

Abstract
Biomarker measurements have become an essential component of oncology drug development, particularly so in this era of targeted therapies. Such measurements ensure that clinical studies are testing our biological hypotheses and can help make the difficult decisions required to choose which drugs to stop developing or de‐prioritise. For those drugs taken forward, biomarker measurements may also help choose the appropriate dose, schedule and patient population. In this review we discuss the intrinsic properties of biological sample based efficacy measurements and how these relate to their implementation in oncology drug development by way of points to consider and examples.
Keywords: Biomarker, Drug, Oncology, Surrogate, Tumour
1. Biomarkers, clinical endpoints and utility
The definitions of a biomarker, clinical endpoint and surrogate endpoint have been generally agreed upon for some time (see Table 1). Furthermore, the perceived importance of biomarkers to drug development has resulted in several recent initiatives and contributions from public health agencies, regulatory authorities and industry (see http://www.biomarkersconsortium.org). A recent paper from Altar et al. (2008) discusses a prototypical process for creating evidentiary standards for biomarkers and diagnostics – a path that quite rightly begins with the purpose of the biomarker. The purpose or “intended use” of a biomarker needs to be the uppermost in the minds of all involved in biomarker development as it dictates timelines, analytical specifications, study design and biomarker success criteria. We propose that a useful biomarker must have the attributes described in Table 1. Briefly, when there is a decision to be made, a useful biomarker is capable of informing the risk/benefit ratio and has some advantages and/or superiority over alternative existing (often cheaper) approaches.
Table 1.
Definitions of biomarkers.
| Term | Definition |
|---|---|
| 1. Biological Marker (Biomarker)∗ | A characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention. |
| 2. Clinical Endpoint∗ | A characteristic or variable that reflects how a patient feels or functions, or how long a patient survives. |
| 3 Surrogate Endpoint∗ | A biomarker intended to substitute for a clinical endpoint. A clinical investigator uses epidemiologic, therapeutic, pathophysiologic, or other scientific evidence to select a surrogate endpoint that is expected to predict clinical benefit, harm, or lack of benefit or harm. |
| 4. Useful Biomarker | • Informs risk/benefit ratio when there is a decision to be made. |
| • Does so in a better/faster/earlier/cheaper way than existing approaches. | |
| • Generally applicable: sample and technology must be available/accessible. | |
| • Has known identity(ies). |
Definitions 1, 2 and 3 from De Gruttola et al. (2001).
Unfortunately, most of us are readily familiar with the type of information that we would like to see from the perspective of a patient or a care‐giver (see Table 2). Clearly the intended use of the biomarker dictates the goal of assay development. For example an assay for particular analyte may be suitable for monitoring the impact of therapy but even an apparently low false positive rate may render the assay wholly inappropriate for an early cancer screening program. It follows therefore that in order to use biomarkers to aid the development of new drugs it is necessary to understand what decisions a pharmaceutical company faces in developing a single candidate drug for an oncology indication and in prioritising limited resources between multiple candidates (see Figure 1). We will discuss the use of biological sample based, efficacy biomarkers in oncology drug development from small first‐time in man (FTIM) to large drug registration studies. However, logistical and ethical considerations mean that non‐ or minimally invasive alternatives should always be considered in parallel (Workman et al., 2006).
Table 2.
Useful information for cancer management.
| Questions from patients and potential patients |
|---|
| • What is my lifetime risk of cancer? |
| • Have I got cancer? |
| • What type of cancer have I got and therefore what is my prognosis? |
| • What treatment is best for me? |
| • Is my treatment working? |
| • Will my cancer come back? |
Figure 1.
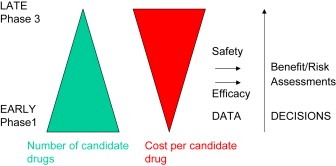
Schematic representing the role of measurements made during drug development. For ultimate drug approval risk/benefit is assessed by regulatory authorities. Prior to that, and providing a drug is judged safe, efficacy estimates are made to in an attempt to guide internal decisions and prioritise finite resources to the most promising candidates.
2. Measurements
Biomarker assays are characterised in terms of their sensitivity, specificity, limit of detection, limit of quantification and variability. We would argue that a thorough consideration of the characteristics listed in Table 3 of both phenotype and candidate biomarker can also save considerable time and effort in a biomarker development program by aiding study design, interpretation and decision making.
Table 3.
