

Abstract
Copy number changes in TOP2A have frequently been linked to ERBB2 (HER2) amplified breast cancers. To study this relationship, copy number changes of ERBB2 and TOP2A were investigated by fluorescence in situ hybridization (FISH) in two cell lines; one characterized by having amplification of both genes and the other by having amplification of ERBB2 and deletion of TOP2A. The characteristics are compared to findings on paired ERBB2 and TOP2A data from 649 patients with invasive breast cancer from a previously published biomarker study. The physical localization of FISH signals in metaphase spreads from cell lines showed that simultaneous amplification is not a simple co‐amplification of a whole amplicon containing both genes. Most gene signals are translocated to abnormal marker chromosomes. ERBB2 genes but not TOP2A genes are present in tandem amplicons, leading to a higher ERBB2 ratio. This observation was confirmed by patient FISH data: among 276 (43% of all patients) abnormal tumors, 67% had different ERBB2 and TOP2A status. ERBB2 amplification with normal TOP2A status was found in 36% of the abnormal tumors (15% of all patients). Simultaneous amplification of both genes was found in 28% of the abnormal tumors (12% of all patients) while TOP2A deletion and ERBB2 amplification was observed in 16% of the abnormal cases (8% of all patients). A small number of tumors had TOP2A amplification (4%) or deletion (6%) without simultaneous changes of the ERBB2 gene. ERBB2 deletion was also observed (5%) but only in tumors with simultaneous TOP2A deletion. The average gene/reference ratio was significantly different: 5.0 for TOP2A but 7.2 for ERBB2 in the amplified tumors (P<0.01). Amplification of the two genes may be caused by different mechanisms, leading to higher level of amplification for ERBB2 compared to TOP2A. In the majority of breast cancer patients, simultaneous aberration of ERBB2 and TOP2A is not explained by simple co‐amplification.
Keywords: TOP2A, ERBB2, HER2, Gene amplification, Gene deletion, Copy number changes, Chromosome aberrations, Breast cancer
1. Introduction
Amplification of ERBB2, commonly known by the alias HER2 or HER‐2/neu, is observed in 18–20% of breast cancers and is used as a selection criterion for HER2 targeted therapies (Wolff et al., 2007). TOP2A, encoding topoisomerase IIα, on 17q21 is located close to ERBB2 on chromosome 17q12 and copy number changes of TOP2A may be associated with a higher sensitivity to topoisomerase poisons, e.g. anthracyclines (Di Leo et al., 2002; Knoop et al., 2005; O'Malley et al., 2009). The proximity of the two genes has lead to much debate on their use as predictive markers for anthracyclines (reviewed in Pritchard et al., 2008). TOP2A aberrations were initially reported in ERBB2 amplified tumors (Järvinen et al., 1999, 2000) and although the genes were shown to be located in different amplicons (Kauraniemi et al., 2003), the proximity of the two genes has led to a conception of co‐amplification of a whole ERBB2 amplicon containing both genes (Harris et al., 2009; Slamon and Press, 2009). This conception is in contrast to other and more complex hypotheses regarding the underlying mechanisms of changes in copy number of particular genes or genomic regions during the cancerous process (Albertson et al., 2003; Tanaka and Yao, 2009). Although amplification of oncogenes and drug resistance genes were originally observed in cell lines harboring double minute chromosomes (DMs) and homogeneously staining regions (HSRs) (Alitalo et al., 1983), amplifications may also be viewed as distributed insertions, either single or in tandem (Albertson, 2006). The original studies on drug resistance in rodent cell lines (Stark et al., 1989) lead to the presumption that DMs/HSRs were the predominant mechanism, but this could not be confirmed when cytogenetic analyses were performed on tumor specimens and cell lines established from breast tumors (Albertson, 2006; Pandis et al., 1998). Many hypotheses have been proposed to explain how genes are amplified or how DMs or HSRs are generated (Singer et al., 2000). The “episome model” argues that circular molecules excised from chromosomes may play an important role in gene amplification (Pauletti et al., 1990). The most popular hypothesis is the breakage‐fusion‐bridge (BFB) cycle model (McClintock, 1941), but conclusive evidence is still lacking after more than 60years (Albertson, 2006; Myllykangas and Knuutila, 2006; Tanaka and Yao, 2009). The BFB model has also been used to explain co‐amplification of ERBB2 and TOP2A (Jacobson et al., 2004), but TOP2A is located outside the ERBB2 amplicon of 280kb (Kauraniemi et al., 2003). More recently, the mechanisms behind amplification have regained interest and several new models have been proposed, e.g. the Hairpin‐capped double strand breaks (Narayanan et al., 2006), the translocation‐excision‐deletion‐amplification mechanism (Van Roy et al., 2006) and the “hot‐spot” mechanism (Kuwahara et al., 2004).
The aim of this study was to investigate the mechanism behind simultaneous amplification of ERBB2 and TOP2A. Demonstration of the exact number and chromosomal localization of the genes by FISH require the use of metaphase spreads. Metaphase spreads are very difficult to obtain from patient samples, but the use of cell lines established from breast cancer specimens enables access to this information. As results obtained on cell lines can be affected by long‐term culturing of the cells, findings always need to be confirmed on patient samples (Lacroix and Leclercq, 2004). In the present study, ERBB2 and TOP2A signals were investigated simultaneously with the centromere 17 signals in breast cancer cell lines, both on metaphase spreads, in interphases and in cut sections of formalin fixed, paraffin‐embedded (FFPE) tissue with the intention of gaining insight in the mechanism behind the gene amplification. Secondly, from a previously published biomarker study, results on ERBB2 and TOP2A from 649 patients participating in the Danish Breast Cancer Cooperative Group (DBCG) trial 89D (Ejlertsen et al., 2007; Knoop et al., 2005; Nielsen et al., 2008) were used to verify the results obtained in cell lines.
