

Abstract
Membrane‐initiated androgen actions have now been acknowledged, even though a specific binding site has not been biochemically characterized yet. Recent data indicate that testosterone–BSA, a non‐permeable testosterone analog, can exert specific actions in breast cancer cell lines, including proper transcriptional effects, independent of the intracellular androgen sites. In the present work we explore the effects of testosterone–BSA in two specifically modified pathways, revealed by early trascriptome analysis, namely the non‐genotropic androgen signaling and the HIF1α pathway. We provide evidence that p38 MAPK and PI3K/Akt/NFκB and/or Rho/Actin pathways are directly involved in testosterone‐induced apoptosis, while the JNK/c‐JUN pathway is involved in membrane site‐initiated transcription. Furthermore we show that membrane‐acting androgens modify the transcription of the erythropoietin receptor (EPOR), leading to erythropoietin‐initiated actions. Interestingly, association of recombinant human erythropoietin (rHuEPO) together with testosterone–BSA protects cells from apoptosis, through discrete signaling events. The effect of testosterone–BSA is exerted through the classical erythropoietin promoter, while rHuEPO decreases the transcription of EPOR acting on a newly identified regulatory/promoter region, upstream of its known promoter. These results suggest a new interaction of membrane‐acting androgen with EPOR and should be taken into account in the pharmaceutical manipulations of breast cancer patients.
Keywords: Breast cancer cell line (T47D, MDA-MB-231); Apoptosis; Signaling; Membrane‐acting androgen; Erythropoietin; Erythropoietin receptor
Abbreviations
- rHuEPO
recombinant human erythropoietin
- mAR
membrane androgen receptors
1. Introduction
Rapid effects of steroids have been recognized (Szego and Davis, 1967); however, in spite of two consensus reports on their characteristics (Falkenstein et al., 2000; Wehling, 1997), discussion concerning their molecular nature still persists. Rapid or membrane‐initiated steroid effects have been considered to take place either after translocation of intracellular steroid‐binding molecules (Pedram et al., 2006), anchored to the plasma membrane by post‐transcriptional modifications (Acconcia et al., 2005, 2004, 2009), or through integral membrane proteins, like G‐protein coupled receptors (GPCR). In addition, an increasing amount of data pinpoints the structural or functional interaction of membrane steroid‐binding elements (including androgen) with growth factor receptors (reviewed in Kampa et al., 2008). Androgens have been reported to exert membrane binding and signaling activity (reviewed in Kaarbo et al., 2009, 2004, 1999, 2007, 2002, 2007, 2006, 2005). In addition, membrane androgen receptors (AR) have been reported to interact with EGFR (Bonaccorsi et al., 2004). Both in breast (Pelekanou et al., 2007) and prostate cancer specimens (Dambaki et al., 2005), membrane androgen binding elements have been described, in the presence of AR‐antagonists, providing histological evidence about distinct molecular forms of membrane androgen sites, resulting in tumor‐reduction (Hatzoglou et al., 2005). Although concrete molecular entities have not been described yet, they might present a homology with classical AR, as they are (partially) recognized by the N‐20 anti‐AR antibody (Kampa et al., 2005; Notas et al., in press). Downstream cascades, involved in membrane androgen‐triggered events, include Ca2+ mobilization (Kampa et al., 2004), FAK/PI3K/Akt/Rac/Actin cytoskeleton rearrangements (Nifli et al., 2005; Papakonstanti et al., 2003), and initiation of a number of major signaling pathways, orienting cells towards apoptosis (reviewed in Kaarbo et al., 2007; Kampa and Castanas, 2006).
In a recent work, we presented early (3h) transcriptomic data suggesting specific testosterone–BSA (an impermeable testosterone analog) early‐regulated genes (Notas et al., in press). Among the pathways modified in two different breast cancer cell lines (T47D and MDA‐MB‐231), androgen mediated signaling and HIF1α pathways emerged. In a previous work, we have reported that the HIF1α‐gene target protein erythropoietin (EPO) and its receptor (EPOR), as well as membrane androgen receptor sites were co‐expressed and correlated in human breast cancer specimens. We have further shown a negative correlation of their expression with patients' survival (Pelekanou et al., 2007). Based on these results, we have analyzed the transcriptional and phenotypic changes induced by testosterone–BSA in the two above mentioned cell lines, in order to better define the mechanism of functional synergy of the two systems.
2. Material and methods
2.1. Transcriptome analysis
For the transcriptome analysis of testosterone–BSA modified genes we have used our previously reported data (Notas et al., in press), (NCBI Gene 113Expression Omnibus (GEO) repository GSE18146). We have analyzed the terms “androgen mediated signaling” (identifying two components: “non‐genotropic androgen signaling” and “regulation of androgen receptor activity”) and “HIF1α network” from the NCI public pathway interaction database (http://pid.nci.nih.gov). We have further extracted the molecule list involved in each pathway and reanalyzed data for testosterone–BSA‐induced changes, with the use of the Genespring V11 (Agilent, Foster City, CA) program, as described previously (Notas et al., in press).
2.2. Cell culture and chemicals
T47D and MDA‐MB‐231 breast cancer cells (DSMZ, Braunschweig, DE and ATCC, LGC Promochem, Middlesex, UK) were cultured in RPMI 1640 and L‐15, respectively (Invitrogen, Life Technologies, Paisley, UK), supplemented with 10% charcoal‐treated fetal bovine serum, at 37°C and 5% CO2 (MDA‐MB‐231 were cultured without CO2). Testosterone 3‐(O‐carboxy‐methyl)‐oxime:BSA (Testosterone–BSA, 10 steroid molecules per molecule BSA), Testosterone–BSA–FITC (3 Testosterone molecules per molecule BSA), and BSA–FITC were from Sigma Hellas (Athens, Greece). Prior to experiments, Testosterone–BSA was charcoal‐treated (3% charcoal and 0.3% dextran, 30min at 4°C), in order to eliminate any non‐complexed steroid. Our previous results (Kampa et al., 2002) and assays of free testosterone in culture media, after variable incubation periods, ensured that no free testosterone and therefore no metabolism of the compound occurs for the time periods used in the present study. Erythropoietin (recombinant human Epoetin alpha, EPREX®, 40,000IU) was from Janssen‐Cilag (Switzerland), while Wortmannin (a PI3K inhibitor), SB203580 (p38 kinase inhibitor), AG490 (JAK2 kinase inhibitor) and SP600125 (JNK inhibitor) were from Calbiochem (San Diego, CA). Cell Dissociation Buffer Enzyme‐Free Hanks'‐based was from Invitrogen. All other chemicals were from Sigma Hellas. EPO molarity was calculated on the basis of a 34kDa molecular weight and an activity of 160,000IU/mg; under these conditions, an activity of ∼533IU/nmol was calculated. Preliminary experiments indicated that neither glycine (up to 5mg/ml) nor polysorbate 80 (up to 0.5mg/ml), used as Epoetin alpha stabilizers, induce any interference in cells.
2.3. Apoptosis assay
Cells were seeded at an initial density of 200,000 cells/well, and were treated with Testosterone–BSA and/or rHuEPO, in the absence, or in the presence of different kinase inhibitors (wortmannin 0.1μM, SP600125 25μM, SB203580 25μM, or AG490 10μM). Apoptosis was quantified by flow cytometry, using Annexin V/PI staining (FITC Annexin V Apoptosis Detection Kit II, BD‐Pharmingen, Franklin Lakes, NJ).
2.4. Quantitative reverse transcription‐PCR (qRT‐PCR)
Total RNA was isolated using Nucleospin II columns (Macheray‐Nagel, Düren, Germany). Quantitative RT‐PCR, after DNase treatment and reverse transcription, was performed using the following primers: EPOR: forward 5′‐GCTGACGCCTAGCGACCT‐3′; reverse 5′AGCCACAGCTGGAAGTTACC‐3′; Actin Forward 5′‐CTTCGTCGCACATTGTGTCT‐3′; reverse 5′‐GACAGCGCCAAGTGAAGC‐3′; Cyclophilin A: forward 5′‐ATGGTCAACCCCACCGTGT‐3′; reverse 5′‐TTCTGCTGTCTTTGGAACTTTGTC‐3′. They were synthesized by VBC Biotech (Vienna, Austria). Real‐time PCR with SYBR Green was performed with DyNAmo SYBR Green qPCR Kit with ROX (Finnzymes, Oy, Finland), using the ABI Prism 7000 Sequence Detection System (Applied Biosystems), with the following program: 95°C for 3min followed by 40 cycles of 95°C for 15s then 60°C for 60s. The changes were normalized according to cyclophylin A mRNA expression. Each set of primers was tested with at least three different RNA samples treated independently.
2.5. Cell signaling protein‐detection assay
Cells were incubated with BSA‐conjugated Testosterone (10−7M) and/or rHuEPO (10−7M) for different time intervals (2, 5, 10, 15, 20, 40 and 60min), washed with ice‐cold TBS and lysed using Universal lysis buffer or lysis buffer A, according to the manufacturer's instructions (Upstate, NY, USA). Detection of cell signaling molecules was performed using Beadlyte Cell Signaling Buffer kit and Cell Signaling Universal Buffer kit according to the manufacturer's protocol (Upstate, NY, USA) using an xMAP Luminex 100 platform (Austin, TX). The following phosphorylated and total signaling molecules were assayed: total and phospho STAT3 (Tyr705), total and phospho STAT5 (Ser727), total and phospho p38 kinase (Thr180/Tyr182), total and phospho JNK kinase (Thr183/Tyr185), phospho c‐jun (Ser73) and activated caspase‐3. Briefly, 25μl of each sample containing 25μg protein were incubated overnight at 4°C in a 96‐well plate with microsphere‐coupled primary antibodies, washed and incubated with signaling molecules‐specific reporter antibodies, followed by streptavidin–phycoerythrin specific labeling according to the manufacturer's protocol and analyzed using the Luminex 100 apparatus. Results were obtained as Median Fluorescence Intensity (MFI).