Properties of biomarker and phenotypic measurements.
| Property | Explanation | Examples and impact |
|---|---|---|
| Dynamism | Measurements can be static or dynamic. | A static biomarker cannot correlate with a dynamic phenotype if the dynamic phenotype cannot predict itself on repeat measurement. Heritable genotype is a static measurement, tumour somatic mutation status is dynamic. Care should be taken to check that dynamic changes during the time elapsed between a biomarker measurement and clinical correlate does not invalidate any potential association. Genotype is a good example of a static (subject level) biomarker which is often proposed to impact on plasma pharmacokinetics a dynamic (subject level) phenotype. Intra‐patient versus inter‐patient reproducibility of pharmacokinetic data will therefore aid an assessment of the likelihood of success of this approach before a single DNA sample has been isolated. |
| Level | Measurements can be made at the level of a thin slice of tissue, single lesion or subject level. | Many candidate predictive tumour markers are measured on single sections of tumour tissue and the results automatically extrapolated as a patient level attribute. Intra‐patient (but inter‐sample and inter‐lesion) correlations of biomarker data can be used to eliminate candidate biomarkers. |
| Average or single molecule detection | Classical assay development methods were established on analytes present at the level of billions of molecules. Recent technologies, especially using nucleic acids, take us down orders of magnitude to levels where stochastic variation must be considered. | The Jak2 mutation assay from Ipsogen is suggested to be used at an input of 25ng of DNA. This corresponds to 10 000 copies of the genome (5000 cells). Therefore the assay may be useful for detecting the presence of high levels of mutant sequence but will not be useful for monitoring residual disease burden once mutant granulocytes drop to a level where the input recommendations preclude the presence of mutant sequence. |
| Univariate versus multivariate markers | Composite measurements should have clear rules to derive a final easy to interpret multivariate index. | The rules governing the combination of target, non‐target and new lesions for determining RECIST measurements are a good example of deriving a single patient level index for decision making. Similarly the development of the Oncotype Dx assay is a good example for molecular biomarkers (Paik et al., 2004). |
| Continuous/categorical | Measurements can be intrinsically continuous or discrete. Continuous measurements can be made discrete via cut‐offs. | Decisions are invariably discrete and therefore care must be taken to provide a clear message with output from continuous markers. |
2.1. Level of attribution
In order to correlate a phenotype and a biomarker, consideration should be given to the level at which the measurement is attributed. The level at which a marker is measured is often not the same level to which it is attributed or acted upon. Germline genetic variants are measured at the level of the individual but the results may have consequences at the level of the family. Conversely, individuals are diagnosed with cancer but conceptually the diagnosis may often be attributed to one of a pair of organs subject to identical genetic and environmental influences (and data from the contra‐lateral “twin” may provide clues as to the impact of these influences). Generally speaking clinical endpoints of interest, such as survival or progression free survival, are usually measured or attributed at the level of the patient or subject. Many biomarker measurements are also at subject level (heritable genotypes, circulating PSA). However, some are attributes of a thin cross‐section of a narrow core of tissue from one of potentially many tumours within an individual. It is rare that efforts are made to validate the assumption that the measurement made at one level can be attributed to another by measuring within subject inter‐biopsy or inter‐lesion variability and yet it is a prerequisite for correlation with subject level measurements and hence utility (Cleator et al., 2006; Kalikaki et al., 2008).
2.2. Dynamism
Conceptually similar to an appreciation of measurement level is consideration of temporal variation. Cancer is a phenotypically and molecularly progressive disease in which tumours evolve over time. Tumour biomarkers are likely to do the same, and even if they do not, the molecular context in which they find themselves almost invariably does. Therefore timing of measurements and sufficient longitudinal granularity can be critical both in assessing or predicting the impact of a therapeutic intervention (see later). The routine assumption of the reproducibility of a patient's response to a therapeutic intervention is elegantly discussed by Senn (2004).
2.3. Technical and biological reproducibility
Similar arguments can be applied to the process validity that leads from data acquisition to subject level attribution of phenotypic or biomarker data. How do the results differ if a different lesion is chosen for measurement of length or biomarker levels? If a different pathologist selects the region of tumour to be macrodissected? If a different panel of experts reviews images or if the same panel reviews them a second time? Such information on phenotypes is vital in setting realistic expectations for the possible sensitivity and specificity of a biomarker as these are unlikely to be higher than the repeat concordance of the phenotypic measurement itself.
2.4. Analyte abundance
Traditional assay development methods apply well to assays in homogenous media where analyte abundance is such that it can be described by the statistics of populations. Historically, we have struggled to make tissue based assays such as tumour immunohistochemistry anything more than semi‐quantitative. Newer, highly sensitive nucleic acid technologies take us into the realms of stochastic variation where a signal may disappear because the analyte is physically not present, rather than reaching the detection limit of an instrument. In such situations extreme care must be taken to discriminate between positive, negative and unknown results (see Hodgson et al., 2002).
2.5. Nature and number of variables
Data may initially be univariate or multivariate, continuous or categorical. Ultimately the goal is to deliver a single “result” or index via a pre‐defined set of rules and although this can remain a continuous value for practical purposes a cut‐off may need to be set as the intended use will usually be to influence a categorical decision. The criteria for RECIST evaluation offer a good example of a multivariate approach to deliver a categorical patient level result and the Oncotype Dx assay is a good example of a multivariate continuous index (Eisenhauer et al., 2009; Paik et al., 2004). In general it is a good idea to maintain data in their continuous form until the final step of data processing as continuous data can always be made categorical but the reverse is not true. Further, we make a clear distinction between data for which the biology is intrinsically categorical (eg genotype) and data which is rendered categorical by the means of measurement such as ER, PR levels – continuous by radioligand and RTPCR and categorical by IHC, respectively (Badve et al., 2008).
