were placed into culture and examined microscopically at various times after seeding for the presence of epithelial foci derived from metastatic cells.
4.13. Immunohistochemistry
Sections of paraffin‐embedded xenograft tissue (4μm) were dewaxed and heated in a pressure cooker in high‐pH EDTA buffer for antigen retrieval. Endogenous peroxidase activity was blocked using H2O2, and sections were incubated in buffer containing normal goat serum to minimize non‐specific protein binding. Sections were incubated with primary antibody (0.5mg/mL rabbit polyclonal IgG anti‐FAK [pY861], Biosource UK #44‐626; or 1.22mg/mL rabbit polyclonal IgG anti paxillin [pY31], Biosource UK #44‐720) for 1h at room temperature, followed by washing and application of the EnVision+ detection system (Dako Denmark A/S, #K4010), with visualization using horseradish peroxidase and diaminobenzidine. Sections were counterstained with hematoxylin and eosin, and dehydrated through alcohol and xylene before mounting. For pharmacodynamic analyses, staining intensity was scored semi‐quantitatively in a blinded fashion on a scale of 0–4.
Acknowledgments
AstraZeneca supported this work, and all authors were employees of AstraZeneca at the time of the work. We thank Drs. Ludovic Otterbein, Wenging Xu, Richard Pauptit and colleagues (Global Structure Chemistry, AstraZeneca, Alderley Park, Macclesfield, UK) who determined the crystal structure of AZD0530, the CDMG (Alderley Park, UK) and Oncodesign (Dijon, France) for in vivo support, and Matt Lewis of Mudskipper Bioscience for medical writing support.
Green Tim P., Fennell Mike, Whittaker Robin, Curwen Jon, Jacobs Vivien, Allen Jack, Logie Armelle, Hargreaves Judith, Hickinson D. Mark, Wilkinson Robert W., Elvin Paul, Boyer Brigitte, Carragher Neil, Plé Patrick A., Bermingham Alun, Holdgate Geoffrey A., Ward Walter H.J., Hennequin Laurent F., Davies Barry R., Costello Gerard F., (2009), Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530, Molecular Oncology, 3, doi: 10.1016/j.molonc.2009.01.002.
References
- Avizienyte, E. , Wyke, A.W. , Jones, R.J. , McLean, G.W. , Westhoff, M.A. , Brunton, V.G. , Frame, M.C. , 2002. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat. Cell Biol.. 4, 632–638. [DOI] [PubMed] [Google Scholar]
- Bolen, J.B. , Veillette, A. , Schwartz, A.M. , DeSeau, V. , Rosen, N. , 1987. Activation of pp60c-src protein kinase activity in human colon carcinoma. Proc. Natl. Acad. Sci. U.S.A.. 84, 2251–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, B. , Bourgeois, Y. , Poupon, M.F. , 2002. Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene. 21, 2347–2356. [DOI] [PubMed] [Google Scholar]
- Boyer, B. , Roche, S. , Denoyelle, M. , Thiery, J.P. , 1997. Src and Ras are involved in separate pathways in epithelial cell scattering. EMBO J.. 16, 5904–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges, A.J. , 2001. Chemical inhibitors of protein kinases. Chem. Rev.. 101, 2541–2572. [DOI] [PubMed] [Google Scholar]
- Carragher, N.O. , Walker, S.M. , Scott Carragher, L.A. , Harris, F. , Sawyer, T.K. , Brunton, V.G. , Ozanne, B.W. , Frame, M.C. , 2006. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene. 25, 5726–5740. [DOI] [PubMed] [Google Scholar]
