

Abstract
Elevated expression of the protocadherin LKC (PCDH24) in HCT116 colon carcinoma cells has been shown to induce contact inhibition, thereby completely abolishing tumor formation in vivo (Carcinogenesis, 2002; 23(7):1139–1148). To clarify the molecular mechanism behind this effect, we performed 2‐DE/MS and DNA microarray analyses in order to compare protein and gene expression patterns of parental HCT116 and PCDH24‐expressing HTC116 derivative cells. The data revealed drastic changes in phenotypic markers between parental and PCDH24‐expressing cells. We found that in PCDH24‐expressing cells β‐catenin, a major player in TCF/lef signaling, is retained in a submembranous location. β‐catenin retention coincided with a subsequent decrease in downstream targets of β‐catenin such as CD44, PLAUR, Myc, cyclin D1 and Met. From these findings we propose a novel model for the suppression of β‐catenin signaling by PCDH24 that leads to contact inhibition.
Keywords: PCDH24, Contact inhibition, βbeta;-catenin, HCT116, Actin, Vimentin
Abbreviations
- qRT‐PCR
quantitative real‐time‐PCR
- LMB
leptomycin B
- 2‐DE/MS
two‐dimensional gel electrophoresis/mass spectrometry
- PLAUR
plasminogen activator urokinase receptor
1. Introduction
Contact inhibition is one of the most drastic phenomena of normal proliferative cells, and loss of this phenomenon is a hallmark of cancers. A number of studies have investigated the participation of several molecules which are thought to play key roles in this process. Some of these molecules have also been identified in stem cell research (Takahashi et al., 2005). Thus, studying contact inhibition promises to contribute to both cancer and stem cell research.
Contact inhibition shares a signaling pathway with anchorage‐dependent growth and tumor suppression. Therefore, this phenomenon has been studied not only in normal cells but also in cancer cells derived from epithelial cells. These studies provided evidence for a pivotal role of β‐catenin in this process (Stockinger et al., 2001; Dietrich et al., 2002).
The cadherin‐mediated assembly of molecules participating in the formation of adherens junctions may inhibit cell proliferation by reducing the levels of β‐catenin available for Wnt signaling (Katoh, 2006). The importance of other molecules involved in contact inhibition, such as p16 (Higashi et al., 1997), p21 (Noseda et al., 2004; Graves et al., 2006), p27 (Levenberg et al., 1999), p53 (Meerson et al., 2004), Pak1 (Zegers et al., 2003), and Merlin (Okada et al., 2005) has also been reported, but the phenomenon could not be fully interpreted by any of these individual signaling pathways alone.
The mammalian protocadherin LKC was first reported in humans by our group, in an effort to isolate a new human protocadherin gene and potential tumor suppressor by a homology search against the Drosophila FAT tumor suppressor (Okazaki et al., 2002; Cho et al., 2006). Subsequently, (Yang et al., 2004) confirmed its downregulation in human nodular hepatocellular carcinoma (NHCC). Human protocadherin LKC is a relatively large membrane protein (1310 amino acids), containing a signal peptide sequence (1–17), seven cadherin repeats (31–107, 129–226, 246–340, 485–577, 590–686, 700–797, 934–1043), a transmembrane domain (1152–1176) and a PDZ recognition sequence (1307–1310). Since Northern blot analysis of various adult human tissues revealed that the 5‐kb transcript was highly expressed in the liver, kidney and colon, we initially designated the gene protocadherin LKC (Protocadherin liver, kidney and colon). Recently the HUGO Gene Nomenclature Committee (HGNC) renamed protocadherin genes using the root symbol PCDH#, and protocadherin LKC was renamed “PCDH24”. The cloning of mouse PCDH24 (protocadherin LKC) was undertaken by the RIKEN Genome Exploration Research Group (Katayama et al., 2005) after our initial cloning of the human gene, but detailed genetic and functional analyses were not performed.
According to our previous work, the saturation density of HCT116 colon cancer cells stably expressing an EGFP‐PCDH24 fusion protein (clones B5 and B6) was reduced to approximately half of that of parental cells, even though no significant differences in the growth rates were observed during the exponential growth phase (Okazaki et al., 2002). The data indicated that the loss of contact inhibition in HCT116 cell had been at least partially reversed by PCDH24 expression.
Our understanding of contact inhibition has grown profoundly, but no information is available about restoring contact inhibition to epithelial cancer cells that have lost that function. To address this issue, we used proteomic and transcriptomic approaches with parental and PCDH24‐expressing HCT116 cells to find candidate key molecules responsible for restoring contact inhibition. We show that downregulation of β‐catenin signaling is one of the major mechanisms leading to contact inhibition by PCDH24 expression.
2. Results and discussion
2.1. Induction of contact inhibition and anchorage‐dependent growth by PCDH24
We previously showed that expression of PCDH24 decreases the saturation density of monolayer cell cultures and suppresses the piling up of colon cancer cells (Okazaki et al., 2002). To extend these findings to the three‐dimensional growth of the cells, we compared the in vivo growth of HCT116 colon cancer cells and of cells of the PCDH24‐expressing derivative clones B5 and B6 in nude mice. While parental HCT116 cells readily formed tumors, tumor formation was completely abolished in cells of PCDH24‐expressing clones (Figure 1A). Since suppression of 3D growth by PCDH24 could be also confirmed in soft agar assays in vitro (Figure 1B), this suppression is only dependent on changes in cell properties, but not on host factors such as neovascularization.
Figure 1.
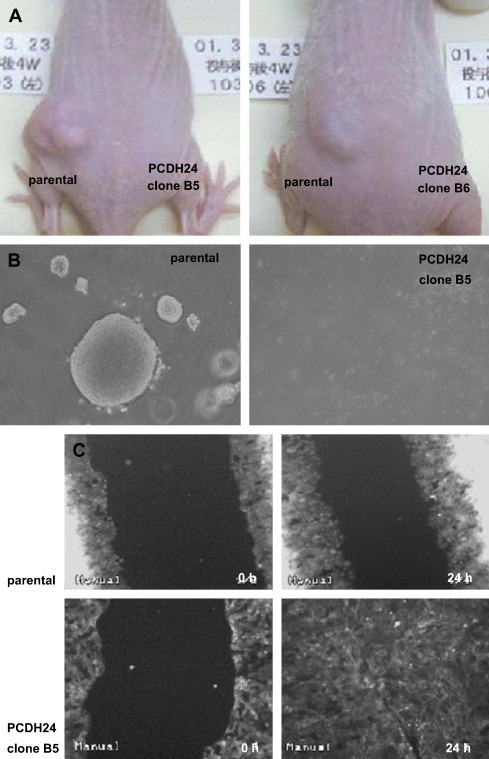
Influence of PCDH24 expression on growth of HCT116 cells. (A) Representative in vivo growth of parental and PCDH24‐expressing HCT116 cells. Each type of cell (3×106cells) was inoculated into the right and left back of the same athymic BALB/cA Jcl‐nu/nu mice (6–7 weeks old, male; CLEA Japan, Inc., Tokyo), and photographs were taken after 4 weeks. The tumor size of parental cells was calculated according to the formula: length (mm)×width (mm)×height (mm)×0.5236 was 206.3±135.9cm3 (n=4). All procedures were approved by the committee on animal care. (B) Representative image of soft agar assays. A total of 3×102cells were plated and the colonies in triplicate wells were counted after 11 days. Data are represented as mean±SD: parental cells (297±18.3), PCDH24‐expression clone B5 (0±0). (C) Effect on cell motility. Photographs were taken before (0h) and 24h (24h) following wound injury.