In summary, it is important to understand the intrinsic biology and assay platform, validate the level to which a measurement may be attributed, take samples at appropriate times and examine how good the biomarker and phenotype are at predicting themselves on repeat biological or technical measurement.
3. Study design
3.1. Use your ESP!
Technical biomarker, phenotypic, clinical relevance and statistical expertise are all required to ensure that precious human samples are only procured for well‐designed studies after ethical approval. Frequently these samples are the result of an altruistic submission to invasive procedures unrelated to the clinical care of the patient from which they are obtained. The number of patients (and associated costs) required for a study requires data upon biomarker variability/prevalence and target effect size. An estimate is also required of what we have referred to as the evaluable sample population or ESP (see Figure 2). The ESP comprises the patients for whom biomarker data is available and analysed. The ESP is a sub‐set of the patients who comply with study procedures (per protocol population), which in turn is a sub‐set of the patients recruited onto a trial (intent to treat population). The ESP must be estimated by considering the whole process from sample access, through logistics and prequalification requirements to assay performance. If the goal is to “bolt‐on” biomarker work without changing study design then the biomarker's evaluability rate, variability and target effect size must be compatible with the existing study design. Brady et al. (2007) demonstrated these principles for gene expression measurements in human hair in the context of a typical phase 1 oncology study. Ultimately, however good they are in model systems the markers of interest must be measurable in the human tissue/fluid of interest.
Figure 2.
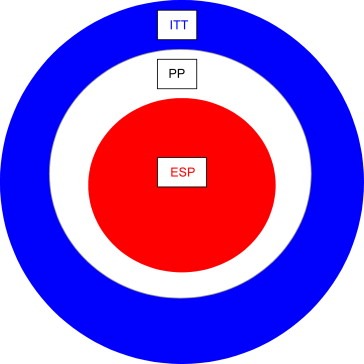
Diminishing returns. The evaluable sample population (ESP) is the population available for linking the effect of drug to a biomarker measurement. The ESP is a sub‐set of the per protocol population, which is itself a sub‐set of the intent to treat (ITT) population. An estimate of the ESP is vital to size a study correctly and must take into account attrition in obtaining samples and in obtaining definitive data from the samples. Even if a study can be correctly powered, significant attrition in obtaining either sample or result suggests an approach that would have difficulty in being translated into clinical practice.
3.2. “Cum on Feel the Noize!” – Noddy Holder & James Lea
It is therefore imperative to discover and develop biomarkers in representative patients and sample types. Take a biomarker whose intended use is a decision based on the output of a study with one or two scientifically excellent and motivated centres of excellence, as is often the case in early efficacy studies. Here one can afford to stipulate strict study inclusion criteria and relatively complex and onerous sample collection procedures. However, a biomarker intended as a patient selection diagnostic, will require different properties, having to routinely return decisions on a local basis at thousands of hospitals worldwide. This biomarker must be robust to the vagaries of variations of clinical practice and be developed in studies which avoid restrictive inclusion and exclusion criteria, non‐robust sample collection, transport and storage requirements. Samples representative of those where the decision‐making data will be derived are essential for assay development. For a candidate predictive diagnostic assay samples from lots of different centres, with all the accompanying noise, are required. AstraZeneca, and others, have proposed a generic suite of studies to qualify a biomarker (Figure 3). Note that the requirement for dynamism to drug is unique to pharmacodynamic markers and that variability may be conceptually replaced by prevalence for a predictive biomarker (see later).
Figure 3.
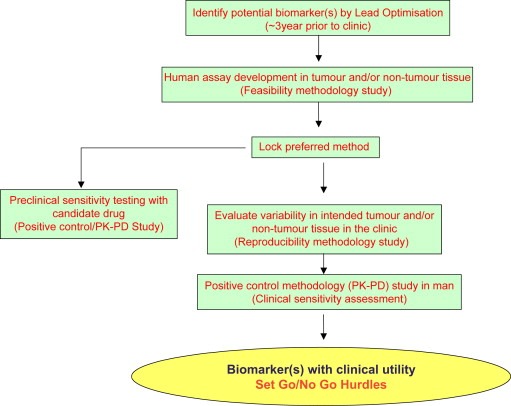
Roadmap for qualification of a biomarker in oncology drug development.
3.3. Where do markers come from?
Broadly speaking a drug's mode of action will suggest candidate molecular biomarker identities. The role of “hypothesis free” “omic” approaches is relatively limited for pharmacodynamic markers but is beginning to be explored for predictive markers. Where biomarker discovery is undertaken strong consideration should be given to phenotypic purity and selection of phenotypic extremes wherever possible. For example, the strongest motivation to discover predictive markers occurs when extraordinarily good responses are seen in a sub‐set of patients.
3.4. “Not everything that matters can be measured, and not everything that is measured matters” – Albert Einstein
At all times explicit consideration and comparison should be given to alternative approaches to avoid “over‐engineering” an expensive biomarker approach where simpler alternatives for decision making exist. Drug development is generally not the arena for biomarker technology development and practitioners are generally looking for precedented approaches where possible.