- Cartwright, C.A. , Eckhart, W. , Simon, S. , Kaplan, P.L. , 1987. Cell transformation by pp60c-src mutated in the carboxy-terminal regulatory domain. Cell. 49, 83–91. [DOI] [PubMed] [Google Scholar]
- Chu, I. , Sun, J. , Arnaout, A. , Kahn, H. , Hanna, W. , Narod, S. , Sun, P. , Tan, C.K. , Hengst, L. , Slingerland, J. , 2007. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 128, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri, B.P. , Paul, R. , Schwartzberg, P.L. , Hood, J.D. , Leng, J. , Cheresh, D.A. , 1999. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell. 4, 915–924. [DOI] [PubMed] [Google Scholar]
- Emaduddin, M. , Bicknell, D.C. , Bodmer, W.F. , Feller, S.M. , 2008. Cell growth, global phosphotyrosine elevation, and c-Met phosphorylation through Src family kinases in colorectal cancer cells. Proc. Natl. Acad. Sci. U.S.A.. 105, 2358–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame, M.C. , 2002. Src in cancer: de-regulation and consequences for cell behaviour. Biochim. Biophys. Acta. 1602, 114–130. [DOI] [PubMed] [Google Scholar]
- Friedl, P. , Wolf, K. , 2003. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer. 3, 362–374. [DOI] [PubMed] [Google Scholar]
- Fu, Y.N. , Yeh, C.L. , Cheng, H.H. , Yang, C.H. , Tsai, S.F. , Huang, S.F. , Chen, Y.R. , 2008. EGFR mutants found in non-small cell lung cancer show different levels of sensitivity to suppression of Src: implications in targeting therapy. Oncogene. 27, 957–965. [DOI] [PubMed] [Google Scholar]
- Golas, J.M. , Arndt, K. , Etienne, C. , Lucas, J. , Nardin, D. , Gibbons, J. , Frost, P. , Ye, F. , Boschelli, D.H. , Boschelli, F. , 2003. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res.. 63, 375–381. [PubMed] [Google Scholar]
- Green, T. , Hennequin, L.F. , Ple, P.A. , Jones, R.J. , Clack, G. , Gallagher, N. , 2005. Pre-clinical and early clinical activity of the highly selective, orally available, dual Src/Abl kinase inhibitor AZD0530. Proc. Am. Assoc. Cancer Res.. 46, abst SY13-3 [Google Scholar]
- Helfrich, B. , Frederick, B. , Raben, D. , Bunn, P.A. , 2006. The dual-specific Src/Abl kinase inhibitor AZD0530 inhibits in vitro growth and induces apoptosis in non-small-cell lung cancer lines. Eur. J. Cancer. (Suppl. 4) 177 [Google Scholar]
- Hennequin, L.F. , Allen, J. , Breed, J. , Curwen, J. , Fennell, M. , Green, T.P. , Lambert-van der, B.C. , Morgentin, R. , Norman, R.A. , Olivier, A. , 2006. N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem.. 49, 6465–6488. [DOI] [PubMed] [Google Scholar]
- Hiscox, S. , Morgan, L. , Green, T.P. , Barrow, D. , Gee, J. , Nicholson, R.I. , 2006. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res. Treat.. 97, 263–274. [DOI] [PubMed] [Google Scholar]
- Huse, M. , Kuriyan, J. , 2002. The conformational plasticity of protein kinases. Cell. 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Irby, R.B. , Mao, W. , Coppola, D. , Kang, J. , Loubeau, J.M. , Trudeau, W. , Karl, R. , Fujita, D.J. , Jove, R. , Yeatman, T.J. , 1999. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet.. 21, 187–190. [DOI] [PubMed] [Google Scholar]
- Irby, R.B. , Yeatman, T.J. , 2002. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res.. 62, 2669–2674. [PubMed] [Google Scholar]
- Jenkins, W.T. , 1991. The pyruvate kinase-coupled assay for ATPases: a critical analysis. Anal. Biochem.. 194, 136–139. [DOI] [PubMed] [Google Scholar]