We next asked whether the change in cellular phenotype resulted from an altered cell motility, and performed a migration assay following wound injury. Unexpectedly, we observed that PCDH24‐expressing cells migrated at a faster rate and were able to close the wound in a shorter time (Figure 1C and Supplementary Videos). Unlike parental cells, PCDH24‐expressing cells did not pile up even after wound closure. This growth pattern is quite similar to that of normal epithelial cells. We therefore defined PCDH24‐expressing clones as revertants which had acquired contact inhibition.
2.2. Molecular changes induced by PCDH24
To comprehensively identify the signaling nodes of this phenomenon at the molecular level, we first used a conventional proteomic approach, applying 2‐DE‐MS/MS analysis. Different expression patterns were observed for 227 out of 683 spots on the gel images using Phoretix 2D Pro software. Subsequent mass spectrometric analysis identified the proteins represented by 124 spots (Figure 2). Obvious quantitative changes were detected in 58 proteins. Among these proteins 14 were upregulated and 44 were downregulated (Figure 2, 1, 2). Interestingly, we detected upregulation of vimentin, a representative marker of mesenchymal cells such as osteoblasts, chondrocytes, myocytes, and adipocytes. Downregulated proteins were more abundant than upregulated proteins. For instance, expression of PCNA, which is a well‐known marker for highly proliferating cells, was a greatly reduced.
Figure 2.
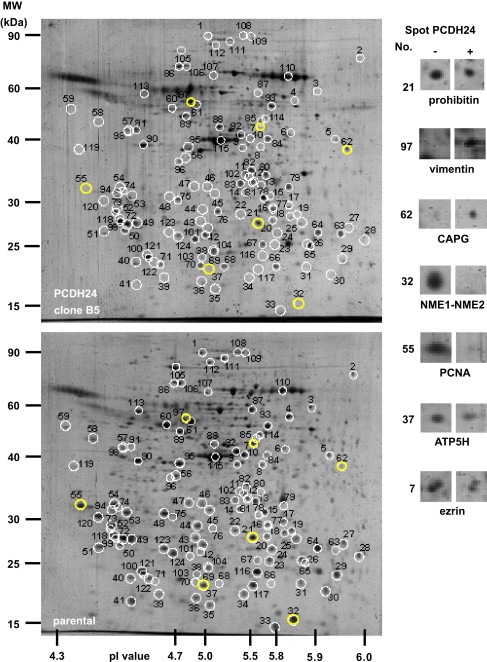
Representative 2‐DE images of total proteins extracted from parental (lower panel) and PCDH24‐expressing HCT116 cells (upper panel). The magnified views of representative up‐ and down‐regulated protein spots (yellow circles in the left) are also indicated (right). Similar results were obtained from three independent experiments with each clone.
Table 1.
Proteins upregulated by PCDH24 expression.
| Spot No.a | Protein name | Accession No. (NCBI) | Predicted | Observed | Sequence coverage (%) | Peptides matched | Mascot score | Fold change | ||
|---|---|---|---|---|---|---|---|---|---|---|
| pI | MW (Da) | pI | MW (Da) | |||||||
| 63 | Alpha‐tubulin | gi|37492 | 5.02 | 50,158 | 5.9 | 25,000 | 5 | 2 | 104 | 3.85 |
| 79 | Annexin A8 | gi|12585137 | 5.56 | 36,843 | 5.7 | 32,000 | 11 | 3 | 191 | 3.58 |
| 76 | Anxa5 protein | gi|13277612 | 4.83 | 35,738 | 4.7 | 31,000 | 8 | 3 | 205 | 1.89 |
| 42 | Dermcidin preproprotein | gi|16751921 | 6.08 | 11,284 | 4.7 | 16,000 | 10 | 1 | 63 | 2.25 |
| 83 | Eukaryotic translation initiation factor 3 | gi|4503513 | 5.38 | 36,502 | 5.7 | 37,000 | 16 | 5 | 284 | 1.36 |
| 62 | Gelsolin‐like capping protein isoform 9 | gi|55597035 | 5.88 | 38,518 | 5.8 | 40,000 | 14 | 3 | 181 | 1.64 |
| 67 | H4 histone family, member A | gi|4504301 | 11.4 | 11,367 | 5.6 | 23,000 | 50 | 5 | 266 | 1.56 |
| 88 | Heterogeneous nuclear ribonucleoprotein F | gi|19527048 | 5.31 | 45,730 | 5.2 | 40,000 | 10 | 3 | 200 | 1.33 |
| 80 | Lactate dehydrogenase B | gi|4557032 | 5.71 | 36,639 | 5.8 | 35,000 | 10 | 4 | 209 | 3.94 |
| 81 | Lactate dehydrogenase B | gi|4557032 | 5.71 | 36,639 | 5.8 | 35,000 | 30 | 9 | 543 | 1.25 |
| 82 | Pyruvate dehydrogenase (lipoamide) beta | gi|18152793 | 6.41 | 38,937 | 5.7 | 35,000 | 12 | 4 | 206 | 1.57 |
| 96 | Tropomyosin 4 | gi|4507651 | 4.67 | 28,522 | 4.6 | 32,000 | 57 | 22 | 1346 | 1.44 |
| 84 | Tubulin, beta 2 | gi|4507729 | 4.78 | 49,907 | 5.7 | 35,000 | 12 | 5 | 285 | 1.57 |
| 97 | Vimentin | gi|62414289 | 5.06 | 53,688 | 5 | 55,000 | 72 | 31 | 292 | 1.19 |
Spot number corresponds to the protein spot number indicated by white circle in Figure 2.
Table 2.
Proteins downregulated by PCDH24 expression.