4. Drug development
4.1. “Prediction is very difficult, especially when it pertains to the future” – Mark Twain
It is useful to divide the use of efficacy biomarkers in oncology drug development into an “early” and a “late” phase. In the early phase stakeholders for decision making are largely internal to a pharmaceutical company, whereas in the late phase many external stakeholders are external to a company, eg regulatory authorities. Clearly a pharmaceutical company needs to manage the risk it carries forwards across its early candidate drug portfolio and make go/no‐go decisions accordingly. The teams responsible for “go” projects will then have to make crucial decisions around patient populations, dose and schedule. It could be cogently argued, however, that external stakeholders should also be included in earlier decision processes if the consequences are that a potentially useful drug fails because it is targeted to the wrong patient group at the wrong dose and schedule.
4.2. Biomarkers for “Early” drug development
The role of biomarkers in early drug development has been extensively discussed (Workman, 2003; Smethurst and Hughes, 2005). Biomarkers deployed at this stage are used to monitor molecular activity as the early part of the pharmacological “audit trail”. Generally speaking the further a drug can be shown to impact on biomarkers leading from proximal mode of action to clinical endpoint the higher the level of confidence in the drug and the better it's claim to development funding. Proof of mechanism biomarkers are drug or drug class specific and confirm that trials are testing the hypothesis that inhibiting a particular target in tumour tissue, rather than the hypothesis that administering a particular dose of drug at a particular frequency, has clinical benefit. An example of a proof of mechanism marker is given in Adjei et al. (2008, Figure 1) where receptor phosphorylation is reduced on drug treatment. In addition to confirming that the drug is reaching and inhibiting its target ideally one would confirm that this inhibition is maintained between doses at a level that correlated with downstream tumour effects in pre‐clinical models. Proximal to proof of mechanism, biomarkers are no longer drug specific and may be used to confirm anticipated downstream effects such as reduced proliferation or increased apoptosis (Greystoke et al., 2008). An example is given in Adjei et al. (2008) where the reduction in receptor phosphorylation is accompanied by a reduction in expression of a proliferation marker.
Practically, such markers demonstrate that a drug is being used at or above its minimally biologically effective dose and may aid decisions around dose and schedule. However, given the unknown or hazy relationship between the magnitude of molecular and clinical response, any dose setting based on molecular efficacy will generally be accompanied by testing of the empirically derived maximum tolerated dose and relief of an obvious symptom, established biomarkers such as PSA or CA125 or simply time on treatment may be the best indication of an efficacious dose.
A new oncology drug will often have a toxicological profile incompatible with testing in healthy human volunteers. Initial trials will typically be run in patients for whom existing treatment options will have been exhausted. Human, pragmatic and scientific issues frequently pull in different directions and all have to be balanced in our drive to extract valuable decision‐making data pertaining to the desired biological action of new pharmacological agents. A frequent compromise is to obtain data from more readily accessible, minimally invasive sources such as blood or hair. In this way valuable longitudinal data can be obtained but ultimately data from surrogate tissues cannot replace data from the tumour itself. Preoperative, longitudinal biopsy studies are scientifically attractive but must be considered carefully as they involve repeated invasive procedures and the participation of patients who may stand a high chance of cure from existing interventions (Dowsett, 2003). Minimally invasive, cheap to run tumour derived circulating marker assays based on simple to perform sample acquisition have the potential to offer the best of both worlds. CA125 and PSA measurements are widely used in ovarian and prostate cancer trials, respectively. Similar measurements have the potential to offer the longitudinal granularity compatible with the dynamism inherent in monitoring the rapidly progressing patients often recruited into early oncology trials. In contrast, although hugely informative, imaging measurements have a weakness in that they must be performed at significant time intervals and a “baseline scan” may actually be performed up to 4 weeks before administration of study drug. In a rapidly progressing patient such timing discrepancies may be crucial to the interpretation of the effect of intervention. Emerging tumour derived circulating markers, such as those of cell death and tumour burden (circulating tumour DNA or cells) may prove useful in providing an early read out of molecular efficacy (Fleischhacker and Schmidt, 2007; Allard et al., 2004). Circulating tumour cells (CTCs) could conceivably serve as a surrogate marker (see later) of clinical benefit although their use may be limited in early, short trials if they are undetectable at baseline in the majority of patients (Cristofanilli et al., 2004, 2005, 2006 July 15, 2005).
Non‐dynamic, disease defining, biomarkers may also be used to select a proof of principle population for “expansion phases” in early trials. An expansion phase is often included in a phase 1 after safety and tolerability are addressed by recruiting a loosely defined patient population to ascending doses of drug. In the expansion phase, performed at a biologically efficacious dose, a tightly defined patient population whose disease is strongly associated with the molecular hypothesis under consideration can provide a strong “no‐go” decision if proof of mechanism is demonstrated, but no benefit accrues to the patients. Examples include the selective recruitment of BRCA mutated and PTEN mutated/Cowden's syndrome cancer patients for the evaluation of PARP and Tor inhibitors respectively (ClinicalTrials.gov Identifier: NCT00647062 and NCT00722449). Such an approach may naturally lead to a “personalised medicine” approach if data are encouraging and a route to market can be found.