2. Results
2.1. Cell line data
From cultures of four breast cancer cell lines, MDA‐231, MDA‐175, MDA‐361 and SKBR3, metaphase spreads were prepared by standard cytogenetic techniques. The number of FISH signals and the localization of the signals were analyzed in 20 interphase nuclei. Karyotype analysis was based on a consensus of four to 15 metaphases from each cell line. The cells from three of the cell lines (MDA‐231, MDA‐361 and SKBR3) were also embedded in paraffin and cut sections from these paraffin blocks were used for the FISH analysis. These cell lines are well characterized and used as control cells for the HercepTest™ (K5204, Dako, Denmark).
The four breast cancer cell lines were screened for TOP2A aberrations (Table 1). Ratios found in metaphase spreads and in whole interphases nuclei showed a good correlation. The number of signals and the localization of the signals were analyzed in detail and illustrated in Figure 1. Normal ratios near 1.0 were found in two of the cell lines, MDA‐231 and MDA‐175. In one of these, the triploid cell line MDA‐231, three copies of apparently normal chromosomes 17 were present (Figure 1A) each with a green CEN‐17 signal and a red TOP2A signal. In the other, the tetraploid cell line MDA‐175, four copies of TOP2A and four copies of CEN‐17 were present, two of which appeared normal (not shown). Loss of TOP2A signals (TOP2A deletion) was found in the triploid cell line, MDA‐361, as four CEN‐17 bearing chromosomes were identified but only one of them had a TOP2A signal (Figure 1B). The tetraploid cell line, SKBR3, showed borderline amplification with predominantly 11 red TOP2A signals and six or seven green CEN‐17 signals (Figure 1C). Estimated by visual inspection none of the chromosomes harboring signals had normal appearance and only four of the TOP2A signals were located in the expected distance from centromere 17.
Table 1.
Description of the ploidy level and localization of TOP2A signals on the chromosomes in four different breast cancer cell lines and TOP2A ratios determined on metaphase spreads, interphase whole nuclei and cut sections of paraffin‐embedded cell lines.
| Cell line | Ploidy level (range of chromosomes) | Description of the chromosomes harboring signals | TOP2A ratio | ||
|---|---|---|---|---|---|
| Meta‐phase | Inter‐phase | Cut sections | |||
| MDA‐231 | Hypotriploid (57–63 chromosomes) | 3 apparently normal chromosomes 17 | 1.02 | 1.05 | 1.04 |
| MDA‐175 | Hypotetraploid (70–94 chromosomes) | 2 normal and 2 abnormal chromosomes 17 | 1.02 | 1.05 | NA |
| MDA‐361 | Hypotriploid (57–66 chromosomes) | 4 green and 1 red signals. No normal chromosome 17 | 0.25 | 0.28 | 0.33 |
| SKBR3 | Hypotetraploid (82–104 chromosomes) | 8–13 red and 6–7 green signal, No normal chromosomes 17 | 1.71 | 1.72 | 2.09 |
Figure 1.
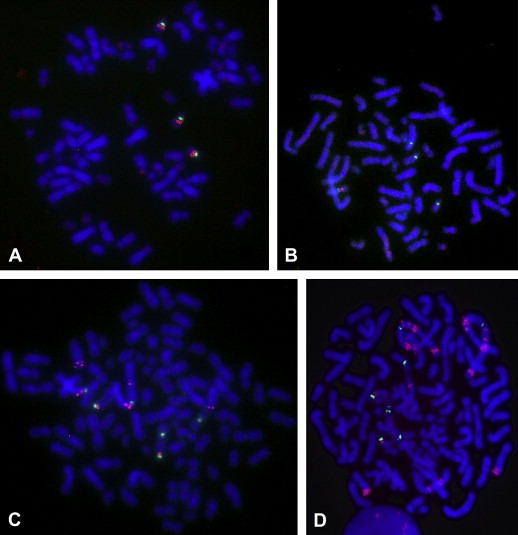
Red TOP2A and green CEN‐17 signals in metaphase spreads from the MDA‐231 cell line with a normal ratio of 1.0 (A) and the MDA‐361 cell line (B) with TOP2A deletion (ratio 0.25). Nuclei are counterstained with blue DAPI. TOP2A (C) and ERBB2 (D) signals in the SKBR3 cell line showing a borderline TOP2A amplification (11 gene signals/ratio 1.7) and a ERBB2 amplification (30 gene signals/ratio 4.6).
Cut sections of paraffin‐embedded cells were available from three of the cell lines and the TOP2A ratio was determined (Table 1). A linear correlation between the TOP2A ratio in truncated nuclei, whole interphases and metaphase spreads from the same cell line was demonstrated. The TOP2A ratio of SKBR3 metaphases was 1.7, but when counted in the truncated nuclei of the paraffin‐embedded cells, SKBR3 had a ratio just above the cutoff of 2.0.