| Spot No.a | Protein name | Accession No. (NCBI) | Predicted | Observed | Sequence coverage (%) | Peptides matched | Mascot score | Fold change | ||
|---|---|---|---|---|---|---|---|---|---|---|
| pI | MW (Da) | pI | MW (Da) | |||||||
| 34 | Adenine phosphoribosyltransferase isoform a | gi|4502171 | 5.78 | 19,608 | 5.5 | 18,000 | 21 | 4 | 231 | 0.23 |
| 111 | Alpha actinin 4 | gi|2804273 | 5.24 | 42,008 | 5.3 | 90,000 | 20 | 20 | 84 | 0.71 |
| 15 | Annexin A3 | gi|1421662 | 5.63 | 36,331 | 5.7 | 31,000 | 50 | 18 | 1178 | 0.22 |
| 37 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit d isoform a ATP5H | gi|5453559 | 5.21 | 18,491 | 5.1 | 20,000 | 24 | 5 | 246 | 0.28 |
| 123 | Calpain, small subunit 1 | gi|4502565 | 5.05 | 28,316 | 4.9 | 27,000 | 17 | 4 | 285 | 0.25 |
| 89 | Calreticulin | gi|11693172 | 4.33 | 47,995 | 4.4 | 50,000 | 24 | 13 | 705 | 0.73 |
| 29 | DJ‐1 protein | gi|31543380 | 6.33 | 19,891 | 6 | 24,000 | 30 | 6 | 243 | 0.16 |
| 27 | Endoplasmic reticulum protein 29 isoform 1 precursor | gi|5803013 | 6.77 | 28,993 | 6.1 | 28,000 | 34 | 8 | 438 | 0.17 |
| 25 | Eukaryotic translation initiation factor 4E | gi|4503535 | 5.79 | 25,097 | 5.8 | 28,000 | 10 | 2 | 136 | 0.65 |
| 2 | Ezrin | gi|21614499 | 5.94 | 69,413 | 6.1 | 70,000 | 22 | 15 | 678 | 0.12 |
| 7 | Ezrin | gi|21614499 | 5.94 | 69,413 | 5.7 | 50,000 | 9 | 6 | 282 | 0.16 |
| 114 | Ezrin | gi|21614499 | 5.94 | 69,413 | 5.6 | 50,000 | 44 | 40 | 121 | 0.29 |
| 11 | F‐actin capping protein alpha‐1 subunit | gi|5453597 | 5.45 | 32,923 | 5.5 | 32,000 | 17 | 4 | 243 | 0.74 |
| 19 | Glutathione‐S‐transferase omega 1 GSTO1 | gi|4758484 | 6.23 | 27,566 | 5.8 | 29,000 | 24 | 5 | 285 | 0.02 |
| 14 | Glyoxalase domain‐containing protein 4 | gi|74718831 | 5.4 | 34,793 | 5.5 | 30,000 | 16 | 5 | 293 | 0.43 |
| 12 | Guanine nucleotide‐binding protein, beta‐1 subunit | gi|6680045 | 5.6 | 37,377 | 5.5 | 32,000 | 16 | 5 | 321 | 0.79 |
| 18 | Hcp beta‐lactamase‐like protein C1orf163 | gi|74760741 | 5.66 | 25,737 | 5.8 | 28,000 | 10 | 3 | 128 | 0.5 |
| 1 | Heat shock 70kDa protein 4 | gi|119582699 | 5.1 | 87,776 | 5 | 90,000 | 27 | 20 | 1200 | 0.01 |
| 8 | Heat shock 90kDa protein 1, beta | gi|256089 | 8.35 | 17,323 | 5.7 | 40,000 | 23 | 17 | 990 | 0.64 |
| 87 | Heat shock 90kDa protein 1, beta | gi|20149594 | 4.97 | 83,264 | 4.9 | 85,000 | 27 | 21 | 1187 | 0.27 |
| 106 | Heat shock 90kDa protein 1, beta | gi|40556608 | 4.97 | 83,281 | 4.9 | 85,000 | 27 | 21 | 1188 | 0.62 |
| 41 | HSPC029 | gi|5114051 | 4.89 | 25,060 | 4.7 | 23,000 | 14 | 3 | 159 | 0.2 |
| 35 | Modifier 2 | gi|53165 | 4.96 | 19,752 | 5.2 | 16,000 | 21 | 4 | 275 | 0.58 |
| 32 | NME1‐NME2 protein | gi|66392203 | 9.06 | 30,137 | 5.8 | 15,000 | 17 | 5 | 255 | 0.14 |
| 21 | Prohibitin | gi|4505773 | 5.57 | 29,804 | 5.6 | 29,000 | 35 | 9 | 589 | 0.81 |
| 55 | Proliferating cell nuclear antigen | gi|4505641 | 4.57 | 28,769 | 4.4 | 32,000 | 25 | 8 | 483 | 0.22 |
| 113 | Prolyl 4‐hydroxylase, beta subunit precursor | gi|20070125 | 4.76 | 57,116 | 5.8 | 25,000 | 35 | 22 | 1201 | 0.58 |
| 51 | Proteasome (prosome, macropain) subunit, alpha type 5 | gi|7106387 | 4.74 | 26,411 | 4.7 | 27,000 | 20 | 4 | 229 | 0.44 |
| 72 | Proteasome (prosome, macropain) subunit, alpha type 5 | gi|7106387 | 4.74 | 26,411 | 4.7 | 27,000 | 8 | 2 | 100 | 0.87 |
| 22 | Proteasome activator subunit 2 isoform 1 | gi|20137004 | 5.54 | 27,057 | 5.4 | 27,000 | 13 | 3 | 202 | 0.48 |
| 44 | Proteasome alpha 3 subunit isoform 1 | gi|4506183 | 5.19 | 28,433 | 5 | 29,000 | 21 | 6 | 295 | 0.35 |
| 23 | Proteasome subunit HsN3 | gi|565651 | 5.72 | 29,192 | 5.7 | 26000 | 12 | 3 | 155 | 0.22 |
| 17 | PSME3 | gi|49456449 | 5.69 | 29,579 | 5.8 | 30,000 | 8 | 2 | 114 | 0.5 |
| 13 | Pyrophosphatase 1 PPA1 | gi|11056044 | 5.54 | 32,660 | 5.5 | 32,000 | 20 | 6 | 302 | 0.73 |
| 36 | RNA binding motif protein 8A | gi|4826972 | 5.5 | 19,889 | 5 | 17,000 | 11 | 2 | 118 | 0.43 |
| 46 | Spermidine synthase | gi|63253298 | 5.3 | 33,825 | 5.3 | 31,000 | 12 | 3 | 135 | 0.43 |
| 47 | Splicing factor, arginine/serine‐rich 1 | gi|119614893 | 10.8 | 25,589 | 5.1 | 32,000 | 21 | 5 | 273 | 0.43 |
| 48 | Splicing factor, arginine/serine‐rich 1 | gi|148669918 | 10.1 | 24,436 | 4.9 | 32,000 | 22 | 5 | 243 | 0.27 |
| 9 | Stomatin (EPB72)‐like 2 | gi|7305503 | 7.71 | 40,014 | 5.6 | 40,000 | 32 | 11 | 711 | 0.77 |
| 118 | Stratifin | gi|16306737 | 4.72 | 27,706 | 4.8 | 23,000 | 18 | 5 | 283 | 0.69 |
| 3 | T‐complex protein 1 isoform a | gi|57863257 | 5.8 | 60,344 | 6.3 | 65,000 | 19 | 10 | 568 | 0.2 |
| 24 | Thioredoxin peroxidase | gi|5453549 | 5.86 | 30,540 | 5.7 | 27,000 | 24 | 6 | 337 | 0.69 |
| 5 | Translation initiation factor eIF3 p44 subunit | gi|3264859 | 6.1 | 35,696 | 6.1 | 40,000 | 18 | 8 | 496 | 0.46 |
| 43 | Trichohyalin | gi|148746195 | 5.73 | 253,925 | 5.2 | 27,000 | 4 | 8 | 351 | 0.43 |
| 53 | Tropomyosin 3 isoform 4 | gi|114155144 | 4.73 | 28,870 | 4.7 | 30,000 | 55 | 13 | 800 | 0.45 |
| 61 | Tubulin, alpha 1C | gi|6678469 | 4.96 | 49,909 | 5 | 55,000 | 25 | 9 | 480 | 0.59 |
| 60 | Tubulin, beta, 2 | gi|5174735 | 4.79 | 49,831 | 4.8 | 54,000 | 37 | 18 | 1122 | 0.45 |
| 109 | Tyrosine 3/tryptophan 5‐monooxygenase activation protein | gi|5803225 | 5.57 | 117,790 | 5.5 | 100,000 | 43 | 12 | 693 | 0.53 |
| 112 | Valosin‐containing protein | gi|6005942 | 4.93 | 33,682 | 5.2 | 80,000 | 18 | 18 | 62 | 0.61 |
| 56 | WD‐40 repeat protein | gi|4519417 | 4.93 | 38,496 | 4.9 | 35,000 | 18 | 6 | 378 | 0.511 |
Spot number corresponds to the protein spot number indicated by white circle in Figure 2.