2.6. EPOR promoter constructs, transient transfection of cells and luciferase assay
The promoter of EPOR was extracted from human genomic DNA, using PCR, with GGTATTCCCAGCACACATTG as forward and GAGTCTTGGATCCTAACATACCC as reverse primers. The PCR product, spanning from −792 to +314 from the open reading frame of EPOR, was digested with BamHI/BtgI or ApaI/BtgI, giving either (a) a 691bp product (−709 to −18) or (b) a 161bp product (−188 to −18), where the 3′ end of both sequences is in the untranslated region of the EPOR gene. pBluescript was digested with SacI/KpnI and a linker including restriction sites for BamHI, ApaI, BtgI and XhoI was added (lpBluescript). lpBluescript was digested with either BamHI/BtgI or ApaI/BtgI and the corresponding pieces of the digested PCR products were added ((a) lpBluescipt and (b) lpBluescipt). Finally (a) lpBluescipt and (b) lpBluescipt were digested with SacI/XhoI and the two pieces were added to the multicloning region of pGL3‐basic vector (pGL3‐(a)EPORprom and pGl3‐(b)EPORprom). The construct was verified with sequencing in an ABI PRISM® 7700 Sequence Detection System.
Transfection of T47D cells with pGL3‐(a)EPORprom and pGl3‐(b)EPORprom was performed with Lipofectamin 2000 (Invitrogen, Carlsband, USA) according to the manufacturer's instructions. 24h later, rHuEPO and Testosterone–BSA (with or without pathway inhibitors) were added and cells were incubated for another 24h. Thereafter protein was collected and assayed with the Dual‐Luciferase Reporter Assay System (Promega, Madisson, WI, USA), in a Berthold luminometer (Pfortzheim, Germany). All experiments were conducted twice in triplicates. In parallel, an empty pGL3‐basic vector was used as a further control. Results were normalized for the corresponding control value, derived from untreated cells.
2.7. Actin cytoskeleton visualization
Cells growing on poly‐l‐lysine coated 22×22mm coverslips were treated with testosterone–BSA (10−7M), EPO (10−7M) or their combination, for 15min. Subsequently, they were fixed in 4% paraformaldehyde for 10min and permeabilized by 0.5% TritonX100 for 10min. They were then incubated with 2% BSA for 15min, followed by rhodamine‐labeled phalloidine staining, for 45min at room temperature, mounted with mounting medium and analyzed using a confocal laser‐scanning module attached to an inverted microscope equipped with an argon–krypton ion laser.
2.8. Statistical analysis
Data were analyzed by the SPSS V16 program, using analysis of variance (with the residual variance used for the determination of a common SEM) or Student's t‐test, as appropriate. A 5% level was used for statistical significance.
3. Results
3.1. Testosterone–BSA modifies early transcription of genes related to androgen signaling and HIF1α networks
Analysis of previously published transcriptomic data indicates that testosterone–BSA, a membrane interacting analog, modifies a number of genes associated with major intracellular pathways, related to androgen signaling. Androgen signaling pathways‐related gene signatures were extracted from our previously published data (Notas et al., in press) and presented in Figure 1A and Supplemental Tables 1 and 2. As shown, a significant number of gene transcripts were modified under the action of testosterone–BSA, in both T47D and MDA‐MB‐231 cells, with the majority of them being related to transcriptional elements.
Figure 1.
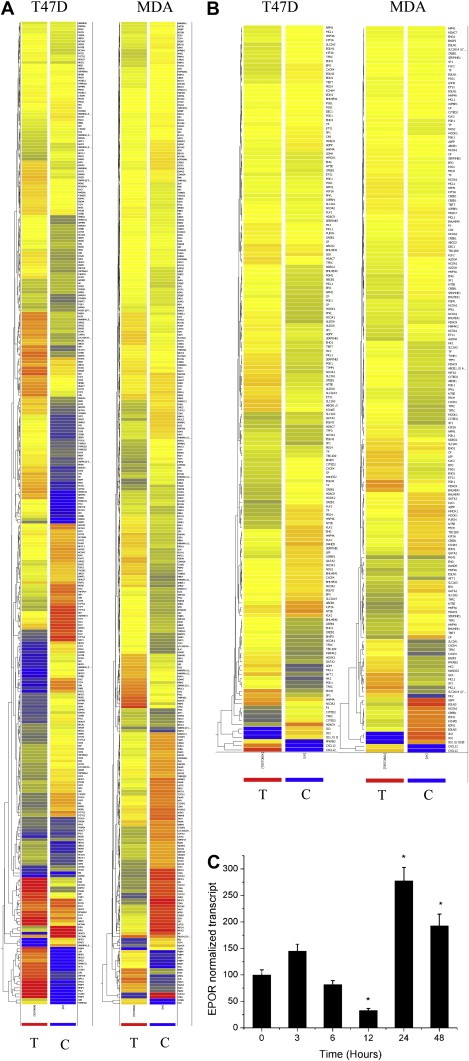
Transcriptional changes of T47D and MDA‐MB‐231 cells, incubated with Testosterone–BSA. Genes, involved in androgen receptor signaling (A) or HIF1α‐related transcription (B) are presented. Control (untreated, C) and Testosterone–BSA‐treated cells (T) are shown. Early transcription (3h) up‐ or down‐regulated genes are presented. In C, EPOR mRNA was assayed by qRT‐PCR in T47D cells. * denotes statistical significance (at least p<0.05).
HIF1α‐target genes display a marked transcriptional modification upon testosterone–BSA stimulation (Figure 1B and Supplemental Tables 3 and 4) (Notas et al., in press). This change confirms previous data of our group (Pelekanou et al., 2007), suggesting also that both membrane androgen sites and HIF1α nuclear localization is evidenced in breast cancer specimens (an example is given in Supplemental Figure 1), suggesting a potential modification of HIF1α‐regulated proteins by testosterone–BSA in the breast. Among genes modified by HIF1α we have spotted Erythropoietin (EPO), which acts through binding with its cognate receptor EPOR. Both EPO and EPOR were equally detected in breast cancer specimens, correlating with membrane androgen binding elements (Pelekanou et al., 2007). Analysis of EPOR transcription as a function of time, in testosterone–BSA‐treated T47D cells (Figure 1C), verified that this androgen analog, acting exclusively at the cell membrane, modifies EPOR transcription, increasing it at long incubation times (≥24h). This might be of potential importance, as recombinant human EPO (rHuEPO) is administered in oncologic patients to palliate for cancer‐related anemia, while a number of reports suggest that this agent might lead to a decreased survival of patients (Khuri, 2007; Yasuda et al., 2003), a result equally reported by us, in a series of human invasive breast carcinoma analysis (Pelekanou et al., 2007).
3.2. Membrane‐acting androgens modify expression and action of non‐genotropic androgen signaling, leading to apoptosis
Recently, a non‐genotropic androgen membrane‐initiated pathway has been released (http://pid.nci.nih.gov). We have therefore extracted gene signatures, related to this pathway and analyzed changes induced by testosterone–BSA (Supplemental Tables 5 and 6). The majority of gene signatures correspond to signaling molecules, expected to be modified after a membrane androgen action. We have therefore investigated further signaling molecule activation (phosphorylation) induced by testosterone–BSA, in T47D cells. As those changes might occur at short time intervals, we have examined minute‐induced changes and analyzed major signaling pathways (PI3K/Akt, JNK/c‐JUN, p38 MAPK, STAT activation) and the subsequent CREB and NFκB activation (through the IκB phosphorylation) (Figure 2A). We verified that testosterone–BSA rapidly induced Akt and IkappaB changes, p38 MAPK activation, followed by a CREB phosphorylation. In addition, a slight but sustained activation of STAT3 was also observed, linking testosterone–BSA to cytokine‐growth factor signaling. We report further a highly increased and sustained active caspase‐3 level, linking testosterone–BSA signaling activation to apoptosis, as reported previously (Kampa et al., 2005). No significant modifications of the c‐Jun, Jnk or STAT5 were observed (not shown). These data confirm previously published membrane‐initiated testosterone signaling (Papakonstanti et al., 2003) and suggest that testosterone–BSA induces changes at the phospho‐proteomic signaling and the transcriptomic level.
Figure 2.