- Jones, R.J. , Avizienyte, E. , Wyke, A.W. , Owens, D.W. , Brunton, V.G. , Frame, M.C. , 2002. Elevated c-Src is linked to altered cell-matrix adhesion rather than proliferation in KM12C human colorectal cancer cells. Br. J. Cancer. 87, 1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson, R. , Kullman-Magnusson, M. , Hamalainen, M.D. , Remaeus, A. , Andersson, K. , Borg, P. , Gyzander, E. , Deinum, J. , 2000. Biosensor analysis of drug–target interactions: direct and competitive binding assays for investigation of interactions between thrombin and thrombin inhibitors. Anal. Biochem.. 278, 1–13. [DOI] [PubMed] [Google Scholar]
- Kmiecik, T.E. , Shalloway, D. , 1987. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 49, 65–73. [DOI] [PubMed] [Google Scholar]
- Lesslie, D.P. , Gallick, G.E. , 2005. Src family kinases as regulators of angiogenesis: therapeutic implications. Curr. Cancer Ther. Rev.. 1, 45–50. [Google Scholar]
- Lombardo, L.J. , Lee, F.Y. , Chen, P. , Norris, D. , Barrish, J.C. , Behnia, K. , Castaneda, S. , Cornelius, L.A. , Das, J. , Doweyko, A.M. , 2004. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem.. 47, 6658–6661. [DOI] [PubMed] [Google Scholar]
- Lozzio, C.B. , Lozzio, B.B. , 1975. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 45, 321–334. [PubMed] [Google Scholar]
- Manley, P.W. , Cowan-Jacob, S.W. , Mestan, J. , 2005. Advances in the structural biology, design and clinical development of Bcr–Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim. Biophys. Acta. 1754, 3–13. [DOI] [PubMed] [Google Scholar]
- Mannervik, B. , 1982. Regression analysis, experimental error, and statistical criteria in the design and analysis of experiments for discrimination between rival kinetic models. Methods Enzymol.. 87, 370–390. [DOI] [PubMed] [Google Scholar]
- Martin, G.S. , 2001. The hunting of the Src. Nat. Rev. Mol. Cell Biol.. 2, 467–475. [DOI] [PubMed] [Google Scholar]
- Metcalf, C.A. , van Schravendijk, M.R. , Dalgarno, D.C. , Sawyer, T.K. , 2002. Targeting protein kinases for bone disease: discovery and development of Src inhibitors. Curr. Pharm. Des.. 8, 2049–2075. [DOI] [PubMed] [Google Scholar]
- Nam, J.S. , Ino, Y. , Sakamoto, M. , Hirohashi, S. , 2002. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin. Cancer Res.. 8, 2430–2436. [PubMed] [Google Scholar]
- Noble, M.E. , Endicott, J.A. , Johnson, L.N. , 2004. Protein kinase inhibitors: insights into drug design from structure. Science. 303, 1800–1805. [DOI] [PubMed] [Google Scholar]
- Oneyama, C. , Hikita, T. , Nada, S. , Okada, M. , 2008. Functional dissection of transformation by c-Src and v-Src. Genes Cells. 13, 1–12. [DOI] [PubMed] [Google Scholar]
- Phillips, K.A. , Parikh, N.U. , Park, S.I. , Kuwai, T. , Nokomura, T. , Green, T.P. , Gallick, G.E. , 2007. Inhibition of colon tumor metastasis in orthotopic nude mouse models with the dual-selective Src/Abl kinase inhibitor, AZD0530. Mol. Cancer Ther.. 6, abst PR-11 [Google Scholar]
- Playford, M.P. , Schaller, M.D. , 2004. The interplay between Src and integrins in normal and tumor biology. Oncogene. 23, 7928–7946. [DOI] [PubMed] [Google Scholar]
- Plé, P.A. , Green, T.P. , Hennequin, L.F. , Curwen, J. , Fennell, M. , Allen, J. , Lambert-van der Brempt, C. , Costello, G. , 2004. Discovery of a new class of anilinoquinazoline inhibitors with high affinity and specificity for the tyrosine kinase domain of c-Src. J. Med. Chem.. 47, 871–887. [DOI] [PubMed] [Google Scholar]