We further examined these differences at the transcriptional level using Affimetrics U133 plus 2.0 DNA microarrays. The data among replicated samples showed high correlation coefficients (parental cells=0.988, PCDH24‐expressing cells=0.952). Comparison of parental cells with PCDH24‐expressing cells revealed 1348 genes that showed a fold change >3 (correlation coefficient=0.546). We confirmed the upregulation of 4 molecules (annexin A8, dermcidin preproprotein, gelsolin‐like capping protein isoform 9, and vimentin) and the downregulation of 17 molecules (annexin A3, ATP synthase, calreticulin, endoplasmic reticulum protein 29, ezrin, glutathione‐S‐transferase omega 1, guanine nucleotide‐binding protein, heat shock 90kDa, NME1‐NME2 protein, prohibitin, PCNA, prolyl 4‐hydroxylase, pyrophosphatase 1, RNA binding motif protein 8A, splicing facto, T‐complex protein 1, and tropomyosin 3), also at the transcriptional level. As in the 2‐DE‐MS/MS analysis, downregulation of gene expression was predominantly observed in PCDH24‐expressing cells.
2.3. PCDH24 induces changes in the expression of phenotypic markers
We found an upregulated expression of vimentin both at the protein and the mRNA levels (132.6‐fold increase over parental HCT116 cells on DNA microarrays). As high level expression of vimentin is indicative of a mesenchymal cellular phenotype, we analyzed other markers that define a cell as epithelial or mesenchymal. DNA microarray analysis revealed the downregulation of E‐cadherin and keratins, which are typical markers for epithelial cells (Venkov et al., 2007). Further qRT‐PCR and Western blot analyses verified these changes (Figure 3A, B). We also performed qRT‐PCR against other epithelial (ZO‐1, β‐catenin) and mesenchymal (actin α 1, collagens) markers; except for β‐catenin other markers were distinctly downregulated. It has been shown that zinc‐finger transcription factors such as Snail1 (Snail), Snail2 (Slug), Twist, EF1/ZEB1, SIP1/ZEB2, and/or E47 inhibit E‐cadherin expression (Barrallo‐Gimeno and Nieto, 2005; Lee et al., 2007). We thus analyzed by qRT‐PCR the possible involvement of these transcription factors in the phenotypic changes induced by PCDH24, as well. We detected a slight but distinct upregulation of Slug, but other transcription factors were markedly downregulated (Figure 3C). These findings are different from those observed in EMTs (epithelial‐mesenchymal transitions), which are characterized by a downregulation of E‐cadherin and an upregulation of vimentin. Therefore, other transcription factors may have more important roles in the phenotypic changes observed in conjunction with PCDH24 expression.
Figure 3.
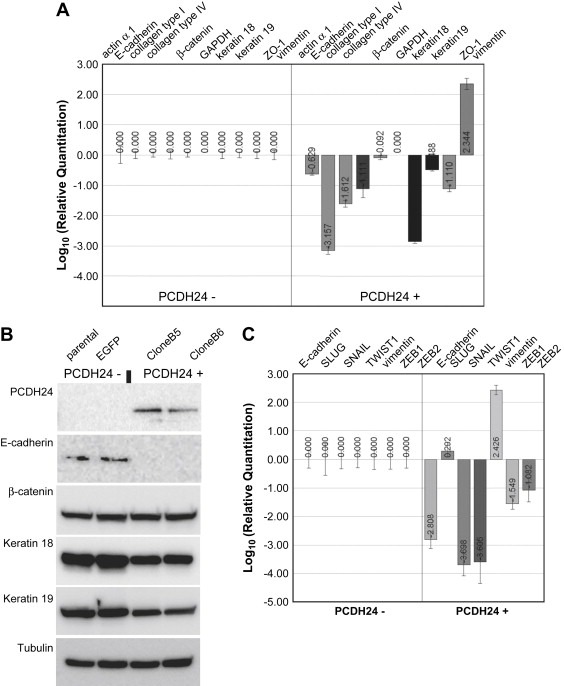
mRNA expression of representative phenotypic markers (A) and EMT‐related transcription factors (C) was analyzed by qRT‐PCR. The relative quantitation of the data is indicated by bar graphs located underneath each marker. (B) Confirmation of the protein expression by Western blotting. Equal amounts of whole cell lysates were loaded on SDS‐PAGE and the transferred membranes were blotted using antibodies against the control (tubulin) and epithelial and mesenchymal markers.
2.4. Down‐regulation of TCF/lef signaling by PCDH24
Careful visual inspection of the data from DNA microarrays revealed a downregulation of Myc (7.2‐fold decrease), cyclin D1 (12.5‐fold decrease), Met (81.2‐fold decrease), CD44 (9.1‐fold decrease), and PLAUR (6.8‐fold decrease) mRNA expression in PCDH24‐expressing HCT116 cells. All these molecules are downstream of TCF/lef signaling (He et al., 1998; Mann et al., 1999; Shtutman et al., 1999; Wielenga et al., 1999; Boon et al., 2002), thereby suggesting an involvement of TCF/lef signaling in the phenotypic changes observed after PCDH24 expression. To verify the array data, we performed Western blotting and qRT‐PCR. Western blotting clearly demonstrated the downregulation of Myc, cyclin D1, and Met protein levels, and qRT‐PCR revealed the downregulation of CD44, Met and PLAUR mRNA levels (Figure 4A, B). In addition, the above mentioned downregulation of another epithelial marker, the keratins, also fits into the pattern, as keratins are known to be downstream of TCF/lef signaling (DasGupta and Fuchs, 1999).
Figure 4.
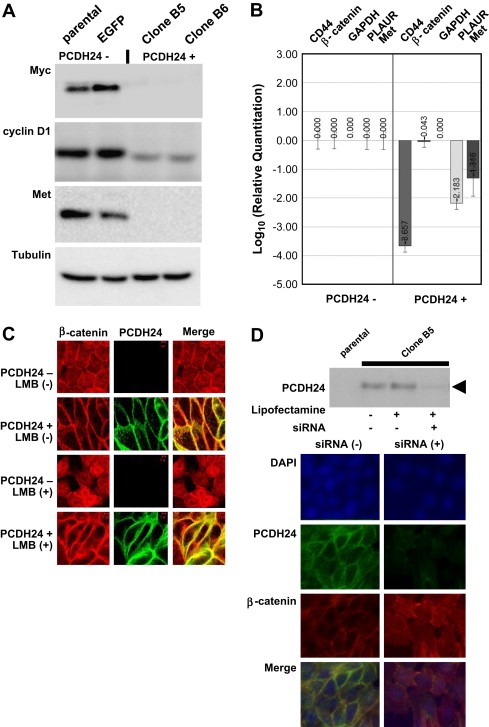
(A) Confirmation of marker protein expression by Western blotting. Equal amounts of whole cell lysates were loaded on SDS‐PAGE and the transferred membranes were blotted using antibodies against Myc, cyclin D1, Met, and control (tubulin). (B) Confirmation of mRNA expression by qRT‐PCR. The mRNA expressions of other downstream molecules of TCF/lef signaling were analyzed by qRT‐PCR. Relative quantitation of the data is indicated by bar graphs located underneath each marker. (C) Immunofluorescence microscopy images of the subcellular localization of β‐catenin. Red and green signals indicate β‐catenin and PCDH24, respectively. (D) PCDH24 knockdown using anti‐EGFP siRNA. PCDH24‐expressing HCT116 cells were seeded in six‐well plates at 24h before siRNA transfection. Anti‐EGFP siRNA was transfected using Lipofectamine 2000 transfection reagent; then Western blotting using anti‐PCDH24 antibody was performed at 24h post‐transfection (upper panel). The subcellular localization of β‐catenin after anti‐EGFP siRNA treatment was analyzed by immunofluorescence microscopy (lower panel). Red and green signals indicate β‐catenin and PCDH24, respectively.