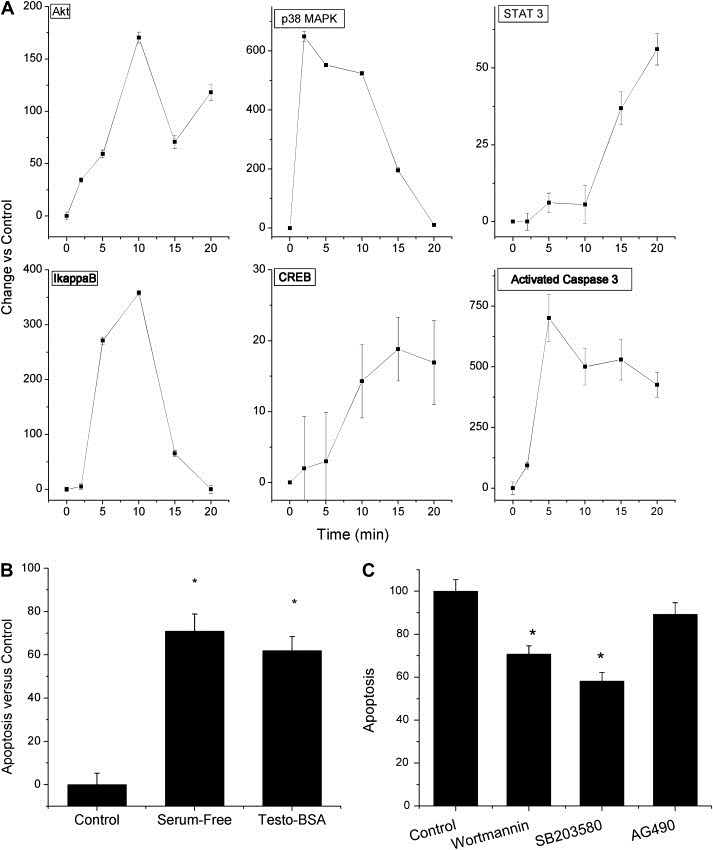
Incubation of T47D cells with Testosterone–BSA (10−7M) induces early signaling events and modifies apoptosis. A. Graphic representation of activated signaling molecules Akt, IkappaB, p38 MAPK, CREB, STAT3 and activated caspase‐3 in T47D cells cultured in serum‐supplemented media with Testosterone–BSA (10−7M) versus control (non‐supplemented) cells, for the indicated periods of time. Figure presents the difference in activation of each signaling molecule as compared to the control (untreated cells). B. Serum‐omission induces a massive apoptosis of T47D cells, after a 24h incubation. A similar pro‐apoptotic effect was equally observed after incubation of cells (in serum‐supplemented conditions) with 10−7M testosterone–BSA. C. The effect of testosterone–BSA (control in the graph) in apoptosis of T47D cells was assayed after a 24h incubation in the presence of wortmannin (PI3K/Akt inhibitor), SB203580 (p38 MAPK inhibitor), or AG490 (JAK2 inhibitor). Data were normalized as per the effect of Testosterone–BSA (control), in the absence of inhibitors. Mean±SEM of three different assays in triplicate, *p<0.05 at least, compared to the no‐inhibitors bars.
The most prominent effects of testosterone–BSA action in both breast and prostate cancer cells concerns actin cytoskeleton changes and apoptosis (Hatzoglou et al., 2005, 2006, 2005), verified equally in our study (Figure 2B). We have therefore assayed the implication of the above mentioned signaling pathways in the pro‐apoptotic effect of testosterone–BSA. Inhibition of PI3K/Akt and p38 MAPK by wortmannin and SB283580 reversed partially the pro‐apoptotic action of testosterone–BSA, suggesting the implication of these two signaling pathways in its pro‐apoptotic effect. In contrast, no significant changes of apoptosis were observed by inhibiting JAK2 kinase by AG490 (considered as an early activator of STATs phosphorylation), which affected apoptosis only by ∼10%, suggesting that the JAK‐STAT pathway might not be a major player in the pro‐apoptotic action of testosterone (Figure 2C). We have further verified that inhibition of JNK by SP600125 does not induce any change in testosterone–BSA‐induced apoptosis, strengthening our hypothesis on the non involvement of this pathway in the pro‐apoptotic effects of the hormone (not shown).
3.3. Membrane‐initiated signaling cascades mediate the effect of testosterone–BSA on EPOR transcription
The net action of membrane‐ and nuclear‐initiating signaling results in discrete transcriptional modifications. In view of the observed modification of HIF1α pathway and the effect of testosterone–BSA on EPOR transcription (Figure 1C), we have examined whether the reported signaling events, initiated by testosterone–BSA could be involved. In this respect, we first analyzed the action of testosterone–BSA on the EPOR gene promoter. An EPOR promoter region has been identified, in front of the transcription initiation site (Noguchi et al., 1991, containing GATA (−184 to −179) and Sp1 (−156 to −151) sites). However, a thorough in silico search upstream of this promoter, revealed another region, at a 500bp distance, containing putative transcription factor‐binding sites, including CREB, ARNT, AP‐1 and AhR. We have therefore produced two different constructs (a short one containing the −188 to −18 sequence‐traditional promoter, termed short promoter, and a second, long promoter, construct containing the −709 to −18bp upstream of the transcription initiation site of the gene), linked to luciferase. Transient transfection of T47D cells with the short construct and application of testosterone–BSA for 24h, revealed that the hormone stimulates luciferase activity (Figure 3A), suggesting a positive effect of membrane‐acting testosterone on EPOR gene transcription, consistent with data presented in Figure 1C. In contrast, a minor effect was observed on the long construct.
Figure 3.
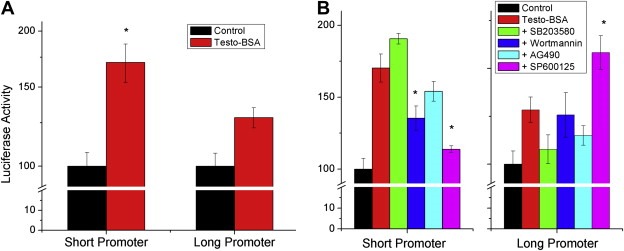
Testosterone–BSA modifies the transcription of EPOR, through a direct effect on its promoter. Two constructs of the EPOR promoter were made: a short one containing the −188 to −18 sequence‐traditional promoter, termed short promoter, and a second, long promoter, construct containing the −709 to −18bp of the putative promoter region, linked to luciferase. A. Effect of Testosterone–BSA (Testo‐BSA, 10−7M, 24h) on the short and the long promoter constructs, in transiently transfected T47D cells. B. Effect of signaling pathways inhibitors (SB203580: p38 MAPK inhibitor; Wortmannin: PI3K/Akt inhibitor; AG490: JAK2 inhibitor; SP600125: Jnk inhibitor) on the transcriptional activity of Testosterone–BSA (10−7M, 24h) on EPOR short and long promoter constructs.
In view of the effect of testosterone–BSA on a number of signaling cascades, we have further assayed whether its transcriptional effect on EPOR could be modified through attenuation of signaling. We have therefore assayed the transcriptional effect of testosterone–BSA on EPOR (through the use of the short and long promoter constructs) after addition of selective inhibitors (Figure 3B). Addition of SB203580 (p38 MAPK inhibitor) or AG490 (JAK2 inhibitor) did not modify significantly luciferase activity of the short promoter, indicating that neither p38 MAP kinase, nor STAT pathways are direct transducers of EPOR membrane‐initiated testosterone transcription. In contrast, wortmannin (PI3K/Akt inhibitor) and especially SP600125 (JNK inhibitor) significantly decreased the effect of testosterone on the short construct. The latter inhibitor was the only identified agent which modified the effect of the hormone on the long construct. In this respect, our data advance two different elements, initiated by testosterone–BSA: PI3K/Akt and p38 MAPK pathways, linked predominantly to apoptosis, and JNK pathway, linked to transcription of target genes.
3.4. Activation of EPOR modifies the pro‐apoptotic actions of testosterone–BSA
The above results indicate that testosterone–BSA membrane‐initiating signaling events result in an increased EPOR transcription. EPOR is present in T47D (as well as in MDA‐MB‐231) cells (Supplemental Figure 2). In this respect, we have further explored whether a functional interaction between membrane androgen sites and EPOR may result, especially in view of the cytoprotective action of rHuEPO (Acs et al., 2004; reviewed in Farrell and Lee, 2004; Jelkmann et al., 2008; Lacombe and Mayeux, 1999; Spivak, 2005; Yasuda et al., 2003). We therefore examined whether rHuEPO could modify events initiated by testosterone–BSA. rHuEPO per se has no effect on apoptosis of breast cancer cells, cultured in the presence of serum (conditions under which testosterone–BSA exerts its pro‐apoptotic action). In contrast, in serum‐deprived cells, rHuEPO reverts apoptosis (not shown), a result previously reported in other cell systems (Acs et al., 2004; reviewed in Farrell and Lee, 2004; Jelkmann et al., 2008; Lacombe and Mayeux, 1999; Spivak, 2005; Yasuda et al., 2003). Interestingly, when cells were co‐incubated with a submaximal concentration of testosterone–BSA (10−7M) and variable concentrations of rHuEPO, a dose‐ and time‐dependent anti‐apoptotic effect was observed (Figure 4B and A respectively), reverting testosterone–BSA pro‐apoptotic effect. This was accompanied by a decrease in the rapid activation of caspase‐3 (Figure 4C). In addition, rHuEPO reverted another specific effect of testosterone–BSA, namely actin cytoskeleton rearrangement (Figure 4E). In this case, addition of rHuEPO (especially in the presence of testosterone–BSA) induced the formation of filopodia and lamelipodia (Figure 4E, arrows). It is further interesting that testosterone–BSA significantly reduced actin mRNA expression. This reduction was rapidly (at 6h) reverted by the addition of rHuEPO, which alone had no effect on actin transcription (Figure 4D). It is to note, that the actin rearrangement effect of the combination rHuEPO+testosterone–BSA did not imply major associated modification of the migratory potential effect of treated cells (not shown).
Figure 4.