ERBB2 (HER2) gene status was investigated in the two cell lines, MDA‐361 and SKBR3, with TOP2A aberrations. Figures 1C,D show the TOP2A and ERBB2 signals, respectively, on metaphase spreads from SKBR3. Figure 2 is based on a detailed analysis of the signals in metaphase spreads of the cell lines and illustrates the marker chromosomes (M) harboring the red and green signals. None of the gene signals are located on a normal chromosome 17. The red gene signals can apparently be found on chromosomes with and without a CEN‐17 signal and gene signals can be translocated independently of centromere signals. One pair of marker chromosomes (M4) has two CEN‐17 signals. Only ERBB2 signals are seen in amplicons of two to four tandem signals while TOP2A signals are localized discretely as single signals. SKBR3 has six amplicons with three ERBB2 genes and three amplicons with two ERBB2 genes. In addition six solitary ERBB2 genes are found, only four of which are located in the normal distance from centromere 17 (Figure 2). None of the TOP2A genes are seen as tandem signals. All six solitary ERBB2 genes are accompanied by a solitary TOP2A gene. Four of the triplet and one of the doublet ERBB2 amplicons contain a solitary TOP2A gene. All 11 TOP2A genes are localized near a ERBB2 gene. However, the ERBB2 gene may be present in one to three copies in the amplicon, leading to a total number of 30 ERBB2 genes. In the MDA‐361 cell line one amplicon with four ERBB2 genes as well as four solitary ERBB2 genes are seen (Figure 2). Three of the four solitary ERBB2 genes are located in an apparently normal distance from the centromere 17, while a CEN‐17 bearing marker chromosome lacks any gene signals. Only one TOP2A gene is retained, appearing on a marker chromosome with both an ERBB2 and a CEN‐17 signal.
Figure 2.
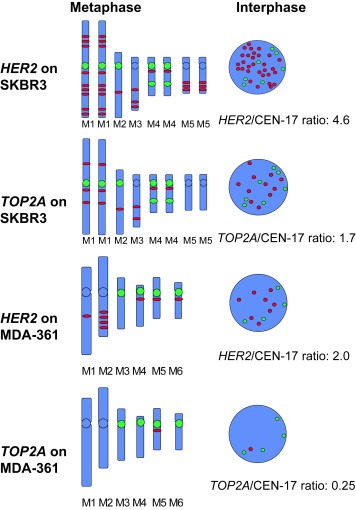
Illustration showing the signal bearing marker chromosomes of the SKBR3 and MCA‐361 cell lines in the TOP2A and ERBB2 assays. M=abnormal marker chromosome.
2.2. Invasive breast cancer tissue data
Data from the TOP2A FISH analysis were available on 773 samples from a previously published biomarker study and on 767 patients all covariates were known. Paired values of TOP2A and ERBB2 (HER2) assays were available from 649 invasive breast cancer samples (Table 2 and Figure 3A). Normal status for both ERBB2 and TOP2A was found in 373 tumors (57%) while 276 tumors (43%) had abnormal status of TOP2A, ERBB2 or both. ERBB2 amplification without simultaneous TOP2A aberration was seen in 36% of the abnormal tumors (15% of all patients). Simultaneous amplification of both genes was found in 28% of the abnormal tumors (12% of all patients) while deletion of both genes was observed in 5% of the abnormal tumors (2% of all patients). ERBB2 deletion was also observed in two tumors with normal TOP2A status. The ERBB2 deletions found in 16 tumors were primarily in HercepTest 0 (nine cases) and 1+ cases (six cases). A single deletion was seen among the 2+ cases and no deletions were seen in the 3+ cases. ERBB2 amplification combined with TOP2A deletion was present in 55 tumors (8% of all patients and 20% of the abnormal tumors). TOP2A amplification was present in 12 (2% of all patients and 4% of the abnormal tumors) tumors with normal ERBB2 status, and TOP2A deletion were present in 16 tumors with normal ERBB2 status (2% of all patients and 6% of the abnormal tumors). Totally, 27% of the tumors (174 tumors) had TOP2A aberrations, 132 in tumors with ERBB2 amplification and 42 in tumors without. Twelve TOP2A amplifications and 30 TOP2A deletions were found in the 42 ERBB2 non‐amplified cases. Thus 24% (42 of 174 tumors) of all TOP2A aberrations are found in ERBB2 non‐amplified cases. In the majority of the abnormal cases, aberrations of both ERBB2 and TOP2A were observed (P<0.001; χ 2‐test) but importantly, 56% of all TOP2A aberrations lead to a TOP2A status (normal/deleted/amplified) that differs from the corresponding HER2 status (normal/amplified).
Table 2.
Relation between TOP2A and ERBB2 FISH findings in 649 breast cancer patients from the DBCG 89D clinical trial.
| ERBB2 deletion (%) | ERBB2 normal (%) | ERBB2 amplification (%) | Total (%) | ERBB2 FISH not available | Total (Knoop et al., 2005) | |
|---|---|---|---|---|---|---|
| TOP2A deletion | 14 (2) | 16 (2) | 55 (8) | 85 (13) | 1 | 86 |
| TOP2A normal | 2 (0) | 373 (57) | 100 (15) | 475 (73) | 114 | 589 |
| TOP2A amplification | 0 (0) | 12 (2) | 77 (12) | 89 (14) | 3 | 92 |
| Total | 16 (2) | 401 (62) | 232 (36) | 649 (100) | 118 | 767 |
Figure 3.