2.5. β‐catenin is a key node in PCDH24‐inducible contact inhibition
Since β‐catenin, an upstream signal for TCF/lef, did not show any apparent changes in the former experiments (Figure 3A,B), a critical question arose regarding the discrepancy between the β‐catenin and the downstream signals. We hypothesized that β‐catenin does not change its expression, but cannot translocate to the nuclei. To verify this hypothesis, we analyzed the subcellular localization of β‐catenin and that β‐catenin was retained in a submembranous region. Absolutely no nuclear accumulation could be observed even after LMB treatment (Figure 4C, lower panel). To provide evidence that PCDH24 causes the submembranous retention of β‐catenin, we analyzed the subcellular localization of β‐catenin after PCDH24 knockdown using anti‐EGFP siRNA. Anti‐EGFP siRNA knocked down EGFP‐fused PCDH24 protein expression by more than 80% (Figure 4D, upper panel). Anti‐EGFP siRNA consequently reduced β‐catenin staining at sites of cell–cell contact, resulting in increased staining of cytoplasmic and nuclear β‐catenin (Figure 4D, lower panel). This result suggests that submembranous retention of β‐catenin depends on PCDH24 expression.
To verify the effect of PCDH24 expression on the transcriptional activity of β‐catenin, we performed a TCF/lef‐reporter gene assay using SW480 cells. SW480 cells, in which the APC gene is mutated, are well known for high transcriptional activities of TCF/lef‐reporter genes (Munemitsu et al., 1995). We first showed that the endogenous PCDH24 expression in these cells was below the level of detection (Figure 5, upper panel). Cotransfection of a wild‐type PCDH24 expression vector with the TCF/lef‐reporter gene abrogated the transcriptional activity of endogenous β‐catenin (Figure 5B).
Figure 5.
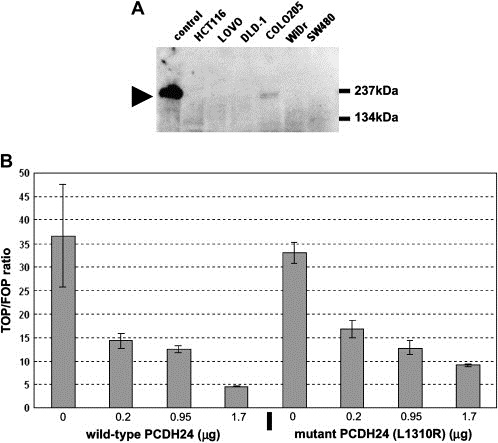
(A) Transcriptional repression of β‐catenin signaling by PCDH24. To perform the reporter assay, we first analyzed PCDH24 expression in SW480 by Western blotting. PCDH24 was not detected, as in other colon cancer cell lines. The name of each cell line is indicated at the top of the panel and the arrow indicates the MW of the PCDH24 protein. These results are in accordance with our previous results by RT‐PCR (Okazaki et al., 2002). (B) Downregulation of TCF/lef signaling by wild‐type and mutant (L1310R) PCDH24 expression in SW480 cells. The ratio of luciferase activities in cells transfected with TOPFLASH versus FOPFLASH reporters was calculated. Means and standard deviations of quadruplicate measures are shown. The amount of PCDH24 expression plasmids was changed from 0.2 to 1.7μg and that is indicated at the bottom of the panel.
Previously, we showed that PCDH24 can interact with MAST205 at the C‐terminal PDZ‐recognized peptide motif (Okazaki et al., 2002). We therefore analyzed the participation of MAST205 in the modulation of β‐catenin transcriptional activity, and introduced an amino acid substitution (Leu to Arg) at the PDZ‐recognized motif to mutant PCDH24 (L1310R), abolishing the interaction of PCDH24 with MAST205. β‐catenin activity was abrogated also by expression of the mutated construct, as with wild‐type PCDH24 (Figure 5B). These results indicate that MAST205 and other PDZ domain‐containing proteins are not involved in this signaling, and that an intracellular domain other than the C‐terminal PDZ‐recognized motif of PCDH24 participates in the repression of β‐catenin activity. We also tried to find a direct interaction between PCDH24 and β‐catenin, but we could not identify the interaction either in immunological (IP‐Western blotting; data not shown) or proteomic (IP‐MS/MS) analyses (Figure 6C). We therefore assume that the submembranous anchoring of β‐catenin arises from a different mechanism than direct interaction with PCDH24.
Figure 6.
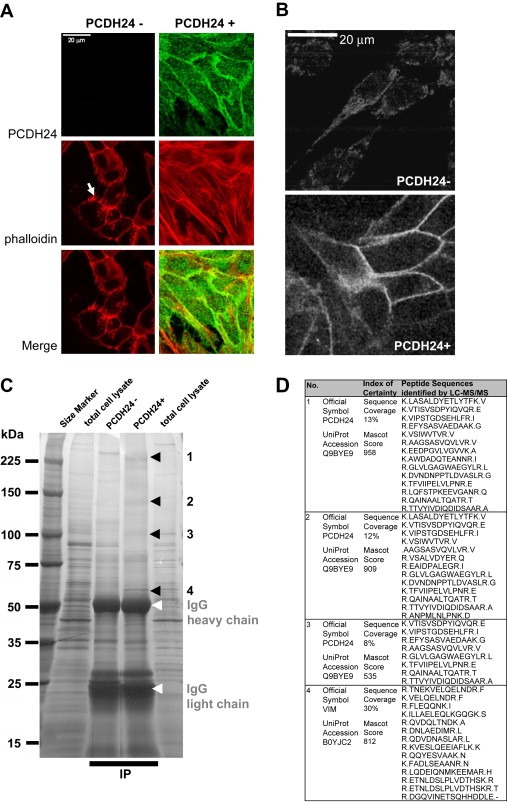
Immunofluorescence analysis of cytoskeletal reorganization by PCDH24‐expression. Distribution of polymerized actin (red) in parental and PCDH24‐expressing HCT116 cells was compared (A). Polymerized actin was mainly observed at the peripheral cytoplasmic protrusion of parental cells (arrow). By contrast, strong actin stress fibers were observed throughout the cellular regions of PCDH24‐expressing cells. Vimentin intermediate filaments also showed a different distribution HCT116 cells and their PCDH24‐expressing derivatives (B). (C) One‐dimensional SDS‐PAGE of anti‐EGFP immunoprecipitates. The immunoprecipitates were resolved by 5–20% 1D‐SDS‐PAGE and then subjected to imidazole‐zinc reverse staining. Excised bands (black arrowheads) were digested with 10μg/ml trypsin for 16h and then subjected to LC‐MS/MS analysis. (D) List of peptides detected by LC‐MS/MS. The resulting MS/MS data were analyzed using the Mascot search engine, and proteins identified with a combined peptide score higher than 500 were considered significant; lower‐scoring proteins were rejected.
2.6. The submembranous retention of β‐catenin not only involves the actin cytoskeleton but also vimentin intermediate filaments
To gain insight into the molecular mechanism of the submembranous retention of β‐catenin, we compared the actin cytoskeleton of PCDH24‐expressing cells with that of parental cells. PCDH24‐expressing cells revealed distinct actin stress fibers which were absent in parental cancer cells (Figure 6A). In these cells, polymerized actin was observed in peripheral cytoplasmic protrusion (Figure 6A, arrow) and in adherens junction. This finding reinforces our previous evidence for a link between the actin cytoskeleton and PCDH24 (Okazaki et al., 2002), like with classical cadherins (Tanoue and Takeichi, 2005). The submembranous retention of β‐catenin thus might be at least partly explained by rigid scaffolding via actin stress fibers. Recent studies on the crosstalk between actin filaments and other major cytoskeleton intermediate filaments such as vimentin have revealed a direct interaction between actin and vimentin (Esue et al., 2006). Vimentin interactions with classical cadherins (Kowalczyk et al., 1998; Kim et al., 2005) and desmosomal cadherins (Garrod et al., 2002) have also been reported. We thus examined the subcelluar localization of vimentin. While vimentin staining in parental cells was weak but distributed throughout the entire cytoplasm that of PCDH24‐expressing cells appeared to be accumulated at the cell periphery (Figure 6B). This finding may be of interest in connection with the effects of PCDH24 and vimentin on the submembranous retention of β‐catenin at the cell periphery.