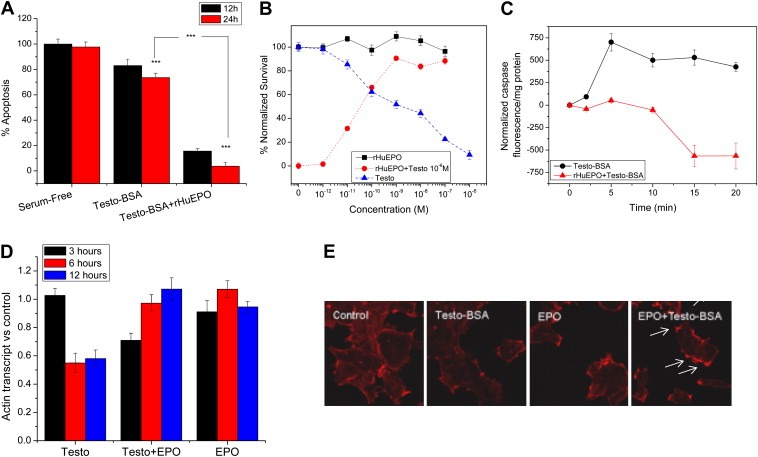
rHuEPO acts as an anti‐apoptotic agent of testosterone–BSA‐induced apoptosis in T47D breast cancer cells. A. Time‐related inhibition of Testosterone–BSA‐induced apoptosis of T47D cells, in the presence of rHuEPO (10−7M of each agent). Mean±SEM of three assays in triplicate. B. rHuEPO has no effect in a serum‐supplemented medium (squares). In contrast, Testosterone–BSA induces a dose‐dependent apoptosis of cells (24h incubation, triangles). Incubation of cells in the presence of 10−7M Testosterone–BSA and variable concentrations of rHuEPO (circles) reverses the pro‐apoptotic effect of Testosterone–BSA, in a dose‐dependent manner. Mean±SEM of three different experiments in triplicate. C. Time‐related changes of active caspase‐3 of T47D cells, in the presence of Testosterone–BSA alone (circles) or combined with an equimolar concentration of rHuEPO (triangles). Each agent was induced at 10−7M. Caspase activation was normalized according to control cells, cultured for the same period. Mean±SEM of three independent experiments, in triplicate. D. Testosterone–BSA induces a profound decrease of actin mRNA, assayed by RT‐PCR. The effect was evident after 6‐h incubation and sustained for at least 12h. This inhibition was time‐dependently reversed by the co‐incubation of cells with Testosterone–BSA and rHuEPO (10−7M each). rHuEPO per se had no effect on actin transcription. E. Testosterone–BSA induces a profound sub‐cortical actin cytoskeleton distribution, stained by rhodamine–phalloidin and visualized in a confocal laser microscope. rHuEPO and especially its association with Testosterone–BSA (10−7M each, 20min) induced the formation of filopodia and lamelipodia (arrows).
3.5. rHuEPO modifies testosterone–BSA signaling
Membrane‐acting androgens were reported to interact functionally with cytokine and growth factor receptors. However, an interaction with EPOR was not previously reported. In this respect and in view of the modification of testosterone–BSA‐induced apoptosis by rHuEPO, we investigated whether rHuEPO modifies also rapid signaling events initiated by testosterone–BSA. PI3K/Akt and IkappaB testosterone–BSA‐induced changes were not altered by the co‐incubation of T47D cells with rHuEPO and testosterone–BSA (not shown), suggesting that this pathway is mainly under the control of membrane‐acting androgen, as reported previously (Nifli et al., 2005; Papakonstanti et al., 2003). However, other major signaling pathways were profoundly modified by the addition of rHuEPO to testosterone–BSA‐treated T47D cells (Supplemental Figure 3). In details, rHuEPO addition delayed p38 MAPK activation, suppressing its early burst found in testosterone–BSA‐treated cells and finally resulted in a massive CREB activation. In addition, it induced a high increase of JNK and c‐Jun. Interestingly, the most prominent change (in addition to the Jnk/c‐Jun) was the switch from STAT3 to STAT5 activation, which is transiently increased by almost 10‐fold. The implication of these signaling cascades in the pro‐/anti‐apoptotic effect of the agents was verified by using pathway‐specific inhibitors (Supplemental Figure 4A). The pro‐survival effect of rHuEPO+Testosterone–BSA combination was reversed by all inhibitors: wortmannin, acting on the Akt/PI3K pathway induced a 3‐fold increase in apoptosis, AG490 induced an almost four‐fold increase in apoptosis, while SB203580 and SP600125 increased apoptosis by 2.5‐fold.
3.6. Transcriptional regulation of EPOR by testosterone–BSA and rHuEPO
In rHuEPO and testosterone–BSA co‐treated T47D cells, the JNK pathway mainly influences apoptosis, while, under testosterone–BSA treatment, this signaling cascade modifies transcription, as reported above. We have therefore assayed transcription of the EPOR gene, using the two constructs as described above and cells incubated with equimolar concentrations of testosterone–BSA and rHuEPO (10−7M). Our data (Figure 5A) indicate that rHuEPO acts preferentially, albeit in a moderate fashion, on a regulatory region upstream of the EPOR promoter, decreasing the gene's transcription. In contrast, testosterone–BSA (acting through JNK signaling, as shown above) acts on the proximal part of the promoter, inducing the transcription of EPOR. However, when T47D cells were co‐incubated with testosterone–BSA and rHuEPO, a positive effect on the proximal part of the promoter was observed (Figure 5A), leading to an enhanced EPOR transcription (Figure 5B). This effect was obvious after 24h incubation (a time point at which the effect of the agents on the promoter regions was assayed). Interestingly, at this time point, testosterone–BSA, in combination with rHuEPO, exerts an enhancement of luciferase activity on the distal part of the promoter too, reverting the effect of rHuEPO, suggesting that both parts may play a role under co‐incubation with testosterone–BSA and rHuEPO. However, EPOR transcript was higher in testosterone–BSA‐treated cells, suggesting that additional elements (such as RNA stability) might be involved in the transcriptional modification of EPOR. In addition, at shorter incubation times (12h), an inhibitory effect of either rHuEPO or testosterone–BSA on EPOR transcription was observed, partially reverted by the co‐administration of both hormones (Figure 5B), while, at 6h, no significant modification of EPOR transcript was observed, suggesting a delayed direct or indirect effect. However, the addition of signaling antagonists revealed a further interesting interaction: all four major pathways (PI3K/Akt, p38 MAPK/NFκB, JNK and Jak/STAT) are involved in the transcription enhancement of EPOR, as their inhibitors decreased the short promoter‐induced luciferase activity. However, the effect of the combination (rHuEPO+testosterone–BSA) on the long promoter construct was inhibited only by the inhibitors of the p38 MAPK/NFκB and JAK2/STAT pathways. In contrast, PI3K/Akt and JNK inhibitors were ineffective, suggestive of a differential regulation of EPOR.
Figure 5.
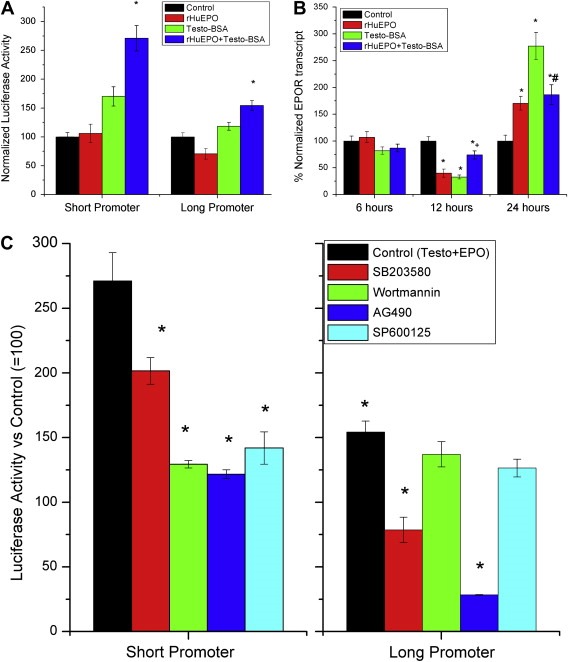
Effect of rHuEPO on the transcriptional effect of Testosterone–BSA on EPOR. A. Effect of rHuEPO, Testosterone–BSA (10−7M) or their equimolar association on luciferase activity in lysates from T47D cells transiently transfected with the short or the long promoter construct, after 24h incubation. *p<0.05 at least, mean±SEM of three assay. B. Quantitative RT‐PCR of EPOR in T47D cells incubated with either agent (10−7M) or their equimolar association, for the indicated time periods. *p<0.05 at least compared to control (non treated cells). +p<0.05 at least compared to testosterone–BSA or rHuEPO‐treated cells, #p<0.05 compared to testosterone–BSA‐treated cells; Mean±SEM of three assays. C. Effect of kinase inhibitors SB203580 (p38 MAPK inhibitor), Wortmannin (pI3K inhibitor), AG490 (JAK2 inhibitor) and SP600125 (JNK inhibitor) on T47D cells, transiently transfected with the short (left panel) or the long (right panel) EPOR promoter transcript and incubated with Testosterone–BSA, or an equimolar association of rHuEPO and Testosterone–BSA (10−7M).