4.3. Biomarkers for “Late” drug development
4.3.1. Surrogate biomarkers
Armed with an idea of dose and schedule, a drugs route to market must be plotted balancing speed, return on investment, clinical need and likelihood of success. True surrogate markers, as defined in Figure 1, hold out huge promise to increase speed to market and bringing forward confidence in a successful outcome. However more compact, efficient and smaller clinical studies can only follow once surrogate status has been demonstrated and accepted. Only a small number of biomarkers have been shown to be surrogate, substitutes for clinical endpoints in some diseases. It is a considerable burden to demonstrate true surrogacy of an endpoint, requiring large randomised clinical trials that capture both the assumed surrogate and clinical outcome (Buyse and Molenberghs, 1998; Molenberghs et al., 2001). Therefore, before a surrogate endpoint can be used in a clinical trial supporting a drug approval by a regulatory agency, there is a requirement for a clinical trial, which is at least as large and uses the same endpoints, to validate the surrogate. In view of this requirement, the use of new biomarkers as substitutes for a clinical outcome(s), for the purpose of directly supporting the drug approval process, is rare and attaining surrogate status and acceptance as a substitute for a registrable clinical endpoint is a non‐trivial goal, beyond the scope of any one pharmaceutical company.
Many surrogate biomarkers in routine use today, however, did not undergo this rigorous evaluation for surrogacy, but have still been used as primary endpoints to support drug approval eg reduction of elevated arterial blood pressure to reflect the reduction in incidence of stroke, congestive cardiac failure, and sub‐sets of cardiovascular death by antihypertensive drugs; serum cholesterol for the reduction of coronary artery disease by the 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors; and response rate of malignant tumours to cytotoxic chemotherapy.
A fuller discussion of surrogacy is beyond the scope of this article and readers are referred to the many excellent reviews of statistical considerations and case studies (eg Prentice, 2007, 2009, [Link], [Link]). In addition to the rigorous statistical definitions, wider acceptance of surrogacy within clinical practice is necessary and attempts to bring private and public sectors into consortia to address the issue of surrogacy are welcome (NIH/FDA Biomarker Consortium).
4.3.2. Patient stratification or personalised medicine: can you please some of the people all of the time?
The goal of a drug company is to maximise patient benefit from a new oncology drug and traditionally this has been attempted in recognised patient populations defined by tumour type, stage and line of therapy. The literature, pre‐clinical data and/or mode of action may suggest a hypothesis that a biomarker may be useful in defining groups of patients who will have a superior benefit/risk profile when treated with a new therapy as opposed to a comparator standard of care. Such markers are described as predictive rather than simply prognostic. Predictive markers are relevant to treatment decisions as the benefit/risk ratio differs if one treatment option is chosen over an alternative.“Does anyone know the way, there's got to be a way, to Block Buster!”(Chinn/Chapman)
For many years the pharmaceutical industry was implicitly accused of preferring to receive payment for treating large populations when only a fraction of patients were likely to receive any benefit. In 2003, the then vice president for Genetics at GSK, Allen Roses, received much attention when he was quoted as saying “The vast majority of drugs – more than 90 per cent – only work in 30 or 50 per cent of the people”. Many factors, not the least of which may be patient compliance and including the optimized schedule, potentially explain this observation and the reality is that, without repeat measures of efficacy within the same individuals, we usually cannot discriminate between a drug that works in 30 or 50 per cent of people 100 per cent of the time and one which works in 100 per cent of the people 30–50% of the time (Senn, 2004; Royal Society Report, 2005).
Despite the early hype and the later caveats “personalised medicine” has been a reality in oncology for decades. Positive hormonal receptor status of tumours indicates utility of hormonal therapy in breast cancer (Harris et al., 2007). Trial design options for developing a “personalised medicine” approach are well covered elsewhere (Drug–Diagnostic Co‐Development Concept paper, 2005; Carroll, 2007; Simon, 2008). For populations defined by established biomarkers patients are recruited on the basis of local practice, however this is performed, as it is essential that the trial will reflect subsequent clinical practice. Little thought is given to recruiting “biomarker negative” patients to the same trial and it is accepted that the goal of the trial is to move towards approval in that established biomarker defined patient population. Any attempt to “improve” the biomarker assays used to identify or label patients in the trial will result in a need to ensure global access to an equivalent test at launch and many of the biomarkers pragmatically and routinely used for “personalised medicine” today are not FDA approved nor standardised in terms of reagents and methods.
A more difficult proposition is presented by a candidate drug with a new mode of action and a strong rationale for patient selection but no relevant predictive biomarker used in clinical practice. The rationale may be based on persuasive pre‐clinical data and/or clinical data demonstrating a striking differential efficacy within an established patient population. Here it may be pertinent to note that a relatively simple scientific rationale probably lies at the heart of most examples of “personalised medicine” in oncology such as the presence, level or existence of functional variants of the drug target. A company is then faced with the prospect of plotting a route to market for a drug in conjunction with developing and commercialising a new predictive biomarker. Drug–diagnostic co‐development has been discussed conceptually (Drug–Diagnostic Co‐Development Concept paper, 2005) and the challenges of reconciling the needs of the high risk/return drug industry with the low risk, high volume, low margin diagnostic industry in a single project are beyond the scope of this scientific review. A drug company's future revenue potentially depends upon their diagnostic partner delivering a test that is geographically and financially accessible to relevant patients. Ultimately such an approach needs to molecularly redefine disease, simultaneously change diagnostic and prescribing practice, such as ultimately occurred for Her2 status and predicting the benefit from therapy with trastuzumab (Harris et al., 2007).
