

Abstract
The KIT mutation D816V is associated with autonomous growth of mast cells (MC) and is detectable in most patients with systemic mastocytosis (SM), including cases with associated hematologic non‐MC‐lineage disease (AHNMD). Recently, KIT D816V was reported to be expressed in patients with acute myeloid leukemia (AML). However, it was not clarified whether these patients have co‐existing occult SM. We investigated neoplastic cells in 101 patients with AML for expression of KIT D816V. In 7/101 patients (6.9%), KIT D816V was detectable. After a thorough histologic, molecular, and biochemical analysis, all 7 cases were found to have an associated SM, leading to the final diagnosis SM‐AML. Microdissected tryptase+ MC displayed KIT D816V in all patients tested, whereas CD34+ blasts exhibited KIT D816V in only 2/4 patients. In one AML patient, SM without KIT D816V was detected. In all other patients, no associated SM was found, and leukemic blasts were negative for KIT D816V. In summary, our data show that KIT D816V in AML is highly associated with co‐existing SM (SM‐AML). Moreover, our data show that AML blasts may lack this transforming target‐mutant, which may be important when considering the use of KIT D816V‐targeting drugs for treatment of patients with KIT D816V‐positive AML.
Keywords: Mastocytosis, KIT D816V, AML, Microdissection
Abbreviations
- AML
acute myeloid leukemia
- ASM
aggressive systemic mastocytosis
- BM
bone marrow
- CMML
chronic myelomonocytic leukemia
- FAB
French–American–British
- FCS
fetal calf serum
- MC
mast cell(s)
- MCL
mast cell leukemia
- PB
peripheral blood
- PBS
phosphate-buffered saline
- RFLP
restriction fragment length polymorphism
- RT
room temperature
- SCF
stem cell factor
- SM
systemic mastocytosis
- TK
tyrosine kinase
1. Introduction
Acute myeloid leukemia (AML) is a life‐threatening hematopoietic stem cell neoplasm characterized by clonal proliferation of myeloid progenitor cells without significant differentiation or maturation (Brunning et al., 2001; Stirewalt and Radich, 2003; Estey and Döhner, 2006). The prognosis and clinical picture in AML vary depending on age, molecular defects, and the specific biological properties of the clone (Stirewalt and Radich, 2003; Mrozek et al., 2004; Estey and Döhner, 2006). During the past few years, a number of molecular markers related to the type of AML and/or the prognosis, have been identified. Several of these markers represent key regulators of growth and survival, and often are growth‐promoting oncoproteins. Constitutively activated tyrosine kinase (TK) receptors are of particular interest as they can serve as therapeutic targets (Mrozek et al., 2004; Estey and Döhner, 2006).
The TK receptor KIT is a well‐characterized oncoprotein that is expressed in diverse hematopoietic cells, including myeloid progenitor cells and mast cells (MC) (Ashman et al., 1991; Sillaber et al., 1991; Simmons et al., 1994; Valent, 1994). The ligand of KIT, stem cell factor (SCF), promotes growth and colony‐formation of myeloid progenitor cells as well as the differentiation of MC (Zsebo et al., 1990; Tsai et al., 1991; Valent et al., 1992; Kirshenbaum et al., 1992). It has also been described that in a group of patients with AML, blast cells express KIT and grow in response to the KIT‐ligand SCF (Bühring et al., 1991; Broudy et al., 1992). A number of previous and more recent studies have shown that certain somatic mutations in KIT can lead to autophosphorylation of the receptor and consecutive autonomous growth of affected cells (Furitsu et al., 1993; Ashman et al., 2000; Feger et al., 2002). The most intriguing example is the KIT mutation D816V that is detectable in neoplastic cells in a vast majority of patients with systemic mastocytosis (SM) (Nagata et al., 2002, 2001, 1996, 1999, 1995). SM is a disease characterized by abnormal growth and accumulation of MC in internal organs (Metcalfe, 1991; Valent, 1996; Valent et al., 2003; Akin and Metcalfe, 2004). In these patients, the KIT mutation D816V is considered to be responsible for abnormal survival and the enhanced accumulation of MC in the bone marrow (BM) and in other organs (Furitsu et al., 1993; Feger et al., 2002; Mayerhofer et al., 2008). In approximately 20–30% of all patients with SM, an associated clonal hematologic non‐MC‐lineage disease (AHNMD) is detected (Travis et al., 1988; Horny et al., 1990; Lawrence et al., 1991; Sperr et al., 2002). In most cases, a myeloid neoplasm such as AML or chronic myelomonocytic leukemia (CMML), is diagnosed (Sperr et al., 2002). In these patients, neoplastic MC (and MC progenitors) usually display KIT D816V (Sotlar et al., 2000; Sperr et al., 2002). A remarkable phenomenon is that in several of these cases, especially in CMML, the mutation is not only detectable in MC but also in the AHNMD‐component of the disease (Sotlar et al., 2000, 2002). By contrast, little is known about the expression of KIT D816V in AML blasts in patients with SM‐AML (Sperr et al., 1998). Moreover, only scattered information is available about the presence of other KIT mutations in AML and other myeloid leukemias.