3.7. The effect of rHuEPO on testosterone–BSA‐induced apoptosis is not restricted in T47D cells
As reported above (Figure 1A and B), testosterone–BSA induces transcriptional changes in both ERα & AR positive (T47D) and ERα & AR negative (MDA‐MB‐231) cells. A question arising is whether the reported interaction of testosterone on the transcriptional regulation of EPOR and the effect of the combination of testosterone–BSA+rHuEPO is cell line‐specific, or is also exerted in other breast cancer cell lines, expressing or not ERα. We have therefore examined the transcriptional modulation of EPOR and the combined effect of both agents on apoptosis of MDA‐MB‐231 cells, which express also EPOR and membrane testosterone sites (Supplemental Figure 2 and Notas et al., in press), but are negative for ERα and intracellular testosterone sites. Testosterone–BSA induces a similar modification of EPOR transcription in MDA‐MB‐231 cells, with an initial significant rise of EPOR transcript at 3h, a return in control levels at 6–12h and a delayed increase at 24h (Figure 6A). However, no significant reduction of transcription at 12h was observed. Furthermore, in MDA‐MB‐231 cells, incubation with testosterone–BSA also induces apoptosis. Here too, rHuEPO reverts the pro‐apoptotic effect of testosterone–BSA (Figure 6B). These data suggest that testosterone–BSA/EPO interactions reflect a more “universal” trait of breast cancer biology, independent of the presence of nuclear androgen or estrogen receptor.
Figure 6.
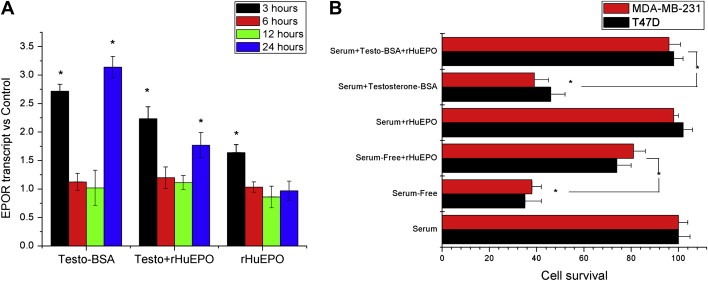
The interaction of rHuEPO with Testosterone–BSA is also exerted in MDA‐MB‐231 breast cancer cells. A. Effect of Testosterone–BSA, rHuEPO or their association on the transcription of EPOR gene, in different time points. B. Comparison of rHuEPO±Testosterone–BSA effect on apoptosis in T47D (black bars) and MDA‐MB‐231 cells (grey bars). Data were normalized as per serum‐supplemented conditions, in the absence of any agent. In both panels, * denotes statistical significance (at least p<0.05); Mean±SEM of three experiments performed in triplicate. All agents were added at a concentration of 10−7M.
4. Discussion
Even though the quest on the molecular nature of membrane androgen binding sites has not resulted, until now, to unanimously accepted results, a whole transcriptome analysis of early changes, induced by testosterone and testosterone–BSA (impermeable) conjugate, reveals a number of genes, uniquely modified by membrane‐acting androgen (Notas et al., in press). This result is suggestive for a specific membrane‐initiated interaction of androgen, independent from that induced by non‐conjugated testosterone and prone for a specific site (not yet identified) of testosterone membrane action in the breast. However, both membrane‐initiated and intracellular (nuclear) androgen actions trigger a massive (albeit different) modification of gene transcription (Notas et al., in press).
Among genes selectively modified by testosterone–BSA the ones involved in androgen receptor pathways and HIF1α transcription can be distinguished, according to standard GO terms and Nature‐curated NCI pathway interaction databases. Here, we have further analyzed these pathways, critical for the survival/apoptosis fate of breast cancer cells. These pathways are appealing in breast cancer biology as androgen can be found in the breast cancer cell environment (reviewed in Dimitrakakis and Bondy, 2009; either circulating or locally produced Spinola et al., 1988), although this result is not unanimously accepted (see below). On the other hand, HIF1α‐initiated pathways lead to modification of a number of tumor properties, through increased angiogenesis and metabolic adaptation, leading to cancer cell proliferation, migration and survival (see Weidemann and Johnson, 2008, for a recent review). EPO transcription is directly modified by HIF1α, while its receptor EPOR, a gene modified by EPO, is crucial for cancer cell survival (Hardee et al., 2007; Lai et al., 2005 and results of the present study). However, data on EPOR transcriptional modulation remain limited.
Testosterone–BSA, being a non‐permeable androgen, exerts transcriptional changes only through initiation of specific intracellular signaling cascades. For this reason, we have investigated major intracellular pathways, namely: (i) the Akt/PI3K/NFκB; (ii) the JAK/STAT; (iii) the JNK/c‐Jun; (iv) the p38 MAPK/CREB. Our data show that PI3K/Akt and p38 MAPK pathways and the JNK/c‐Jun pathway are linked to the pro‐apoptotic and transcriptional action of testosterone–BSA respectively. In contrast, the JAK/STAT pathway has no effect. p38 MAPK has four different isoforms (named α–δ), with specific actions on cell proliferation, differentiation and apoptosis (Zarubin and Han, 2005). Interestingly, a recent report indicates that p38α is the dominant isoform of p38 in both cell lines (T47D and MDA‐MB‐231) and breast tumors (Chen et al., 2009). These authors further report that p38α activation induces cell proliferation, while its inhibition results in cell cycle arrest, with no effect on apoptosis. However, these data were obtained in non‐challenged cells. In contrast, here we report that, after testosterone–BSA stimulation, p38 MAPK is directly involved in the pro‐apoptotic effect of the agent. Chen et al. (2009) suggest that this is due to a functional interaction of activated p38 MAPK with the p53 protein, with the wild type protein inducing apoptosis, while its mutated forms lead to cell proliferation. T47D cells contain a mutated form of p53 (Lim et al., 2009) and therefore, early activation of p38 should protect cells from apoptosis. However, other mechanisms, including a GADD45 p38/JNK interaction (reviewed by Liebermann and Hoffman, 2008; Yin et al., 2004) might be also involved. In this respect, our data join the current concept that p38 MAPK pathway may function as a tumor suppressor through regulating Ras‐dependent and ‐independent proliferation, transformation, invasion and cell death (Loesch and Chen, 2008).
In contrast to the above pathways, JNK/c‐Jun activation by testosterone–BSA induces changes in the transcription of target genes. Here, we have focused on the transcription of EPOR, an element not previously reported as a target of androgen‐dependent transcription and of potential importance in breast cancer, albeit not widely investigated for the moment. We report that EPOR promoter has two distinct elements: a proximal one, previously described (Noguchi et al., 1991), containing putative GATA and Sp1 binding sites and a distal one, at 500bp upstream, revealed in the present study. There are compelling data to indicate that the Sp1‐dependent activation and repression of target promoters is modulated by its interactions with a repertoire of heterogeneous transcriptional regulators; a partial list of Sp1‐interacting proteins includes the Mediator complex, c‐Myc, Ah, Arnt, ER, AR, GATAs, p53, MEF2C, Smad2–4, Msx1, Rb, NFkB, NF‐YA, VHL, MyoD, E2F, NFAT‐1 and YY1 (reviewed in Solomon et al., 2008), linking the effect of testosterone–BSA to transcriptional activation of EPOR. In addition, although the c‐Jun implication in the AP‐1 complex of steroid receptors has been reported since the '90s (Karin et al., 1993), our data provide further evidence for a new interaction which should be further investigated.
The enhanced transcription of EPOR by testosterone–BSA led us to investigate the possible interaction of membrane‐acting androgen with rHuEPO in breast cancer cells. Our data converge to surprising effects: co‐incubation of cells with testosterone–BSA and rHuEPO results in a reversion of the pro‐apoptotic effect of testosterone–BSA, a withdrawal of the inhibitory effect of testosterone on actin transcription and actin cytoskeleton cellular distribution and a further increase of EPOR transcript. This result provides a mechanistic explanation of intriguing data reported previously from our group, that patients with a co‐expression of EPOR and membrane androgen sites have a decreased survival (Pelekanou et al., 2007). In addition, rHuEPO modifies profoundly some of the signaling pathways triggered by testosterone–BSA. In details, rHuEPO induces: (i) a massive transient increase of the JNK/c‐Jun pathway, (ii) a switch of STAT3 to STAT5 activation and (iii) a delayed activation and an anti‐apoptotic switch on p38 MAPK, acting as a cytoprotective agent and enhancing cell survival. The absence of the PI3K/Akt/NFκB pathway modification, is indicative of its exclusive control by testosterone–BSA. Interestingly enough, under these conditions, all four pathways are linked to the anti‐apoptotic action of the combination, as well as to transcriptional modifications, acting preferentially on the proximal EPOR promoter. While thorough changes take place on actin cytoskeleton distribution, they are not followed by corresponding migration modulation, supporting our hypothesis that apoptosis is the main cancer cell function affected by these agents. For the first time, these data integrate EPOR to potential cytokine/growth factor receptors interacting with membrane androgen (see Kampa et al., 2008 for a review).
In a recent report, Stelacci et al. described an interaction of EPOR with membrane glucocorticoid receptors, affecting maturation of proerythroblasts. In this study, the authors show that the antagonistic effect of dexamethasone on rHuEPO induced STAT5 phosphorylation and transcriptional activation. In contrast, the two agents did not compete for inhibition of apoptosis (Stellacci et al., 2009). Herein however, we describe for the first time a functional cross‐talk between membrane‐acting androgen and EPOR, which does not take place at the membrane level, but is mediated through specific signal transduction mediators, reverting the pro‐apoptotic action of testosterone–BSA in a dose‐ and time‐dependent manner. A further paradox reported here is the pro‐/anti‐apoptotic switch of p38 MAPK. However, such a switch has been previously reported in both cancer and non‐malignant cells and has been associated with tyrosine kinase receptors, like IGFR‐1 (Thornton and Rincon, 2009). One of the mechanisms proposed for this effect was the phosphorylation of caspase‐3, compatible with our findings. Indeed, the combination of testosterone–BSA with rHuEPO induced a pronounced decrease of activated caspase‐3, following a similar time course with p38 activation, or the interaction of p38 MAPK with JNK/GADD45 or p53, as discussed above.