2.7. Vimentin is a potential binding partner of PCDH24
To gain insight into the link between PCDH24 and vimentin, we tried to identify the endogenous binding partners of PCDH24 in HCT116 cells. SDS‐PAGE followed by immunoprecipitation revealed several specific bands against the control immunoprecipitates from parental HCT116 cells (Figure 6C). Subsequent MS/MS analysis identified three major bands at approximately 250, 130 and 100kDa as EGFP‐fused PCDH24 itself and its derivatives, respectively (Figure 6D). We found an additional distinct band of approximately 60kDa (Figure 6C, arrowhead 4). This band was identified as vimentin by ensuing MS/MS analysis (Figure 6D). Therefore, at least overexpressed PCDH24 can induce vimentin expression and interact with vimentin in HCT116 colon cancer cell lines. Although the primary amino acid sequence of the intracellular domain of PCDH24 has no similarity to that of previously identified vimentin‐binding proteins in the plakin family (desmoplakin, BPAG1, ACF/microtubule actin cross‐linking factor, envoplakin, periplakin, and epiplakin) (Karashima and Watt, 2002; Jang et al., 2005; Spurny et al., 2007), the PCDH24 protein may have a tertiary structure that allows vimentin‐binding, or may bind indirectly via other vimentin‐binding proteins.
β‐catenin has a dual role in epithelial cells: one is cell–cell adhesion involving the cadherin complex, another one is transcriptional regulation upon entry into the nucleus (Zhou and Hung, 2005). PCDH24 has no effect on the total protein amount of β‐catenin, but is a distinctly negative regulator for nuclear translocation and the subsequent transcriptional activation. Outside–inside signals of contact inhibition are transduced by other molecules such as p16 (Higashi et al., 1997), p21 (Noseda et al., 2004; Graves et al., 2006), p27 (Levenberg et al., 1999), p53 (Meerson et al., 2004), Pak1 (Zegers et al., 2003), and Merlin (Okada et al., 2005). Hence, we may have observed only one aspect of PCDH24‐inducible contact inhibition and further detailed analyses regarding such signals are necessary. However, inhibition of β‐catenin‐TCF/lef signaling seems to be one of the major causes of PCDH24‐inducible contact inhibition, and reorganization of actin and vimentin intermediate filaments also plays an important role in this process.
3. Materials and methods
3.1. Cell culture
SW480 and HCT116 cells (including the derivative clones B5 and B6) were cultured in DMEM (Dulbecco's modified Eagle's minimum essential medium) supplemented with 10% (V/V) heat‐inactivated fetal bovine serum in a humidified CO2 incubator at 37°C. To evaluate the nuclear accumulation of β‐catenin, cells were treated with 4ng/ml of leptomycin B (LMB) for 16h. For soft agar assays, 1.2% and 0.6% of agarose gels (NuSieve® GTG® Agarose: Lonza, Valais, Switzerland) were used as base and top agars in 35‐mm dishes, respectively. The cells were trypsinized, centrifuged, resuspended, and 3×102cells were plated. The surface was kept wet by addition of a small amount of growth medium. After 11 days, colonies were photographed and counted.
3.2. Microscopic observation
Axiovert S100 (Carl Zeiss, Jena, Germany) was used for phase‐contrast and fluorescence images at low magnification, and the acquired images were converted to electronic files by a DP Controller (Version1.2.1: Olympus, Tokyo, Japan). Confocal microscopic analysis using LSM510 (Version1.5: Carl Zeiss) was performed according to a previous report (Okazaki et al., 2002). Rabbit anti‐PCDH24 polyclonal (1:2000) and anti‐β catenin monoclonal (Transduction Laboratories, Lexington, KY, USA) (1:400) antibodies were used as primary antibodies. Alexa™ 594‐conjugated goat anti‐rabbit (1:200) and Alexa™ 488‐conjugated goat anti‐mouse antibodies (Molecular Probes, Eugene, OR, USA) (1:200) were used as secondary antibodies. Anti‐vimentin antibody (ab58462; Abcam, Cambridge, UK) (1:1000) and Alexa™ 488 Phalloidin (Molecular Probes) were used for the observation of cytoskeletal reorganization. For the migration assay, cell movement was monitored for 24h using a time‐lapse videomicroscope BZ‐8000 (KEYENCE, Osaka, Japan).
3.3. 2‐DE/MS analysis
We performed 2‐DE principally according to Toda's protocol (Toda and Kimura, 1997). Protein spots were visualized by staining with Deep Purple™ Total Protein Stain (GE Healthcare Life Sciences, Uppsala, Sweden), and the images were captured by FLA3000GF (Fujifilm, Tokyo, Japan). Phoretix 2D Pro software (Nonlinear Dynamics Ltd, Newcastle, UK) was used for subsequent 2‐D gel image analysis. The mean fold changes were calculated using Multi Gauge software version 3.0 (Fujifilm). Spots with distinct changes were excised manually and the proteins were identified by ion trap mass spectrometer LCQ (Thermo Finnigan, CA, USA) as previously described (Koga et al., 2004b). Proteins identified with a combined peptide score of higher than 50 were considered significantly as present, and lower‐scoring proteins were rejected. Some of the spots (spot No. 19, 24, 97, 104, 112, 114) were also identified by MALDI‐TOF analysis according to the previously described method (Koga et al., 2004a) and the data are shown in tables.
3.4. RNA extraction and microarray analysis
Total RNA was extracted with the use of an RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The quantities and qualities of the RNAs were measured by electrophoresis using an Agilent 2100 Bioanalyzer and RNA 6000 Nano Kit (Agilent Technologies, Palo Alto, CA). The experimental procedures for GeneChip® Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA, USA) followed those provided by the manufacturer. Raw intensity data for each experiment was first normalized by Robust Multichip Analysis (RMA, Affymetrix). The data for each experiment was log2 transformed and then used for the calculation of Z scores. Z scores were calculated by subtracting the overall average gene intensity (within a single experiment) from the raw intensity data for each gene, and dividing that result by the SD of all of the measured intensities, according to the previously established formula (Cheadle et al., 2003). Further statistical analyses were performed by ANOVA (Kerr et al., 2000). All the raw data described in this study were deposited in the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE10650.
3.5. Real‐time quantitative RT‐PCR (qRT‐PCR)
Sets of primers were designed to amplify from 100‐ to 150‐bp fragments within the genes extracted from DNA microarrays (see Table S1 in the Supplementary material) using the Primer Express Software (Applied Biosystems, Foster City, CA). The qRT‐PCR was performed using SuperScript III Platinum SYBR Green One‐Step qRT‐PCR kit (Invitrogen) and a 7500 Fast Real‐Time PCR system (Applied Biosystems). Levels of gene expression in each sample were determined with the comparative Ct method, using the GAPDH gene as an endogenous control. All qRT‐PCR experiments were performed in quadruplicate and the results are represented as mean±SD.