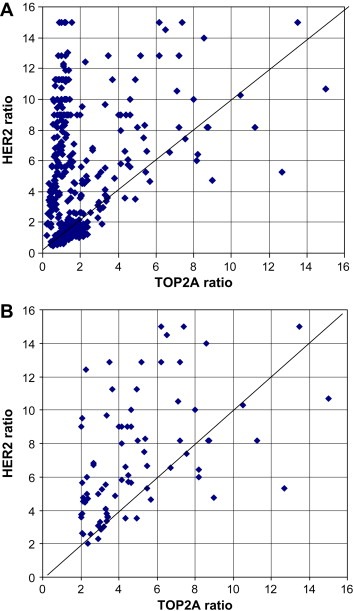
ERBB2 ratio plotted against TOP2A ratio in 649 tumors where both ratios were available (A) and in the 77 tumors with TOP2A and ERBB2 amplification (B).
The paired ratios of 77 cases with simultaneous amplification of ERBB2 and TOP2A is shown in Figure 3B, and only 22 cases (29%) had similar ratios (defined as a difference in ratios <1.0). In 47 cases the HER2 ratio was higher than the TOP2A ratio (from 1.0 to 10.2 higher) while only eight cases had higher TOP2A ratio (from 1.0 to 7.4 higher). The HER2 ratio was significantly higher (P<0.01; McNemar's test) than the TOP2A ratio and the average ratio was 7.2 for HER2 and 5.0 for TOP2A. The average ratio among all 649 tumors was 3.3 for HER2 and 1.6 for TOP2A; however this difference represents the combined contribution from both a higher frequency of HER2 amplified cases and a higher degree of amplification. In the HER2 assay 398 (61%) of the 649 tumors had more than four gene signals per nucleus while only 96 (15%) of the tumors had more than four TOP2A signals per nucleus. Tumors containing more than 10 signals per nucleus were seen in 202 (33%) of the 649 HER2 tests and only in 29 (4%) of the 649 TOP2A tests.
3. Discussion
The metaphase and interphase results from the SKBR3 and MDA‐361 cell lines in this study are not coherent with amplification of the entire genomic region harboring ERBB2 and TOP2A being a predominant mechanism. SKBR3 has ERBB2 amplification with a ratio of 4.3 and TOP2A amplification with a ratio approximately half of this value. Although clearly polysomic for chromosome 17 (six to seven copies are present in the metaphases), the gene signals and the centromere 17 signals in SKBR3 does not undergo genetic changes as a whole unit, but rather behave as independent events. Gene signals are observed on abnormal chromosomes with and without centromere 17 signals. The most obvious explanation for this is that one or more translocations are involved in the genetic changes leading to the aberrant karyotype of SKBR3. Both SKBR3 and MDA‐361 contained amplicons with two to four ERBB2 genes. However, these amplicons either did not contain any or only a single copy of the TOP2A gene, but our observation only covers a total of four cell lines and cannot exclude the existence of one or more cell line with tandem amplicons of TOP2A.
Our cell line data indicates that at least two different mechanisms are involved. The ERBB2 tandem amplicons seems to be generated independently of the amplicons with solitary genes. The most favorable mechanism for generation of the ERBB2 tandem amplicons is the “hot‐spot” mechanism (Kuwahara et al., 2004). The identification of a fragile site between the ERBB2 and its neighbor gene, GRB7, could explain why TOP2A is not included in the amplicon (Kuwahara et al., 2004). At least 13 translocations must have occurred in the SKBR3 cell line, so the “hot‐spot” mechanism, although explaining the ERBB2 tandem amplicons is not sufficient to explain all the observed genomic changes. The FISH analysis of both cell lines demonstrated translocation of a larger segment containing both genes, occasionally followed by deletion of sequences telomeric to the amplified sequences on the derivative chromosomes in line with the translocation‐excision‐deletion‐amplification mechanism (Van Roy et al., 2006). In line with our observations, Rogalla et al. (2002) studied the breast cancer cell line EFM‐19 and demonstrated five ERBB2 and six centromere 17 signals all appearing on different chromosomes (Rogalla et al., 2002). Detailed analysis of the EFM‐19 cell line showed that the distribution of signals was caused by a fragmentation of chromosome 17 leading to amplification of small parts of the chromosome as well as to extended losses of other parts of the chromosome. Combining this finding with our results suggests that HER2 and centromere 17 belong to the small parts that are amplified while TOP2A belong to the parts that are preferentially lost.
Among the 649 patients in our clinical study 77 had simultaneous amplification of ERBB2 and TOP2A, but in agreement with our cell line experiments the ratio was different in 55 (71%) of these tumors. TOP2A deletion was observed in 85 (13%) tumors and a simultaneous deletion of ERBB2 was only found in 14 (16%), while 55 (65%) had amplification of ERBB2. Among tumors with normal ERBB2 status, 16 TOP2A deletions and 12 TOP2A amplifications were found showing that TOP2A aberration can occur independently of the ERBB2 amplification. Overall the number of ERBB2 signals was much higher than the TOP2A signals. More than 10 ERBB2 signals per nucleus were seen in 202 (33%) of the 649 tumors compared to only 29 (4%) tumors with more than 10 TOP2A signals per nucleus. In patient samples ERBB2 amplification was often seen as clusters that could represent amplicons with tandem signals. These clusters were rarely seen in the TOP2A assay. Further, the generally higher level of ERBB2 amplification leads to higher HER2 (ERBB2) ratios in the paired values.