Recently, KIT D816V has been reported to be expressed in leukemic cells in patients with AML (Cairoli et al., 2005; Nanri et al., 2005; Goemans et al., 2005; Cammenga et al., 2005; Schnittger et al., 2006). However, in most of these studies, AML patients were not investigated thoroughly for the presence of a co‐existent (occult) SM by histology or immunohistochemistry using histomorphological or biochemical criteria. We have recently shown that at least in some of these KIT D816V+ cases, the excess of AML blasts can mask a concomitant (thus ‘occult’) SM even when the bone marrow is examined histologically (Bernd et al., 2004).
The aim of the present study was to examine a cohort of AML patients for mutations in KIT, and to ask whether expression of KIT D816V can occur in the absence of (independent from) a co‐existing SM.
2. Patients and methods
2.1. Patients
Bone marrow or/and peripheral blood (PB) samples obtained from 101 patients with AML (observation period 1988–2006) were investigated. Median age was 58 years (range 15–89 years), and the female:male ratio 1:0.9. Diagnoses were established using criteria of the French–American–British (FAB) study group (Bennett et al., 1985). Accordingly, patients were diagnosed to have AML M0 (n=7), AML M1 (n=17), AML M2 (n=9), AML M3 (n=4), AML M4 (n=23), AML M4eo (n=5), AML M5 (n=13), AML M6 (n=4), AML M7 (n=1), and secondary AML (n=18). In patients with SM‐AML, the WHO type of AML (Brunning et al., 2001) is also provided. The patients' characteristics are summarized in 1, 2A, 2B. In 86 patients, BM was investigated; and in 15 patients, only PB was available for mutation analysis. Each patient gave written informed consent before BM or PB was obtained.
Table 1.
Characteristics of patients with AML
| AML (FAB) | N | % | Female (N) | Male (N) | Median age at diagnosis (years) | Median WBC (x 109/L) | Median HGB (g/dL) | Median PLT (×109/L) | % PB blasts, median (range) | % BM blasts, median (range) | Median serum tryptase level, ng/mL (range) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M0 | 7 | 6.9 | 4 | 3 | 55.0 | 21.2 | 8.5 | 35.5 | 50.5 (6.0–85.0) | 55.5 (25.0–83.0) | 25.6 (3.9–203) |
| M1 | 17 | 16.8 | 9 | 8 | 61.0 | 22.0 | 8.9 | 61.0 | 59.0 (64.0–93.0) | 87.0 (64.0–93.0) | 11.2 (0–775) |
| M2 | 9 | 8.9 | 1 | 8 | 57.0 | 17.9 | 8.7 | 74.0 | 30.0 (17.0–78.0) | 43.0 (30.0–82.0) | 41.6 (5.3–496) |
| M3 | 4 | 4.0 | 2 | 2 | 42.5 | 38.2 | 10.0 | 34.0 | 66.5 (38.0–93.0) | 85.5 (80.0–90.0) | 48.8 (12.4–122) |
| M4 | 23 | 22.8 | 15 | 8 | 61.0 | 22.2 | 8.8 | 47.0 | 18.0 (1.0–83.0) | 49.5 (30.0–90.0) | 7.9 (4–124) |