In brief, analysis of pathways, uniquely modified by membrane‐acting androgen, resulted in some interesting conclusions: (i) that distinct signaling pathways are involved in the pro‐apoptotic and the transcriptional effects of the hormone; (ii) that a transcriptional enhancement of EPOR occurs in cells treated with testosterone–BSA; (iii) finally, activation of EPOR, in testosterone–BSA‐treated cells modifies profoundly signaling cascades and reverts the pro‐apoptotic action of the agent.
A major question raised by our data is whether the reported results may have a potential biological relevance, especially in the light of the ambiguity still dominating androgen profile in breast cancer. In this study, we used concentrations of rHuEPO and testosterone–BSA (10−7M) much higher than those physiologically circulating or exogenously administered. However, as presented in Figure 4, the apparent IC50s of both testosterone–BSA and rHuEPO on apoptosis (∼0.7nM) are compatible with their reported interactions with the corresponding binding sites (Alexaki et al., 2006; Farrell and Lee, 2004; Nicolas Diaz‐Chico et al., 2007). The choice of a higher concentration (∼IC80) was made, in order to obtain a (sub)maximal effect and decipher the underlying mechanisms.
An additional question requiring clarification is whether androgen (circulating, locally synthesized or exogenously administered) may exist in the vicinity of breast cancer cells. Interestingly, while circulating levels of testosterone and DHT are five‐ to ten‐fold higher in men than women, their metabolites is less than two‐fold higher in men, suggesting that local androgen synthesis and action in women may be more significant than usually thought (Dimitrakakis and Bondy, 2009). Androgen is secreted by both ovaries and adrenals, throughout the menstrual cycle, and in menopause, constituting a dominant hormone throughout a woman's entire life (Nicolas Diaz‐Chico et al., 2007). On the other hand, the mammary gland is capable of synthesizing both estradiol and testosterone, as verified in normal tissue, cancer specimens and cancer cell lines (Miki et al., 2009, 1995, 1991, 1996, 2009, 2008, 2008). However, data on intracellular concentration of androgens are controversial, due mainly to divergent assay methodologies. In addition, intracellular dynamics of androgen, their metabolites and gene‐expression potential depend on additional elements, such as their affinity for AR and their dimerization and coactivator recruitment. While still debatable, the diversity between serum and tissue concentration of testosterone, DHT and estradiol, seems to be age dependent, and displays variation between normal and cancerous breast (Recchione et al., 1995, 1991, 1996, 2009).
The correlation of circulating androgens and breast cancer risk has been studied in several epidemiological studies, with debatable results (reviewed in Dimitrakakis and Bondy, 2009). This could be due to several factors: most studies have been designed on the basis of assays compatible with higher androgen concentrations found in males, without taking into account circadian variability and standardized times of blood sampling. In addition, most studies focused on circulating testosterone, without considering variability of SHBG, or peripheral conversion of precursors like DHEA, or endocrine therapies (like oral contraceptives and hormone replacement therapies), triggering a negative feedback in hypophysis and steroidogenesis, which may influence androgen circulating levels. Therefore, until now, the controversy of the beneficial or indifferent effect of androgen on breast cancer risk still persists (Dimitrakakis and Bondy, 2009, 2009, 2007, 2008, 2009, 2009). Our data provide another potential target of androgen in the breast, namely the cell membrane. Indeed, membrane‐acting androgen induce apoptosis (reviewed in Kampa et al., 2008) at concentrations compatible to their locally present in the breast, while both animal and human intervention studies have shown that administration of androgen combined with anti‐estrogens were more effective than that of anti‐estrogen alone, in breast cancer patients (Baum et al., 2002).
Another potential source of androgen in the breast could result from aromatase inhibitor neoadjuvant therapy. However, only a limited number of research groups have worked in this field. Spinola et al. reported that treatment with an aromatase inhibitor markedly elevated intratumoral androgen concentrations in rat mammary tumors (Spinola et al., 1988), while Suzuki et al. disclosed that aromatase expression was inversely associated with intratumoral DHT concentration in breast carcinoma and suppressed DHT production from precursors (Suzuki et al., 2008, 2007, 2008). If such a local production of androgen could be confirmed, after aromatase inhibitor therapy, our data could provide an additional hint on its beneficial effect in breast cancer therapy.
The skepticism on androgen breast effect is not limited to the ligands, their origin, bioavailability and metabolism, but extends to their cognate androgen receptor (AR). While traditionally considered as a “male” receptor, accumulating evidence reveals the quasi‐omnipresence in the breast. AR expression is abundant in normal mammary epithelium and in the majority of breast cancer specimens and cell lines (Agoff et al., 2003; Dimitrakakis and Bondy, 2009; Nicolas Diaz‐Chico et al., 2007). They are preferentially co‐expressed with ER (Niemeier et al., 2009; Park et al., 2009), but they may also be found in ER‐negative tumors. Agrawal et al. have shown that AR expression is associated with better adjuvant therapy response and prognosis after mastectomy (Agrawal et al., 2008). In contrast, AR have been related to lymph node invasion (Brys et al., 2002; see Nicolas Diaz‐Chico et al., 2007, for review), a result equally verified in breast cancer cell lines, where AR is associated with loss of E‐cadherin and potentiation of the Epithelial–Mesnchymal Transition (Liu et al., 2008). In recent reports AR hyperexpression was linked with the emergence of Tamoxifen resistance and impaired patient survival (De Amicis et al., in press), while an interaction of AR with ER was equally shown (Peters et al., 2009). The perplexity of the AR effect gets an additional dimension by the revelation of the membrane binding sites and data presented here: membrane androgen sites have been detected in the majority of breast carcinomas (Pelekanou et al., 2007), and their activation induces apoptosis both in breast and in prostate tumors (Kampa et al., 2008). However, as shown here, they may also interact with growth factor receptors, including EPOR, as shown here and thus modify their action. Therefore, additional work is needed in this controversial field, which might provide new clues and possibilities in breast cancer management.
5. Conclusions
Our data indicate that membrane‐acting androgen induce changes in the transcription and the activation of signaling molecules involved in major intracellular signaling cascades, leading to apoptosis and transcriptional modifications. Among genes modified by testosterone–BSA action is EPOR, the cognate receptor of erythropoietin, one of the major targets of the HIF1α pathway. Additionally, the association of rHuEPO and testosterone–BSA reverts the pro‐apoptotic effect of the later, through a modification of signaling events. This functional cross‐talk provides a mechanistic insight explaining (at least in part) an unexpected finding we have reported previously: the detrimental effect on breast cancer patients' survival, whose tumors co‐expressed both EPOR and membrane androgen sites (Pelekanou et al., 2007). These data should be taken into account in the therapeutic intervention of patients.
Supporting information
Supplementary Figure Legends
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Figure 4
Supplemental Table 1
Supplemental Table 2
Supplemental Table 3
Supplemental Table 4
Supplemental Table 5
Supplemental Table 6
Acknowledgements
Work partially supported by a grant from the Greek Ministry of Health (KESY 128926/30/2005).
Supplementary data 1.
1.1.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.molonc.2010.01.004.
Pelekanou Vassiliki, Notas George, Sanidas Elias, Tsapis Andreas, Castanas Elias, Kampa Marilena, (2010), Testosterone membrane‐initiated action in breast cancer cells: Interaction with the androgen signaling pathway and EPOR, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.01.004.
Contributor Information
Vassiliki Pelekanou, Email: vasiliki.pelekanou@ulb.ac.be.
Elias Castanas, Email: castanas@med.uoc.gr.
Marilena Kampa, Email: kampa@med.uoc.gr.