In contrast to the high incidence of amplifications in cancer cells, the number of reports that have carefully scrutinized the occurrence and possible role of accompanying deletions is still small. In addition to 16 cases with TOP2A deletion in tumors with normal ERBB2 status, the present study also demonstrates 16 cases of ERBB2 deletions, mostly associated with TOP2A deletion. The ERBB2 deleted cases are primarily found to have HercepTest 0 status; however these cases may be false negative. It has been suggested (Koscielny et al., 1998) that low gene expression of HER2 is associated with very poor prognosis and that the cases with extremely low gene expression should be considered abnormal. With the current definition, ERBB2 deletions are grouped together with the ERBB2 normal gene copy variants. However, the phenomenon is rarely reported and may not affect the clinical use of HER2 assessment in breast cancer patients. In the present study 2.5% of the patients had ERBB2 deletion in line with a frequency of 1–6% reported by other studies (Bartlett et al., 2008; Bouchalova et al., 2006; Hicks et al., 2005).
TOP2A amplifications and deletions have been observed with variable frequencies, especially in the HER2 negative patients. Our study identified 33% TOP2A amplifications and 24% TOP2A deletions among the ERBB2 amplified cases. Among the ERBB2 non‐amplified cases 3% TOP2A amplifications and 7% TOP2A deletions were identified. Overall we have previously reported 12% amplifications and 11% deletions among all patients in the study (Knoop et al., 2005). Studies that only include HER2 positive cases find 35–50% TOP2A amplifications and 13–42% TOP2A deletions (Di Leo et al., 2002; Hicks et al., 2005; Järvinen et al., 2000). However, some studies omit detection of deletions due to lack of a reference probe (Arriola et al., 2007; Tanner et al., 2006). In studies including both HER2 positive and negative patients the frequency of TOP2A deletions is 6–17% (Bartlett et al., 2008; Knoop et al., 2005; O'Malley et al., 2009; Olsen et al., 2004) and almost the same frequency is found for amplifications. The lowest frequency of TOP2A amplification (2%) is reported in a series of 53 patients with a very high proportion (43%) of HER2 positive cases (Kuo et al., 2007) and the highest frequency of TOP2A deletion is 17% (Bartlett et al., 2008) reported in a patient series with relatively low frequency of ERBB2 amplification (21%). This high discrepancy illustrates the need of validated methods, identical cutoff levels and standardized scoring of signals for comparison between different studies. TOP2A deletions have been shown to be equally important for the clinical outcome as TOP2A amplifications (Knoop et al., 2005; Nielsen et al., 2008; O'Malley et al., 2009), but deletions are much more difficult to assess than amplifications if non‐validated methods are used. The present study uses a TOP2A method that has been validated prior to premarket approval by the authorities
Based on cell line studies, it was initially proposed (Järvinen et al., 1998, 1999) that TOP2A deletions would lead to lower topoisomerase IIα protein level and thus less sensitivity to anthracyclines. This hypothesis has been based on the presumption that deletion should lead to decrease of protein. The observed deletions only involve one allele, thus leaving the other allele which may be functional, mutated or inactivated. The only study (Corzo et al., 2007) comparing FISH results with both mRNA and protein measurements has revealed that TOP2A deletions are not associated with absence of mRNA and protein. TOP2A amplifications and deletions may not be two different ends of a continuum, but rather be considered as an abnormal state to be distinguished from the normal state as suggested by Koscielny et al. (1998) for HER2 protein. This would be in line with the observation on the two largest published investigations (Knoop et al., 2005; O'Malley et al., 2009) that both the prognostic and predictive value of TOP2A is similar for deletions and amplifications. Total absence of TOP2A protein is expected if both alleles are lost, but homozygotic deletion is a very rare condition, only described for the PTEN gene in prostate cancer (McCall et al., 2008).
In conclusion, the results on patients with invasive breast cancer supports the finding in cell lines that simple co‐amplification is not the main mechanism leading to patients with both ERBB2 and TOP2A aberrations and the term simultaneous amplification is more precise. ERBB2 amplification is of higher level than TOP2A amplification.
4. Experimental procedures
4.1. Tumor tissue from invasive breast cancer
The design of the original clinical trial and the biological sub‐study has been described previously in detail (Ejlertsen et al., 2007; Knoop et al., 2005; Nielsen et al., 2008). The Danish Breast Cancer Cooperative Group prepared the original protocol (DBCG trial 89D) as well as the biomarker sub‐study protocol (DBCG 89D/TOP2A) and The Danish National Committee on Biomedical Research Ethics approved both before activation. DBCG trial 89D was an open‐label randomized, phase III, trial comparing cyclophosphamide/epirubicin/5‐fluorouracil (CEF) against cyclophosphamide/methotrexate/5‐fluorouracil (CMF). The biomarker analysis was performed according to REMARK (McShane et al., 2005).
From January 1990 to January 1998, 1224 patients were randomized in DBCG trial 89D and 980 of these were recruited in Denmark. Archival paraffin embedded tissue blocks from 806 Danish patients enrolled in the trial were collected between September 2001 and August 2002 from the study sites and stored centrally. From the previously published biomarker study, data from TOP2A FISH analysis were available on 773 samples and on 767 patients all covariates were known. Paired HER2 (ERBB2) FISH results were available from 649 of these patients. The HER2 status on the remaining cases was determined by HER2 protein analysis. The main reason for the reduced number of HER2 FISH data was a decision by the study group not to analyze the remaining cases with HercepTest score 0 after the first 100 samples were analyzed without the finding of a single amplification.