3.6. Western blotting
Western blotting with several antibodies was performed according to the method previously described (Koga et al., 2004b). The following antibodies were used for the first antibody: anti‐E‐cadherin (67A4; InnoGenex, San Ramon, CA, USA), anti‐keratin 18 (10830‐1‐AP; Protein Tech Group Inc., Chicago, IL, USA), anti‐keratin 19 (10712‐1‐AP; Protein Tech Group Inc.), anti‐α‐tubulin (DM1A; Calbiochem, Darmstadt, Germany), anti‐Myc (sc‐764; Santa cruz), and anti‐β catenin (Transduction Laboratories), anti‐Met (sc‐161; Santa cruz), anti‐cyclin D1 (DCS‐6, Thermo Fisher Scientific, Fremont, CA).
3.7. Construction of mutant PCDH24 expression plasmid and reporter gene assays
The construction of pCEP‐PCDH24 has already been described (Okazaki et al., 2002). The pCEP‐PCDH24‐L1310R bearing an amino acid substitution (amino acid 1310 Leu to Arg) was generated using the GeneEditor™ in vitro site‐directed mutagenesis system (Promega, Madison). SW480 cells (2×104 per well) were seeded into 24‐well plates and transfected the following day by a FuGENE6 transfection reagent (Roche, Nutley, NJ, USA) with a TOPFLASH or FOPFLASH reporter gene (Upstate Biotechnology, Lake Placid, NY, USA) and PCDH24‐expression plasmids. After 24h, cells were harvested and lysed; then luciferase activity was determined using the Dual‐Luciferase® assay system (Promega). The total amount of DNA was kept constant (2μg) by supplementation with 50ng of the pRL‐TK Renilla luciferase construct (Promega) as a control to normalize the transfection efficiency. The luciferase activities were first normalized by pRL‐TK Renilla luciferase; then normalized TOPFLASH luciferase activity was divided by that of FOPFLASH to calculate the TOP/FOP ratio.
3.8. siRNA experiment
Parental and PCDH24‐expressing cells (clone B5) were seeded in a Lab Tech chamber slide (Nalgene Nunc, Naperville, IL, USA) at 24h before siRNA transfection. Synthetic anti‐EGFP siRNA (Stealth™ RNAi GFP Reporter Control, #No. 12935‐145, Invitrogen, San Diego, CA, USA) was transfected using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacture's detailed instructions. After 24h, 4ng/ml of LMB was added to the medium to enhance the nuclear staining. Immunohistochemical analysis at 48h post‐transfection was performed using anti‐PCDH24 and anti‐β catenin antibodies as the primary antibodies according to Section 3.2. The suppression levels of the PCDH24 by anti‐EGFP siRNAs were analyzed using Western blot using anti‐PCDH24 antibody.
3.9. Mass spectrometry analysis of immunoprecipitated samples
Approximately 0.4g of HCT116 cells (the derivative PCDH24‐expressing clone B5 and the control EGFP‐expressing clone) was homogenized with CelLytic M (Sigma, St. Louis, MO, USA) containing 0.5% Protease Inhibitor Cocktail (Sigma) and then subjected to immunoprecipitation with anti‐GFP polyclonal antibody (ab6556, Abcam). The resulting precipitates were recovered in 20μl of 2×SDS sample buffer containing 0.2M DTT by boiling for 10min. The supernatant was resolved by 5–20% 1D‐SDS‐PAGE and then subjected to imidazole‐zinc reverse staining. All excised bands were digested with 10μg/ml trypsin (Promega) for 16h. After dilution with 1% TFA, the resulting peptide mixture was subjected to LC‐MS/MS analysis as previously described (Koga et al., 2004b). The resulting MS/MS data were analyzed using the Mascot search engine (Matrix Science, London, UK). Two independent experiments were performed, and proteins identified with a combined peptide score higher than 500 were considered significantly present.
Supporting information
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
We are grateful to Dr. Wolfgang Deppert (Heinrich‐Pette‐Institut für Experimentelle Virologie und Immunologie, Tumor Virology, Hamburg, Germany) for editing the manuscript. This study was supported by a grant from the Genome Network Project of the Ministry of Education, Culture, Sports, Science and Technology, Japan. This study was also supported by grants from the Kazusa DNA Research Institute.
Supplementary material 1.
1.1.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molonc.2008.10.005.
References
- Barrallo-Gimeno, A. , Nieto, M.A. , 2005. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 132, 3151–3161. [DOI] [PubMed] [Google Scholar]
- Boon, E.M. , van der Neut, R. , van de Wetering, M. , Clevers, H. , Pals, S.T. , 2002. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Cancer Res.. 62, 5126–5128. [PubMed] [Google Scholar]
- Cheadle, C. , Vawter, M.P. , Freed, W.J. , Becker, K.G. , 2003. Analysis of microarray data using Z score transformation. J. Mol. Diagn.. 5, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, E. , Feng, Y. , Rauskolb, C. , Maitra, S. , Fehon, R. , Irvine, K.D. , 2006. Delineation of a fat tumor suppressor pathway. Nat. Genet.. 38, 1142–1150. [DOI] [PubMed] [Google Scholar]
- DasGupta, R. , Fuchs, E. , 1999. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 126, 4557–4568. [DOI] [PubMed] [Google Scholar]
- Dietrich, C. , Scherwat, J. , Faust, D. , Oesch, F. , 2002. Subcellular localization of beta-catenin is regulated by cell density. Biochem. Biophys. Res. Commun.. 292, 195–199. [DOI] [PubMed] [Google Scholar]
- Esue, O. , Carson, A.A. , Tseng, Y. , Wirtz, D. , 2006. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem.. 281, 30393–30399. [DOI] [PubMed] [Google Scholar]
- Garrod, D.R. , Merritt, A.J. , Nie, Z. , 2002. Desmosomal cadherins. Curr. Opin. Cell Biol.. 14, 537–545. [DOI] [PubMed] [Google Scholar]
- Graves, T.G. , Harr, M.W. , Crawford, E.L. , Willey, J.C. , 2006. Stable low-level expression of p21WAF1/CIP1 in A549 human bronchogenic carcinoma cell line-derived clones down-regulates E2F1 mRNA and restores cell proliferation control. Mol. Cancer. 5, 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, T.C. , Sparks, A.B. , Rago, C. , Hermeking, H. , Zawel, L. , da Costa, L.T. , Morin, P.J. , Vogelstein, B. , Kinzler, K.W. , 1998. Identification of c-MYC as a target of the APC pathway. Science. 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Higashi, H. , Suzuki-Takahashi, I. , Yoshida, E. , Nishimura, S. , Kitagawa, M. , 1997. Expression of p16INK4a suppresses the unbounded and anchorage-independent growth of a glioblastoma cell line that lacks p16INK4a. Biochem. Biophys. Res. Commun.. 231, 743–750. [DOI] [PubMed] [Google Scholar]
- Jang, S.I. , Kalinin, A. , Takahashi, K. , Marekov, L.N. , Steinert, P.M. , 2005. Characterization of human epiplakin: RNAi-mediated epiplakin depletion leads to the disruption of keratin and vimentin IF networks. J. Cell Sci.. 118, 781–793. [DOI] [PubMed] [Google Scholar]
- Karashima, T. , Watt, F.M. , 2002. Interaction of periplakin and envoplakin with intermediate filaments. J. Cell Sci.. 115, 5027–5037. [DOI] [PubMed] [Google Scholar]