References
- Acconcia, F. , Ascenzi, P. , Bocedi, A. , Spisni, E. , Tomasi, V. , Trentalance, A. , Visca, P. , Marino, M. , 2005. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol. Biol. Cell.. 16, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acconcia, F. , Ascenzi, P. , Fabozzi, G. , Visca, P. , Marino, M. , 2004. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem. Biophys. Res. Commun.. 316, 878–883. [DOI] [PubMed] [Google Scholar]
- Acs, G. , Chen, M. , Xu, X. , Acs, P. , Verma, A. , Koch, C.J. , 2004. Autocrine erythropoietin signaling inhibits hypoxia-induced apoptosis in human breast carcinoma cells. Cancer Lett.. 214, 243–251. [DOI] [PubMed] [Google Scholar]
- Agoff, S.N. , Swanson, P.E. , Linden, H. , Hawes, S.E. , Lawton, T.J. , 2003. Androgen receptor expression in estrogen receptor-negative breast cancer. Immunohistochemical, clinical, and prognostic associations. Am. J. Clin. Pathol.. 120, 725–731. [DOI] [PubMed] [Google Scholar]
- Agrawal, A.K. , Jelen, M. , Grzebieniak, Z. , Zukrowski, P. , Rudnicki, J. , Nienartowicz, E. , 2008. Androgen receptors as a prognostic and predictive factor in breast cancer. Folia Histochem. Cytobiol.. 46, 269–276. [DOI] [PubMed] [Google Scholar]
- Alexaki, V.I. , Charalampopoulos, I. , Kampa, M. , Nifli, A.P. , Hatzoglou, A. , Gravanis, A. , Castanas, E. , 2006. Activation of membrane estrogen receptors induce pro-survival kinases. J. Steroid. Biochem. Mol. Biol.. 98, 97–110. [DOI] [PubMed] [Google Scholar]
- Alexaki, V.I. , Charalampopoulos, I. , Panayotopoulou, M. , Kampa, M. , Gravanis, A. , Castanas, E. , 2009. Dehydroepiandrosterone protects human keratinocytes against apoptosis through membrane binding sites. Exp. Cell. Res.. 315, 2275–2283. [DOI] [PubMed] [Google Scholar]
- Baum, M. , Budzar, A.U. , Cuzick, J. , Forbes, J. , Houghton, J.H. , Klijn, J.G. , Sahmoud, T. , 2002. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 359, 2131–2139. [DOI] [PubMed] [Google Scholar]
- Benten, W.P. , Guo, Z. , Krucken, J. , Wunderlich, F. , 2004. Rapid effects of androgens in macrophages. Steroids. 69, 585–590. [DOI] [PubMed] [Google Scholar]
- Benten, W.P. , Lieberherr, M. , Stamm, O. , Wrehlke, C. , Guo, Z. , Wunderlich, F. , 1999. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Mol. Biol. Cell.. 10, 3113–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaccorsi, L. , Muratori, M. , Carloni, V. , Marchiani, S. , Formigli, L. , Forti, G. , Baldi, E. , 2004. The androgen receptor associates with the epidermal growth factor receptor in androgen-sensitive prostate cancer cells. Steroids. 69, 549–552. [DOI] [PubMed] [Google Scholar]
- Brys, M. , Wojcik, M. , Romanowicz-Makowska, H. , Krajewska, W.M. , 2002. Androgen receptor status in female breast cancer: RT-PCR and Western blot studies. J. Cancer Res. Clin. Oncol.. 128, 85–90. [DOI] [PubMed] [Google Scholar]
- Chen, L. , Mayer, J.A. , Krisko, T.I. , Speers, C.W. , Wang, T. , Hilsenbeck, S.G. , Brown, P.H. , 2009. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, J. , Watkins, S.C. , Walker, W.H. , 2007. Testosterone activates mitogen-activated protein kinase via Src kinase and the epidermal growth factor receptor in sertoli cells. Endocrinology. 148, 2066–2074. [DOI] [PubMed] [Google Scholar]
- Dambaki, C. , Kogia, C. , Kampa, M. , Darivianaki, K. , Nomikos, M. , Anezinis, P. , Theodoropoulos, P.A. , Castanas, E. , Stathopoulos, E.N. , 2005. Membrane testosterone binding sites in prostate carcinoma as a potential new marker and therapeutic target: study in paraffin tissue sections. BMC Cancer. 5, 148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Amicis, F., Thirugnansampanthan, J., Cui, Y., Selever, J., Beyer, A., Parra, I., Weigel, N.L., Herynk, M.H., Tsimelzon, A., Lewis, M.T., et al., Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. in press. [DOI] [PMC free article] [PubMed]
- Dimitrakakis, C. , Bondy, C. , 2009. Androgens and the breast. Breast Cancer Res.. 11, 212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenstein, E. , Norman, A.W. , Wehling, M. , 2000. Mannheim classification of nongenomically initiated (rapid) steroid action(s). J. Clin. Endocrinol. Metab.. 85, 2072–2075. [DOI] [PubMed] [Google Scholar]
- Farrell, F. , Lee, A. , 2004. The erythropoietin receptor and its expression in tumor cells and other tissues. Oncologist. 9, (Suppl. 5) 18–30. [DOI] [PubMed] [Google Scholar]
- Hall, S.A. , Araujo, A.B. , Kupelian, V. , Maserejian, N.N. , Travison, T.G. , 2009. Testosterone and breast cancer. J. Sex. Med. [DOI] [PubMed] [Google Scholar]
- Hardee, M.E. , Cao, Y. , Fu, P. , Jiang, X. , Zhao, Y. , Rabbani, Z.N. , Vujaskovic, Z. , Dewhirst, M.W. , Arcasoy, M.O. , 2007. Erythropoietin blockade inhibits the induction of tumor angiogenesis and progression. PLoS ONE. 2, e549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzoglou, A. , Kampa, M. , Kogia, C. , Charalampopoulos, I. , Theodoropoulos, P.A. , Anezinis, P. , Dambaki, C. , Papakonstanti, E.A. , Stathopoulos, E.N. , Stournaras, C. , 2005. Membrane androgen receptor activation induces apoptotic regression of human prostate cancer cells in vitro and in vivo. J. Clin. Endocrinol. Metab.. 90, 893–903. [DOI] [PubMed] [Google Scholar]
- Heinlein, C.A. , Chang, C. , 2002. The roles of androgen receptors and androgen-binding proteins in nongenomic androgen actions. Mol. Endocrinol.. 16, 2181–2187. [DOI] [PubMed] [Google Scholar]
- Jelkmann, W. , Bohlius, J. , Hallek, M. , Sytkowski, A.J. , 2008. The erythropoietin receptor in normal and cancer tissues. Crit. Rev. Oncol. Hematol. 67, (1) 39–61. [DOI] [PubMed] [Google Scholar]
- Kaarbo, M. , Klokk, T.I. , Saatcioglu, F. , 2007. Androgen signaling and its interactions with other signaling pathways in prostate cancer. Bioessays. 29, 1227–1238. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Castanas, E. , 2006. Membrane steroid receptor signaling in normal and neoplastic cells. Mol. Cell. Endocrinol.. 246, 76–82. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Kogia, C. , Theodoropoulos, P.A. , Anezinis, P. , Charalampopoulos, I. , Papakonstanti, E.A. , Stathopoulos, E.N. , Hatzoglou, A. , Stournaras, C. , Gravanis, A. , 2006. Activation of membrane androgen receptors potentiates the antiproliferative effects of paclitaxel on human prostate cancer cells. Mol. Cancer Ther.. 5, 1342–1351. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Nifli, A.P. , Charalampopoulos, I. , Alexaki, V.I. , Theodoropoulos, P.A. , Stathopoulos, E.N. , Gravanis, A. , Castanas, E. , 2005. Opposing effects of estradiol- and testosterone-membrane binding sites on T47D breast cancer cell apoptosis. Exp. Cell Res.. 307, 41–51. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Papakonstanti, E.A. , Alexaki, V.I. , Hatzoglou, A. , Stournaras, C. , Castanas, E. , 2004. The opioid agonist ethylketocyclazocine reverts the rapid, non-genomic effects of membrane testosterone receptors in the human prostate LNCaP cell line. Exp. Cell Res.. 294, 434–445. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Papakonstanti, E.A. , Hatzoglou, A. , Stathopoulos, E.N. , Stournaras, C. , Castanas, E. , 2002. The human prostate cancer cell line LNCaP bears functional membrane testosterone receptors that increase PSA secretion and modify actin cytoskeleton. FASEB J.. 16, 1429–1431. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Pelekanou, V. , Castanas, E. , 2008. Membrane-initiated steroid action in breast and prostate cancer. Steroids. 73, 953–960. [DOI] [PubMed] [Google Scholar]
- Karin, M. , Yang-Yen, H.F. , Chambard, J.C. , Deng, T. , Saatcioglu, F. , 1993. Various modes of gene regulation by nuclear receptors for steroid and thyroid hormones. Eur. J. Clin. Pharmacol.. 45, (Suppl. 1) S9–S15. discussion S43–4 [DOI] [PubMed] [Google Scholar]
- Khuri, F.R. , 2007. Weighing the hazards of erythropoiesis stimulation in patients with cancer. N. Engl. J. Med.. 356, 2445–2448. [DOI] [PubMed] [Google Scholar]
- Lacombe, C. , Mayeux, P. , 1999. The molecular biology of erythropoietin. Nephrol. Dial Transpl.. 14, (Suppl. 2) 22–28. [DOI] [PubMed] [Google Scholar]
- Lai, S.Y. , Childs, E.E. , Xi, S. , Coppelli, F.M. , Gooding, W.E. , Wells, A. , Ferris, R.L. , Grandis, J.R. , 2005. Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene. 24, 4442–4449. [DOI] [PubMed] [Google Scholar]