4.2. FISH analysis
TOP2A and ERBB2 gene aberrations were identified by FISH (K5333 TOP2A FISH pharmDx™ Kit and K5331 HER2 FISH pharmDx™ Kit, Dako, Denmark) according to the manufactures instructions. Both kits are validated by the manufacturer for obtaining premarket approval by the FDA (US Food and Drug Administration). Further details are described elsewhere (Nielsen et al., 2004, 2008, 2004). The TOP2A probe covers a 228kb segment containing the TOP2A gene and two flanking genes on each side. The TOP2A gene is located 679kb from the ERBB2 gene, not overlapping the 280kb ERBB2 amplicon. The ERBB2 probe covers a 218kb segment containing 5 flanking genes. With respect to TOP2A and ERBB2 gene aberrations the ratio was calculated as the number of signals for the gene probes divided by the number of signals for the centromere 17 reference probe. Cases were scored as ERBB2 or TOP2A FISH amplified when the ratio was ≥2.0. Cases were scored as deleted when the ratio was <0.8. A previously conducted pilot study (Nielsen et al., 2008; Olsen et al., 2004) has shown that two different counting methods gave identical results: either the signals were counted in 60 nuclei or a total of 60 red gene probe signals were counted along with the green reference probe signals in the same nuclei. The latter method was used in the DBCG 89D/TOP2A study and has the advantage, that the highest number of cells will be counted in deleted cases, normal cases and cases around the cutoff, while the lowest number of cells will be counted in the amplified cases. Amplified cases are often obvious to identify just by looking in the microscope, but are more time consuming to evaluate if 60 nuclei should be scored.
4.3. Statistical analysis
Correlation between HER2 (ERBB2) and TOP2A status were tested by non‐parametric χ 2‐test and McNemar's test. P‐values are two‐tailed. Statistical analyses were done with the SAS 9·1 program package.
Acknowledgement
We thank Anwhar Al‐Abdulla and Birgit Gering for counting the embedded cell lines and Merete Mathiassen for excellent technical assistance preparing the metaphase spreads of the cell lines. The present study was supported by grants from the Danish Ministry of Health and from the Danish Research Council.
Nielsen Kirsten Vang, Müller Sven, Møller Susanne, Schønau Andreas, Balslev Eva, Knoop Ann S., Ejlertsen Bent, (2010), Aberrations of ERBB2 and TOP2A genes in breast cancer, Molecular Oncology, 4, doi: 10.1016/j.molonc.2009.11.001.
References
- Albertson, D.G. , 2006. Gene amplification in cancer. Trends Genet.. 22, (8) 447–455. [DOI] [PubMed] [Google Scholar]
- Albertson, D.G. , Collins, C. , McCormick, F. , Gray, J.W. , 2003. Chromosome aberrations in solid tumors. Nat. Genet.. 34, (4) 369–376. [DOI] [PubMed] [Google Scholar]
- Alitalo, K. , Schwab, M. , Lin, C.C. , Varmus, H.E. , Bishop, J.M. , 1983. Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc. Natl. Acad. Sci. USA. 80, (6) 1707–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriola, E. , Rodriguez-Pinilla, S.M. , Lambros, M.B. , Jones, R.L. , James, M. , Savage, K. , Smith, I.E. , Dowsett, M. , Reis-Filho, J.S. , 2007. Topoisomerase II alpha amplification may predict benefit from adjuvant anthracyclines in HER2 positive early breast cancer. Breast Cancer Res. Treat. 106, (2) 181–189. [DOI] [PubMed] [Google Scholar]
- Bartlett, J.M. , Munro, A. , Cameron, D.A. , Thomas, J. , Prescott, R. , Twelves, C.J. , 2008. Type 1 receptor tyrosine kinase profiles identify patients with enhanced benefit from anthracyclines in the BR9601 adjuvant breast cancer chemotherapy trial. J. Clin. Oncol. 26, (31) 5027–5035. [DOI] [PubMed] [Google Scholar]
- Bouchalova, K. , Trojanec, R. , Kolar, Z. , Cwiertka, K. , Cernakova, I. , Mihal, V. , Hajduch, M. , 2006. Analysis of ERBB2 and TOP2A gene status using fluorescence in situ hybridization versus immunohistochemistry in localized breast cancer. Neoplasma. 53, (5) 393–401. [PubMed] [Google Scholar]
- Corzo, C. , Bellosillo, B. , Corominas, J.M. , Salido, M. , Coll, M.D. , Serrano, S. , Albanell, J. , Sole, F. , Tusquets, I. , 2007. Does polysomy of chromosome 17 have a role in ERBB2 and topoisomerase IIalpha expression? Gene, mRNA and protein expression: a comprehensive analysis. Tumour Biol.. 28, (4) 221–228. [DOI] [PubMed] [Google Scholar]
- Di Leo, A. , Gancberg, D. , Larsimont, D. , Tanner, M. , Jarvinen, T. , Rouas, G. , Dolci, S. , Leroy, J.Y. , Paesmans, M. , Isola, J. , 2002. HER-2 amplification and topoisomerase IIalpha gene aberrations as predictive markers in node-positive breast cancer patients randomly treated either with an anthracycline-based therapy or with cyclophosphamide, methotrexate, and 5-fluorouracil. Clin. Cancer Res.. 8, (5) 1107–1116. [PubMed] [Google Scholar]