- Katayama, S. , Tomaru, Y. , Kasukawa, T. , Waki, K. , Nakanishi, M. , Nakamura, M. , Nishida, H. , Yap, C.C. , Suzuki, M. , Kawai, J. , Suzuki, H. , Carninci, P. , Hayashizaki, Y. , Wells, C. , Frith, M. , Ravasi, T. , Pang, K.C. , Hallinan, J. , Mattick, J. , Hume, D.A. , Lipovich, L. , Batalov, S. , Engstrom, P.G. , Mizuno, Y. , Faghihi, M.A. , Sandelin, A. , Chalk, A.M. , Mottagui-Tabar, S. , Liang, Z. , Lenhard, B. , Wahlestedt, C. , 2005. Antisense transcription in the mammalian transcriptome. Science. 309, 1564–1566. [DOI] [PubMed] [Google Scholar]
- Katoh, M. , 2006. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther.. 5, 1059–1064. [DOI] [PubMed] [Google Scholar]
- Kerr, M.K. , Martin, M. , Churchill, G.A. , 2000. Analysis of variance for gene expression microarray data. J. Comput. Biol.. 7, 819–837. [DOI] [PubMed] [Google Scholar]
- Kim, Y.J. , Sauer, C. , Testa, K. , Wahl, J.K. , Svoboda, R.A. , Johnson, K.R. , Wheelock, M.J. , Knudsen, K.A. , 2005. Modulating the strength of cadherin adhesion: evidence for a novel adhesion complex. J. Cell Sci.. 118, 3883–3894. [DOI] [PubMed] [Google Scholar]
- Koga, H. , Shimada, K. , Hara, Y. , Nagano, M. , Kohga, H. , Yokoyama, R. , Kimura, Y. , Yuasa, S. , Magae, J. , Inamoto, S. , Okazaki, N. , Ohara, O. , 2004. A comprehensive approach for establishment of the platform to analyze functions of KIAA proteins: generation and evaluation of anti-mKIAA antibodies. Proteomics. 4, 1412–1416. [DOI] [PubMed] [Google Scholar]
- Koga, H. , Yuasa, S. , Nagase, T. , Shimada, K. , Nagano, M. , Imai, K. , Ohara, R. , Nakajima, D. , Murakami, M. , Kawai, M. , Miki, F. , Magae, J. , Inamoto, S. , Okazaki, N. , Ohara, O. , 2004. A comprehensive approach for establishment of the platform to analyze functions of KIAA proteins II: public release of inaugural version of InGaP database containing gene/protein expression profiles for 127 mouse KIAA genes/proteins. DNA Res.. 11, 293–304. [DOI] [PubMed] [Google Scholar]
- Kowalczyk, A.P. , Navarro, P. , Dejana, E. , Bornslaeger, E.A. , Green, K.J. , Kopp, D.S. , Borgwardt, J.E. , 1998. VE-cadherin and desmoplakin are assembled into dermal microvascular endothelial intercellular junctions: a pivotal role for plakoglobin in the recruitment of desmoplakin to intercellular junctions. J. Cell Sci.. 111, (Pt 20) 3045–3057. [DOI] [PubMed] [Google Scholar]
- Lee, J.D. , Silva-Gagliardi, N.F. , Tepass, U. , McGlade, C.J. , Anderson, K.V. , 2007. The FERM protein Epb4.1l5 is required for organization of the neural plate and for the epithelial-mesenchymal transition at the primitive streak of the mouse embryo. Development. 134, 2007–2016. [DOI] [PubMed] [Google Scholar]
- Levenberg, S. , Yarden, A. , Kam, Z. , Geiger, B. , 1999. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene. 18, 869–876. [DOI] [PubMed] [Google Scholar]
- Mann, B. , Gelos, M. , Siedow, A. , Hanski, M.L. , Gratchev, A. , Ilyas, M. , Bodmer, W.F. , Moyer, M.P. , Riecken, E.O. , Buhr, H.J. , Hanski, C. , 1999. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc. Natl. Acad. Sci. U.S.A.. 96, 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerson, A. , Milyavsky, M. , Rotter, V. , 2004. p53 mediates density-dependent growth arrest. FEBS Lett.. 559, 152–158. [DOI] [PubMed] [Google Scholar]
- Munemitsu, S. , Albert, I. , Souza, B. , Rubinfeld, B. , Polakis, P. , 1995. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl. Acad. Sci. U.S.A.. 92, 3046–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseda, M. , Chang, L. , McLean, G. , Grim, J.E. , Clurman, B.E. , Smith, L.L. , Karsan, A. , 2004. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression. Mol. Cell. Biol.. 24, 8813–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, T. , Lopez-Lago, M. , Giancotti, F.G. , 2005. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol.. 171, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki, N. , Takahashi, N. , Kojima, S. , Masuho, Y. , Koga, H. , 2002. Protocadherin LKC, a new candidate for a tumor suppressor of colon and liver cancers, its association with contact inhibition of cell proliferation. Carcinogenesis. 23, 1139–1148. [DOI] [PubMed] [Google Scholar]
- Shtutman, M. , Zhurinsky, J. , Simcha, I. , Albanese, C. , D'Amico, M. , Pestell, R. , Ben-Ze'ev, A. , 1999. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U.S.A.. 96, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurny, R. , Abdoulrahman, K. , Janda, L. , Runzler, D. , Kohler, G. , Castanon, M.J. , Wiche, G. , 2007. Oxidation and nitrosylation of cysteines proximal to the intermediate filament (IF)-binding site of plectin: effects on structure and vimentin binding and involvement in IF collapse. J. Biol. Chem.. 282, 8175–8187. [DOI] [PubMed] [Google Scholar]
- Stockinger, A. , Eger, A. , Wolf, J. , Beug, H. , Foisner, R. , 2001. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J. Cell Biol.. 154, 1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K. , Murakami, M. , Yamanaka, S. , 2005. Role of the phosphoinositide 3-kinase pathway in mouse embryonic stem (ES) cells. Biochem. Soc. Trans.. 33, 1522–1525. [DOI] [PubMed] [Google Scholar]
- Tanoue, T. , Takeichi, M. , 2005. New insights into Fat cadherins. J. Cell Sci.. 118, 2347–2353. [DOI] [PubMed] [Google Scholar]
- Toda, T. , Kimura, N. , 1997. Standardization of protocol for Immobiline 2-D PAGE and construction of 2-D PAGE protein database on World Wide Web home page. Jpn. J. Electroph.. 41, 13–19. [Google Scholar]
- Venkov, C.D. , Link, A.J. , Jennings, J.L. , Plieth, D. , Inoue, T. , Nagai, K. , Xu, C. , Dimitrova, Y.N. , Rauscher, F.J. , Neilson, E.G. , 2007. A proximal activator of transcription in epithelial–mesenchymal transition. J. Clin. Invest.. 117, 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielenga, V.J. , Smits, R. , Korinek, V. , Smit, L. , Kielman, M. , Fodde, R. , Clevers, H. , Pals, S.T. , 1999. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol.. 154, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L.Y. , Wang, W. , Peng, J.X. , Yang, J.Q. , Huang, G.W. , 2004. Differentially expressed genes between solitary large hepatocellular carcinoma and nodular hepatocellular carcinoma. World J. Gastroenterol.. 10, 3569–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegers, M.M. , Forget, M.A. , Chernoff, J. , Mostov, K.E. , ter Beest, M.B. , Hansen, S.H. , 2003. Pak1 and PIX regulate contact inhibition during epithelial wound healing. Embo J.. 22, 4155–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, B.P. , Hung, M.C. , 2005. Wnt, hedgehog and snail: sister pathways that control by GSK-3beta and beta-Trcp in the regulation of metastasis. Cell Cycle. 4, 772–776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data