The “ideal” novel predictive biomarker process is outlined in Table 4. A predictive hypothesis is generated from the literature or pre‐clinical data and strengthened by data from pre‐clinical models. In the next phase the biomarker(s) must be measured in samples representative of the intention to treat population to estimate evaluability and prevalence rates. Given the data on biomarker evaluability and prevalence and estimate of effect size it is then possible to design a phase 2 trial to examine the benefit/risk ratio in all potential patients within an existing indication and compare it with the benefit/risk ratio in a sub‐set of patients as defined by the candidate predictive biomarker. The phase 2 trial is effectively converted from a tool to estimate the likelihood of success in phase 3 to a tool to additionally estimate the predictive value of a biomarker and a case to invest in it's conversion to a diagnostic test.
Table 4.
Simplified overview of drug/diagnostic co‐development.
| Drug phase | Drug/biomarker interaction | Predictive biomarker/diagnostic activities |
|---|---|---|
| Pre‐clinical | Link to drug sensitivity for biomarker discovery and strength of hypothesis. | |
| Phase 1 | No meaningful data usually available. | Biomarker validation on relevant clinical samples: evaluability and prevalence. |
| Phase 2 trial against comparator in biomarker +ve and −ve patients | Indication of clinical utility of biomarker. Role of predictive biomarker in Phase 3 trial design and case for co‐development. | Biomarker status of trial participants by research use only (RUO) assay |
| Preparation for commercial launch | Conversion of RUO assay to final format. | |
| Phase 3 | Confirmation of clinical utility of biomarker. | Biomarker status of trial participants by diagnostic assay |
To date most co‐development projects have not followed this “idealised” route but it is hoped that as the industries learn to work together then success stories will become more frequent. Recent data with the EGFR inhibitor gefitinib from the IPASS clinical trial has demonstrated the tremendous potential value to patients of predictive biomarkers. In the IPASS patient population EGF receptor mutation status was strikingly shown to be predictive of benefit from treatment with gefitinib (Mok, 2008).
The remarkable results from the IPASS study may be in part due to the contemporaneous measurement of biomarker and outcome generally only afforded in a first‐line setting for tumour based tests. On longer timescales “predictive” marker levels or categories in a diagnostic biopsy may or may not be maintained in the metastatic tumours, subject to the selective pressure of several prior therapies, of a typical patient enrolled in a study with a candidate drug. Furthermore a measurement may be “static” with respect to the appropriate time‐period under consideration but may be superseded by other events, eg a mutation present in the EGFR of a diagnostic tumour sample may still be present in the tumour which gets treated several years later but it's relevance to outcome may be altered because of the acquisition of additional mutations under selective pressure of treatment in the intervening period (Costa et al., 2008). Furthermore, the ESP in the IPASS trial, which has one of the highest ESPs in a phase III study, comprised 35% of patients again highlighting a real need for a tumour derived circulating biomarker test that could (i) deliver a test to guide treatment for all patients and (ii) deliver a test result reflective of tumour status at the time of treatment choice.
5. Summary
It is an exciting time to be involved with biomarker research in oncology drug development. It is beholden on scientists in the area to maintain a continual dialogue with customers; those who must take decisions on the basis of our data. A clear line of sight to the intended use of biomarker data will help us make the best possible use of precious human samples and maximise the chances of success of the most promising therapeutic approaches.
Acknowledgements
The authors would like to express their thanks to the patients and all in the clinical, academic and industrial communities with whom they have worked together over the past years.
Hodgson Darren R., Whittaker Robin D., Herath Athula, Amakye Dereck, Clack Glen, (2009), Biomarkers in oncology drug development, Molecular Oncology, 3, doi: 10.1016/j.molonc.2008.12.002.