- Ejlertsen, B. , Mouridsen, H.T. , Jensen, M.B. , Andersen, J. , Cold, S. , Edlund, P. , Ewertz, M. , Jensen, B.B. , Kamby, C. , Nordenskjold, B. , 2007. Improved outcome from substituting methotrexate with epirubicin: results from a randomised comparison of CMF versus CEF in patients with primary breast cancer. Eur. J. Cancer. 43, (5) 877–884. [DOI] [PubMed] [Google Scholar]
- Harris, L.N. , Broadwater, G. , Abu-Khalaf, M. , Cowan, D. , Thor, A.D. , Budman, D. , Cirrincione, C.T. , Berry, D.A. , Winer, E.P. , Hudis, C.A. , 2009. Topoisomerase II{alpha} amplification does not predict benefit from dose-intense cyclophosphamide, doxorubicin, and fluorouracil therapy in HER2-amplified early breast cancer: results of CALGB 8541/150013. J. Clin. Oncol. 27, (21) 3430–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks, D.G. , Yoder, B.J. , Pettay, J. , Swain, E. , Tarr, S. , Hartke, M. , Skacel, M. , Crowe, J.P. , Budd, G.T. , Tubbs, R.R. , 2005. The incidence of topoisomerase II-alpha genomic alterations in adenocarcinoma of the breast and their relationship to human epidermal growth factor receptor-2 gene amplification: a fluorescence in situ hybridization study. Hum. Pathol. 36, (4) 348–356. [DOI] [PubMed] [Google Scholar]
- Jacobson, K.K. , Morrison, L.E. , Henderson, B.T. , Blondin, B.A. , Wilber, K.A. , Legator, M.S. , O'Hare, A. , Van Stedum, S.C. , Proffitt, J.H. , Seelig, S.A. , 2004. Gene copy mapping of the ERBB2/ TOP2A region in breast cancer. Genes Chromosomes Cancer. 40, (1) 19–31. [DOI] [PubMed] [Google Scholar]
- Järvinen, T.A. , Holli, K. , Kuukasjarvi, T. , Isola, J.J. , 1998. Predictive value of topoisomerase IIalpha and other prognostic factors for epirubicin chemotherapy in advanced breast cancer. Br. J. Cancer. 77, (12) 2267–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen, T.A.H. , Tanner, M. , Bärlund, M. , Borg, Å. , Isola, J. , 1999. Characterization of topoisomerase IIα gene amplification and deletion in breast cancer. Genes Chromosomes Cancer. 26, 142–150. [PubMed] [Google Scholar]
- Järvinen, T.A. , Tanner, M. , Rantanen, V. , Barlund, M. , Borg, A. , Grenman, S. , Isola, J. , 2000. Amplification and deletion of topoisomerase IIα associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am. J. Pathol. 156, (3) 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauraniemi, P. , Kuukasjarvi, T. , Sauter, G. , Kallioniemi, A. , 2003. Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am. J. Pathol. 163, (5) 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoop, A.S. , Knudsen, H. , Balslev, E. , Rasmussen, B.B. , Overgaard, J. , Nielsen, K.V. , Schonau, A. , Gunnarsdottir, K. , Olsen, K.E. , Mouridsen, H. , 2005. Retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil: Danish Breast Cancer Cooperative Group. J. Clin. Oncol. 23, (30) 7483–7490. [DOI] [PubMed] [Google Scholar]
- Koscielny, S. , Terrier, P. , Spielmann, M. , Delarue, J.C. , 1998. Prognostic importance of low c-erbB2 expression in breast tumors. J. Natl. Cancer Inst. 90, (9) 712 [DOI] [PubMed] [Google Scholar]
- Kuo, S.J. , Wang, B.B. , Chang, C.S. , Chen, T.H. , Yeh, K.T. , Lee, D.J. , Yin, P.L. , Chen, M. , 2007. Comparison of immunohistochemical and fluorescence in situ hybridization assessment for HER-2/neu status in Taiwanese breast cancer patients. Taiwan J. Obstet. Gynecol. 46, (2) 146–151. [DOI] [PubMed] [Google Scholar]
- Kuwahara, Y. , Tanabe, C. , Ikeuchi, T. , Aoyagi, K. , Nishigaki, M. , Sakamoto, H. , Hoshinaga, K. , Yoshida, T. , Sasaki, H. , Terada, M. , 2004. Alternative mechanisms of gene amplification in human cancers. Genes Chromosomes Cancer. 41, (2) 125–132. [DOI] [PubMed] [Google Scholar]
- Lacroix, M. , Leclercq, G. , 2004. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res. Treat. 83, (3) 249–289. [DOI] [PubMed] [Google Scholar]
- McCall, P. , Witton, C.J. , Grimsley, S. , Nielsen, K.V. , Edwards, J. , 2008. Is PTEN loss associated with clinical outcome measures in human prostate cancer? Br. J. Cancer. 99, (8) 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock, B. , 1941. The stability of broken ends of chromosomes in Zea mays . Genetics. 26, (2) 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane, L.M. , Altman, D.G. , Sauerbrei, W. , Taube, S.E. , Gion, M. , Clark, G.M. , 2005. Reporting recommendations for tumor marker prognostic studies (REMARK). J. Natl. Cancer Inst. 97, (16) 1180–1184. [DOI] [PubMed] [Google Scholar]