- Liebermann, D.A. , Hoffman, B. , 2008. Gadd45 in stress signaling. J. Mol. Signal. 3, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, L.Y. , Vidnovic, N. , Ellisen, L.W. , Leong, C.O. , 2009. Mutant p53 mediates survival of breast cancer cells. Br. J. Cancer. 101, 1606–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.N. , Liu, Y. , Lee, H.J. , Hsu, Y.H. , Chen, J.H. , 2008. Activated androgen receptor downregulates E-cadherin gene expression and promotes tumor metastasis. Mol. Cell. Biol.. 28, 7096–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch, M. , Chen, G. , 2008. The p38 MAPK stress pathway as a tumor suppressor or more?. Front Biosci.. 13, 3581–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, Y. , Suzuki, T. , Sasanoa, H. , 2009. Intracrinology of sex steroids in ductal carcinoma in situ (DCIS) of human breast: comparison to invasive ductal carcinoma (IDC) and non-neoplastic breast. J. Steroid Biochem. Mol. Biol.. 114, 68–71. [DOI] [PubMed] [Google Scholar]
- Nicolas Diaz-Chico, B. , German Rodriguez, F. , Gonzalez, A. , Ramirez, R. , Bilbao, C. , Cabrera de Leon, A. , Aguirre Jaime, A. , Chirino, R. , Navarro, D. , Diaz-Chico, J.C. , 2007. Androgens and androgen receptors in breast cancer. J. Steroid Biochem. Mol. Biol.. 105, 1–15. [DOI] [PubMed] [Google Scholar]
- Niemeier, L.A. , Dabbs, D.J. , Beriwal, S. , Striebel, J.M. , Bhargava, R. , 2009. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol. [DOI] [PubMed] [Google Scholar]
- Nifli, A.P. , Bosson-Kouame, A. , Papadopoulou, N. , Kogia, C. , Kampa, M. , Castagnino, C. , Stournaras, C. , Vercauteren, J. , Castanas, E. , 2005. Monomeric and oligomeric flavanols are agonists of membrane androgen receptors. Exp. Cell Res.. 309, 329–339. [DOI] [PubMed] [Google Scholar]
- Noguchi, C.T. , Bae, K.S. , Chin, K. , Wada, Y. , Schechter, A.N. , Hankins, W.D. , 1991. Cloning of the human erythropoietin receptor gene. Blood. 78, 2548–2556. [PubMed] [Google Scholar]
- Notas, G., Pelekanou, V., Castanas, E., Kampa, M. Conjugated and non-conjugated androgens differentially modulate specific early gene transcription in breast cancer in a cell-specific manner. Steroids, in press. doi:10.1016/j.steroids.2009.1010.1004. [DOI] [PubMed]
- Papakonstanti, E.A. , Kampa, M. , Castanas, E. , Stournaras, C. , 2003. A rapid, nongenomic, signaling pathway regulates the actin reorganization induced by activation of membrane testosterone receptors. Mol. Endocrinol.. 17, 870–881. [DOI] [PubMed] [Google Scholar]
- Park, S. , Koo, J. , Park, H.S. , Kim, J.H. , Choi, S.Y. , Lee, J.H. , Park, B.W. , Lee, K.S. , 2009. Expression of androgen receptors in primary breast cancer. Ann. Oncol. [DOI] [PubMed] [Google Scholar]
- Pedram, A. , Razandi, M. , Kim, J.K. , O'Mahony, F. , Lee, E.Y. , Luderer, U. , Levin, E.R. , 2009. Developmental phenotype of a membrane only estrogen receptor alpha (MOER) mouse. J. Biol. Chem.. 284, 3488–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedram, A. , Razandi, M. , Levin, E.R. , 2006. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol.. 20, 1996–2009. [DOI] [PubMed] [Google Scholar]
- Pelekanou, V. , Kampa, M. , Kafousi, M. , Dambaki, K. , Darivianaki, K. , Vrekoussis, T. , Sanidas, E. , Tsiftsis, D.D. , Stathopoulos, E.N. , Castanas, E. , 2007. Erythropoietin and its receptor in breast cancer: correlation with steroid receptors and outcome. Cancer Epidemiol. Biomark. Prev.. 16, 2016–2023. [DOI] [PubMed] [Google Scholar]
- Peters, A.A. , Buchanan, G. , Ricciardelli, C. , Bianco-Miotto, T. , Centenera, M.M. , Harris, J.M. , Jindal, S. , Segara, D. , Jia, L. , Moore, N.L. , 2009. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res.. 69, 6131–6140. [DOI] [PubMed] [Google Scholar]
- Recchione, C. , Venturelli, E. , Manzari, A. , Cavalleri, A. , Martinetti, A. , Secreto, G. , 1995. Testosterone, dihydrotestosterone and oestradiol levels in postmenopausal breast cancer tissues. J. Steroid Biochem. Mol. Biol.. 52, 541–546. [DOI] [PubMed] [Google Scholar]
- Secreto, G. , Recchione, C. , Ballerini, P. , Callegari, L. , Cavalleri, A. , Attili, A. , Fariselli, G. , Moglia, D. , Del Prato, I. , 1991. Accumulation of active androgens in breast cyst fluids. Eur. J. Cancer. 27, 44–47. [DOI] [PubMed] [Google Scholar]
- Secreto, G. , Venturelli, E. , Bucci, A. , Piromalli, D. , Fariselli, G. , Galante, E. , 1996. Intratumour amount of sex steroids in elderly breast cancer patients. An approach to the biological characterization of mammary tumours in the elderly. J. Steroid Biochem. Mol. Biol.. 58, 557–561. [DOI] [PubMed] [Google Scholar]
- Secreto, G. , Venturelli, E. , Meneghini, E. , Greco, M. , Ferraris, C. , Gion, M. , Zancan, M. , Fabricio, A.S. , Berrino, F. , Cavalleri, A. , 2009. Testosterone and biological characteristics of breast cancers in postmenopausal women. Cancer Epidemiol. Biomark. Prev.. 18, 2942–2948. [DOI] [PubMed] [Google Scholar]
- Shibuya, R. , Suzuki, T. , Miki, Y. , Yoshida, K. , Moriya, T. , Ono, K. , Akahira, J. , Ishida, T. , Hirakawa, H. , Evans, D.B. , 2008. Intratumoral concentration of sex steroids and expression of sex steroid-producing enzymes in ductal carcinoma in situ of human breast. Endocr. Relat. Cancer. 15, 113–124. [DOI] [PubMed] [Google Scholar]
- Shufelt, C.L. , Braunstein, G.D. , 2008. Testosterone and the breast. Menopause Int.. 14, 117–122. [DOI] [PubMed] [Google Scholar]
- Shufelt, C.L. , Braunstein, G.D. , 2009. Safety of testosterone use in women. Maturitas. 63, 63–66. [DOI] [PubMed] [Google Scholar]
- Sieri, S. , Krogh, V. , Bolelli, G. , Abagnato, C.A. , Grioni, S. , Pala, V. , Evangelista, A. , Allemani, C. , Micheli, A. , Tagliabue, G. , 2009. Sex hormone levels, breast cancer risk, and cancer receptor status in postmenopausal women: the ORDET cohort. Cancer Epidemiol. Biomark. Prev.. 18, 169–176. [DOI] [PubMed] [Google Scholar]
- Solomon, S.S. , Majumdar, G. , Martinez-Hernandez, A. , Raghow, R. , 2008. A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sci.. 83, 305–312. [DOI] [PubMed] [Google Scholar]
- Spinola, P.G. , Marchetti, B. , Merand, Y. , Belanger, A. , Labrie, F. , 1988. Effects of the aromatase inhibitor 4-hydroxyandrostenedione and the antiandrogen flutamide on growth and steroid levels in DMBA-induced rat mammary tumors. Breast Cancer Res. Treat.. 12, 287–296. [DOI] [PubMed] [Google Scholar]
- Spivak, J.L. , 2005. The anaemia of cancer: death by a thousand cuts. Nat. Rev. Cancer. 5, 543–555. [DOI] [PubMed] [Google Scholar]
- Stellacci, E. , Di Noia, A. , Di Baldassarre, A. , Migliaccio, G. , Battistini, A. , Migliaccio, A.R. , 2009. Interaction between the glucocorticoid and erythropoietin receptors in human erythroid cells. Exp. Hematol.. 37, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Miki, Y. , Akahira, J. , Moriya, T. , Ohuchi, N. , Sasano, H. , 2008. Aromatase in human breast carcinoma as a key regulator of intratumoral sex steroid concentrations. Endocr. J.. 55, 455–463. [DOI] [PubMed] [Google Scholar]
- Suzuki, T. , Miki, Y. , Moriya, T. , Akahira, J. , Hirakawa, H. , Ohuchi, N. , Sasano, H. , 2007. In situ production of sex steroids in human breast carcinoma. Med. Mol. Morphol.. 40, 121–127. [DOI] [PubMed] [Google Scholar]
- Suzuki, T. , Miki, Y. , Ohuchi, N. , Sasano, H. , 2008. Intratumoral estrogen production in breast carcinoma: significance of aromatase. Breast Cancer. 15, 270–277. [DOI] [PubMed] [Google Scholar]
- Szego, C.M. , Davis, J.S. , 1967. Adenosine 3′,5′-monophosphate in rat uterus: acute elevation by estrogen. Proc. Natl. Acad. Sci. U S A. 58, 1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, T.M. , Rincon, M. , 2009. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int. J. Biol. Sci.. 5, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling, M. , 1997. Specific non-genomic effects of steroid hormones. Annu. Rev. Physiol.. 59, 365–393. [DOI] [PubMed] [Google Scholar]
- Weidemann, A. , Johnson, R.S. , 2008. Biology of HIF-1alpha. Cell Death Differ.. 15, 621–627. [DOI] [PubMed] [Google Scholar]
- Yasuda, Y. , Fujita, Y. , Matsuo, T. , Koinuma, S. , Hara, S. , Tazaki, A. , Onozaki, M. , Hashimoto, M. , Musha, T. , Ogawa, K. , 2003. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis. 24, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Yin, F. , Bruemmer, D. , Blaschke, F. , Hsueh, W.A. , Law, R.E. , Herle, A.J. , 2004. Signaling pathways involved in induction of GADD45 gene expression and apoptosis by troglitazone in human MCF-7 breast carcinoma cells. Oncogene. 23, 4614–4623. [DOI] [PubMed] [Google Scholar]
- Zarubin, T. , Han, J. , 2005. Activation and signaling of the p38 MAP kinase pathway. Cell Res.. 15, 11–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Figure 4
Supplemental Table 1
Supplemental Table 2
Supplemental Table 3
Supplemental Table 4
Supplemental Table 5
Supplemental Table 6