References
- Adjei, A.A. , Cohen, R.B. , Franklin, W. , Morris, C. , Wilson, D. , Molina, J.R. , Hanson, L.J. , Gore, L. , Chow, L. , Leong, S. , Maloney, L. , Gordon, G. , Simmons, H. , Marlow, A. , Litwiler, K. , Brown, S. , Poch, G. , Kane, K. , Haney, J. , Eckhardt, S.G. , 2008 May 1. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J. Clin. Oncol.. 26, (13) 2139–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard, W.J. , Matera, J. , Miller, M.C. , Repollet, M. , Connelly, M.C. , Rao, C. , Tibbe, A.G. , Uhr, J.W. , Terstappen, L.W. , 2004 Oct 15. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer. Res.. 10, (20) 6897–6904. [DOI] [PubMed] [Google Scholar]
- Altar, C.A. , Amakye, D. , Bounos, D. , Bloom, J. , Clack, G. , Dean, R. , Devanarayan, V. , Fu, D. , Furlong, S. , Hinman, L. , Girman, C. , Lathia, C. , Lesko, L. , Madani, S. , Mayne, J. , Meyer, J. , Raunig, D. , Sager, P. , Williams, S.A. , Wong, P. , Zerba, K. , 2008. Feb. A prototypical process for creating evidentiary standards for biomarkers and diagnostics. Clin. Pharmacol. Ther. 83, (2) 368–371. [DOI] [PubMed] [Google Scholar]
- Badve, S.S. , Baehner, F.L. , Gray, R.P. , Childs, B.H. , Maddala, T. , Liu, M.L. , Rowley, S.C. , Shak, S. , Perez, E.A. , Shulman, L.J. , 2008 May 20. Estrogen- and progesterone-receptor status in ECOG 2197: comparison of immunohistochemistry by local and central laboratories and quantitative reverse transcription polymerase chain reaction by central laboratory. J. Clin. Oncol.. 26, (15) 2473–2481. Erratum in: J. Clin. Oncol. Jul 10;26(20):3472. Perez, Edith D [corrected to Perez, Edith A] [DOI] [PubMed] [Google Scholar]
- Baker, S.G. , 2006 April 19. Surrogate endpoints: wishful thinking or reality?. J. Natl. Cancer Inst.. 98, (8) [DOI] [PubMed] [Google Scholar]
- Bouxsein, M.L. , Delmas, P.D. , 2008. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. JBMR. 23, (8) 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyse, M. , Molenberghs, G. , 1998. Criteria for the validation of surrogate endpoints in randomized experiments. Biometrics. 54, 1014–1029. [PubMed] [Google Scholar]
- Brady, G. , Hurley, P. , Grimes, E. , Herath, A. , Wilson, C. , Moore, J. , Potten, C. , Graham, A. , Haggerty, C. , Vinsun, D. , Hodgson, D. , 2007. Hair is a viable surrogate tissue for routine measurement of gene expression in clinical studies [abstract]. Proceedings of the 98th Annual Meeting of the American Association for Cancer Research; 2007 Oct 22–26; San Francisco. AACR; California (CA) 7 (Abstract no. C44) [Google Scholar]
- Carroll, K.J. , 2007 Oct–Dec. Biomarkers in drug development: friend or foe? A personal reflection gained working within oncology. Pharm. Stat.. 6, (4) 253–260. [DOI] [PubMed] [Google Scholar]
- Cleator, S.J. , Powles, T.J. , Dexter, T. , Fulford, L. , Mackay, A. , Smith, I.E. , Valgeirsson, H. , Ashworth, A. , Dowsett, M. , 2006. The effect of the stromal component of breast tumours on prediction of clinical outcome using gene expression microarray analysis. Breast Cancer Res.. 8, (3) R32 (Epub 2006 Jun 21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, D.B. , Nguyen, K.S. , Cho, B.C. , Sequist, L.V. , Jackman, D.M. , Riely, G.J. , Yeap, B.Y. , Halmos, B. , Kim, J.H. , Jänne, P.A. , 2008 Nov 1. Effects of erlotinib in EGFR mutated non-small cell lung cancers with resistance to gefitinib. Clin. Cancer Res.. 14, (21) 7060–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofanilli, M. , Hayes, D. , 2004. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med.. 351, 781–791. [DOI] [PubMed] [Google Scholar]
- Cristofanilli, M. , Hayes, D. , 2005. Circulating tumor cells: a novel prognostic factor for newly diagnosed breast cancer. J. Clin. Oncol.. 23, (7) 1420–1430. [DOI] [PubMed] [Google Scholar]
- Drug–Diagnostic Co-Development Concept paper, 2005. FDA. Available from: <http://www.fda.gov/cder/genomics/pharmacoconceptfn.pdf>
- De Gruttola, V.G. , Clax, P. , DeMets, D.L. , Downing, G.J. , Ellenberg, S.S. , Friedman, L. , Gail, M.H. , Prentice, R. , Wittes, J. , Zeger, S.L. , 2001 Oct. Considerations in the evaluation of surrogate endpoints in clinical trials. Summary of a National Institutes of Health Workshop. Control Clin. Trials. 22, (5) 485–502. [DOI] [PubMed] [Google Scholar]
- Dowsett, M. , 2003 Jan. Preoperative models to evaluate endocrine strategies for breast cancer. Clin. Cancer Res.. 9, (1 Pt 2) 502S–510S. [PubMed] [Google Scholar]