- Myllykangas, S. , Knuutila, S. , 2006. Manifestation, mechanisms and mysteries of gene amplifications. Cancer Lett.. 232, (1) 79–89. [DOI] [PubMed] [Google Scholar]
- Narayanan, V. , Mieczkowski, P.A. , Kim, H.M. , Petes, T.D. , Lobachev, K.S. , 2006. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell. 125, (7) 1283–1296. [DOI] [PubMed] [Google Scholar]
- Nielsen, K.V. , Müller, S. , Poulsen, T.S. , Gabs, S. , Schonau, A. , 2004. Combined use of PNA and DNA for fluorescence in situ hybridization (FISH). In Nielsen P.E., Peptide Nucleic Acids: Protocols and Applications. second ed Horizon Bioscience; Norfolk: 227–260. [Google Scholar]
- Nielsen, K.V. , Ejlertsen, B. , Moller, S. , Jorgensen, J.T. , Knoop, A. , Knudsen, H. , Mouridsen, H.T. , 2008. The value of TOP2A gene copy number variation as a biomarker in breast cancer: update of DBCG trial 89D. Acta Oncol. 47, (4) 725–734. [DOI] [PubMed] [Google Scholar]
- O'Malley, F.P. , Chia, S. , Tu, D. , Shepherd, L.E. , Levine, M.N. , Bramwell, V.H. , Andrulis, I.L. , Pritchard, K.I. , 2009. Topoisomerase II alpha and responsiveness of breast cancer to adjuvant chemotherapy. J. Natl. Cancer Inst. 101, (9) 615–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen, K.E. , Knudsen, H. , Rasmussen, B.B. , Balslev, E. , Knoop, A. , Ejlertsen, B. , Nielsen, K.V. , Schonau, A. , Overgaard, J. , 2004. Amplification of HER2 and TOP2A and deletion of TOP2A genes in breast cancer investigated by new FISH probes. Acta Oncol. 43, (1) 35–42. [DOI] [PubMed] [Google Scholar]
- Pandis, N. , Teixeira, M.R. , Adeyinka, A. , Rizou, H. , Bardi, G. , Mertens, F. , Andersen, J.A. , Bondeson, L. , Sfikas, K. , Qvist, H. , 1998. Cytogenetic comparison of primary tumors and lymph node metastases in breast cancer patients. Genes Chromosomes Cancer. 22, (2) 122–129. [PubMed] [Google Scholar]
- Pauletti, G. , Lai, E. , Attardi, G. , 1990. Early appearance and long-term persistence of the submicroscopic extrachromosomal elements (amplisomes) containing the amplified DHFR genes in human cell lines. Proc. Natl. Acad. Sci. USA. 87, (8) 2955–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, K.I. , Messersmith, H. , Elavathil, L. , Trudeau, M. , O'Malley, F. , Dhesy-Thind, B. , 2008. HER-2 and topoisomerase II as predictors of response to chemotherapy. J. Clin. Oncol. 26, (5) 736–744. [DOI] [PubMed] [Google Scholar]
- Rogalla, P. , Helbig, R. , Drieschner, N. , Flohr, A.M. , Krohn, M. , Bullerdiek, J. , 2002. Molecular-cytogenetic analysis of fragmentation of chromosome 17 in the breast cancer cell line EFM-19. Anticancer Res.. 22, (4) 1987–1992. [PubMed] [Google Scholar]
- Singer, M.J. , Mesner, L.D. , Friedman, C.L. , Trask, B.J. , Hamlin, J.L. , 2000. Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proc. Natl. Acad. Sci. USA. 97, (14) 7921–7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon, D.J. , Press, M.F. , 2009. Alterations in the TOP2A and HER2 genes: association with adjuvant anthracycline sensitivity in human breast cancers. J. Natl. Cancer Inst. 101, (9) 615–618. [DOI] [PubMed] [Google Scholar]
- Stark, G.R. , Debatisse, M. , Giulotto, E. , Wahl, G.M. , 1989. Recent progress in understanding mechanisms of mammalian DNA amplification. Cell. 57, (6) 901–908. [DOI] [PubMed] [Google Scholar]
- Tanaka, H. , Yao, M.C. , 2009. Palindromic gene amplification—an evolutionarily conserved role for DNA inverted repeats in the genome. Nat. Rev. Cancer. 9, (3) 216–224. [DOI] [PubMed] [Google Scholar]
- Tanner, M. , Isola, J. , Wiklund, T. , Erikstein, B. , Kellokumpu-Lehtinen, P. , Malmstrom, P. , Wilking, N. , Nilsson, J. , Bergh, J. , 2006. Topoisomerase IIalpha gene amplification predicts favorable treatment response to tailored and dose-escalated anthracycline-based adjuvant chemotherapy in HER-2/neu-amplified breast cancer: Scandinavian Breast Group Trial 9401. J. Clin. Oncol. 24, (16) 2428–2436. [DOI] [PubMed] [Google Scholar]
- Van Roy, N. , Vandesompele, J. , Menten, B. , Nilsson, H. , De Smet, E. , Rocchi, M. , De Paepe, A. , Pahlman, S. , Speleman, F. , 2006. Translocation-excision-deletion-amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes Chromosomes Cancer. 45, (2) 107–117. [DOI] [PubMed] [Google Scholar]
- Wolff, A.C. , Hammond, M.E. , Schwartz, J.N. , Hagerty, K.L. , Allred, D.C. , Cote, R.J. , Dowsett, M. , Fitzgibbons, P.L. , Hanna, W.M. , Langer, A. , 2007. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. 25, (1) 118–145. [DOI] [PubMed] [Google Scholar]