- Rosen, N. , Bolen, J.B. , Schwartz, A.M. , Cohen, P. , DeSeau, V. , Israel, M.A. , 1986. Analysis of pp60c-src protein kinase activity in human tumor cell lines and tissues. J. Biol. Chem.. 261, 13754–13759. [PubMed] [Google Scholar]
- Serrels, A. , Macpherson, I.R. , Evans, T.R. , Lee, F.Y. , Clark, E.A. , Sansom, O.J. , Ashton, G.H. , Frame, M.C. , Brunton, V.G. , 2006. Identification of potential biomarkers for measuring inhibition of Src kinase activity in colon cancer cells following treatment with dasatinib. Mol. Cancer Ther.. 5, 3014–3022. [DOI] [PubMed] [Google Scholar]
- Shor, A.C. , Keschman, E.A. , Lee, F.Y. , Muro-Cacho, C. , Letson, G.D. , Trent, J.C. , Pledger, W.J. , Jove, R. , 2007. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res.. 67, 2800–2808. [DOI] [PubMed] [Google Scholar]
- Song, L. , Morris, M. , Bagui, T. , Lee, F.Y. , Jove, R. , Haura, E.B. , 2006. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res.. 66, 5542–5548. [DOI] [PubMed] [Google Scholar]
- Sullivan, J.E. , Holdgate, G.A. , Campbell, D. , Timms, D. , Gerhardt, S. , Breed, J. , Breeze, A.L. , Bermingham, A. , Pauptit, R.A. , Norman, R.A. , 2005. Prevention of MKK6-dependent activation by binding to p38alpha MAP kinase. Biochemistry. 44, 16475–16490. [DOI] [PubMed] [Google Scholar]
- Summy, J.M. , Gallick, G.E. , 2003. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev.. 22, 337–358. [DOI] [PubMed] [Google Scholar]
- Talamonti, M.S. , Roh, M.S. , Curley, S.A. , Gallick, G.E. , 1993. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J. Clin. Invest.. 91, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S.M. , Brugge, J.S. , 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol.. 13, 513–609. [DOI] [PubMed] [Google Scholar]
- Trevino, J.G. , Summy, J.M. , Lesslie, D.P. , Parikh, N.U. , Hong, D.S. , Lee, F.Y. , Donato, N.J. , Abbruzzese, J.L. , Baker, C.H. , Gallick, G.E. , 2006. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am. J. Pathol.. 168, 962–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valles, A.M. , Beuvin, M. , Boyer, B. , 2004. Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. J. Biol. Chem.. 279, 44490–44496. [DOI] [PubMed] [Google Scholar]
- Vultur, A. , Buettner, R. , Kowolik, C. , Liang, W. , Smith, D. , Boschelli, F. , Jove, R. , 2008. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol. Cancer Ther.. 7, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedge, S.R. , Kendrew, J. , Hennequin, L.F. , Valentine, P.J. , Barry, S.T. , Brave, S.R. , Smith, N.R. , James, N.H. , Dukes, M. , Curwen, J.O. , 2005. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res.. 65, 4389–4400. [DOI] [PubMed] [Google Scholar]
- Wilkinson, R.W. , Odedra, R. , Heaton, S.P. , Wedge, S.R. , Keen, N.J. , Crafter, C. , Foster, J.R. , Brady, M.C. , Bigley, A. , Brown, E. , 2007. AZD1152, a selective inhibitor of aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res.. 13, 3682–3688. [DOI] [PubMed] [Google Scholar]
- Wolf, K. , Mazo, I. , Leung, H. , Engelke, K. , von Andrian, U.H. , Deryugina, E.I. , Strongin, A.Y. , Brocker, E.B. , Friedl, P. , 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol.. 160, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]