- Eisenhauer, E.A. , Therasse, P. , Bogaerts, J. , Schwartz, L.H. , Sargent, D. , Ford, R. , Dancey, J. , Arbuck, S. , Gwyther, S. , Mooney, M. , Rubinstein, L. , Shankar, L. , Dodd, L. , Kaplan, R. , Lacombe, D. , Verweij, J. , 2009. Jan. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer. 45, (2) 228–247. [DOI] [PubMed] [Google Scholar]
- Fleischhacker, M. , Schmidt, B. , 2007 Jan. Circulating nucleic acids (CNAs) and cancer—a survey. Biochim. Biophys. Acta. 1775, (1) 181–232. (Epub 2006 Oct 7) [DOI] [PubMed] [Google Scholar]
- Greystoke, A. , Cummings, J. , Ward, T. , Simpson, K. , Renehan, A. , Butt, F. , Moore, D. , Gietema, J. , Blackhall, F. , Ranson, M. , Hughes, A. , Dive, C. , 2008 May. Optimisation of circulating biomarkers of cell death for routine clinical use. Ann. Oncol.. 19, (5) 990–995. [DOI] [PubMed] [Google Scholar]
- Harris, L. , Fritsche, H. , Mennel, R. , Norton, L. , Ravdin, P. , Taube, S. , Somerfield, M.R. , Hayes, D.F. , Bast, R.C. , American Society of Clinical Oncology, 2007 Nov 20. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol.. 25, (33) 5287–5312. [DOI] [PubMed] [Google Scholar]
- Hayes, Cristofanilli, M. , 2006 July 15. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin. Cancer Res.. 12, (14) [DOI] [PubMed] [Google Scholar]
- Hodgson, D.R. , Foy, C.A. , Partridge, M. , Pateromichelakis, S. , Gibson, N.J. , 2002 May. Development of a facile fluorescent assay for the detection of 80 mutations within the p53 gene. Mol. Med.. 8, (5) 227–237. [PMC free article] [PubMed] [Google Scholar]
- Kalikaki, A. , Koutsopoulos, A. , Trypaki, M. , Souglakos, J. , Stathopoulos, E. , Georgoulias, V. , Mavroudis, D. , Voutsina, A. , 2008 Aug 26. Comparison of EGFR and K-RAS gene status between primary tumours and corresponding metastases in NSCLC. Br. J. Cancer. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Chines, A.A. , Meredith, M.P. , 2004. Statistical validation of surrogate endpoints: is bone density a valid surrogate for fracture?. J. Musculoskelet. Neuronal Interact.. 4, (1) 64–74. [PubMed] [Google Scholar]
- Mok, 2008. Available from: <http://www.esmo.org/fileadmin/media/presentations/977/2123/3_Tony%20Mok_2008%2009%2010%20IPASS%20ESMO%20press%20conference_v3.ppt.pdf>
- Molenberghs, G. , Geys, H. , Buyse, M. , 2001. Evaluation of surrogate endpoints in randomised experiments with mixed discrete and continuous outcomes. Stat. Med.. 20, 3023–3038. [DOI] [PubMed] [Google Scholar]
- NIH/FDA Biomarker Consortium. Available from: <http://www.biomarkersconsortium.org>.
- Paik, S. , Shak, S. , Tang, G. , Kim, C. , Baker, J. , Cronin, M. , Baehner, F.L. , Walker, M.G. , Watson, D. , Park, T. , 2004 Dec 30. Multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med.. 351, (27) 2817–2826. (Epub 2004 Dec 10) [DOI] [PubMed] [Google Scholar]
- Prentice, R.L. , 1989. Surrogate endpoints in clinical trials: definitions and operational criteria. Stat. Med.. 8, 431–440. [DOI] [PubMed] [Google Scholar]
- Response evaluation criteria in solid tumors (RECIST): new guidelines. Med. Pediatr. Oncol.. 37, (1) 1–3. [DOI] [PubMed] [Google Scholar]
- Royal Society Report, 2005. Personalised medicines: hopes and realities. Available from: <http://royalsociety.org/displaypagedoc.asp?id=15874>.
- Senn, S. , 2004 Oct 23. Individual response to treatment: is it a valid assumption?. BMJ. 329, (7472) 966–968. (Review) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, R. , 2008 Oct 1. The use of genomics in clinical trial design. Clin. Cancer Res.. 14, (19) 5984–5993. [DOI] [PubMed] [Google Scholar]
- Smethurst, D. , Hughes, A. , 2005. Staged risk management of candidate drug efficacy. Int. J. Pharm. Med.. 19, (4) 227 [Google Scholar]
- Stopeck, A. , Cristofanilli, M. , 2005. Circulating tumor cells - not serum tumor markers - predict survival in metastatic breast cancer Journal of Clinical Oncology, 2005 ASCO Annual Meeting Proceedings. Vol 23, No. 16S, Part I of II (June 1 Supplement): 9516 [Google Scholar]
- Weir, C.J. , Walley, R.J. , 2006. Statistical evaluation of biomarkers as surrogate endpoints: a literature review. Stat. Med.. 25, 183–230. [DOI] [PubMed] [Google Scholar]
- Workman, P. , Aboagye, E.O. , Chung, Y.L. , Griffiths, J.R. , Hart, R. , Leach, M.O. , Maxwell, R.J. , McSheehy, P.M. , Price, P.M. , Zweit, J. , Cancer Research UK Pharmacodynamic/Pharmacokinetic Technologies Advisory Committee, 2006 May 3. Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J. Natl. Cancer Inst.. 98, (9) 580–598. (Review) [DOI] [PubMed] [Google Scholar]
- Workman, P. , 2003. How much gets there and what does it do?: the need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Curr. Pharm. Des.. 9, (11) 891–902. (Review) [DOI] [PubMed] [Google Scholar]