| M4eo | 5 | 5.0 | 4 | 1 | 38.0 | 17.0 | 8.5 | 17.0 | 56.0 (33.0–77.0) | 49.0 (31.0–80.0) | 54.0 (4.5–254) |
| M5 | 13 | 12.9 | 8 | 5 | 63.5 | 59.7 | 9.9 | 100.0 | 11.0 (1.0–38.0) | 52.0 (30.0–75.0) | 4.8 (3.5–45) |
| M6 | 4 | 4.0 | 0 | 4 | 55.5 | 3.0 | 8.7 | 21.5 | 2.0 (0.0–7.0) | 36.0 (14.0–85.0) | 8.8 (1.9–89.3) |
| M7 | 1 | 1.0 | 0 | 1 | 69.0 | 14.1 | 7.5 | 109.0 | 29.5 (6.0–53.0) | 41.0 (25.0–47.0) | 91.6 |
| sAML | 18 | 17.8 | 10 | 8 | 59.5 | 4.6 | 9.9 | 101.0 | 14.0 (0.0–90.0) | 48.0 (16.0–77.0) | 285.0 (20.2–305) |
| all pts | 101 | 100 | 43 | 58 | 58.0 | 17.9 | 9.0 | 56.0 | 22.0 (0.0–96.0) | 60.0 (14.0–93) | 11.4 (0–775) |
AML indicates acute myeloid leukemia; FAB, French–American–British classification; N, number; WBC, white blood count; HGB, hemoglobin; PLT, platelet count; PB, peripheral blood; BM, bone marrow; sAML, secondary acute myeloid leukemia; pts, patients.
Table 2A.
Characteristics and diagnosis of patients with SM‐AML at various times of investigation.
| Pts | Diagnosis | Date (Year) | Age | Sex | AML (FAB) | AML (WHO) | WBC (×109/L) | % Blasts PB | HGB (g/dL) | PLT (×109/L) | Typical skin lesions* | Serum tryptase (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | AML | 1988 | 65 | f | M4 | ND | 4400 | 13 | 10.2 | 102,000 | No | ND |
| #1 | SM‐MDS | 1995 | 72 | f | – | – | 2400 | 0 | 8.9 | 195,000 | Yes | 101 |
| #1 | SM‐AML | 1996 | 73 | f | M4 | ND | 14,410 | 28 | 7.5 | 16,000 | Yes | ND |
| #2 | ISM | 1993 | 55 | f | – | – | ND | 0 | ND | ND | Yes | ND |
| #2 | SM‐AML | 1995 | 57 | f | M0 | ND | 1260 | 4 | 7.4 | 123,000 | Yes | 101 |
| #2 | SM‐AML | 1996 | 59 | f | M0 | ND | 13,850 | 65 | 9.0 | 85,000 | Yes | ND |
| #3 | ASM | 2001 | 59 | m | – | – | 6750 | 0 | 9.6 | 115,000 | No | 285 |
| #3 | MCL | 2002 | 60 | m | – | – | 13,450 | 0 | 9.1 | 53,000 | No | 393 |
| #3 | ASM‐RARS | 2005 | 62 | m | – | – | 11,070 | 3 | 8.9 | 54,000 | No | 341 |
| #3 | ASM‐AML | 2005 | 63 | m | sec. AML | AML, therapy related | 3730 | 13 | 7.6 | 15,000 | No | 191 |
| #4 | ASM‐CMML | 2000 | 52 | f | – | – | 7250 | 0 | 10.2 | 86,000 | No | 294 |
| #4 | ASM‐AML | 2002 | 54 | f | M1 | AML without maturation | 27,590 | 76 | 10.5 | 49,000 | No | 1510 |
| #4 | ASM‐CMML | 2002 | 54 | f | – | – | 2240 | 0 | 10.2 | 102,000 | No | 127 |
| #5 | ISM‐AML | 2002 | 57 | m | M6 | Acute erythroid leukemia | 6200 | 7 | 10.2 | 9000 | No | ND |
| #6 | ASM‐CMML | 2001 | 53 | m | – | – | 27,950 | 0 | 8.8 | 54,000 | No | 610 |
| #6 | ASM‐AML | 2003 | 55 | m | sec. AML | AML with multi‐lineage dysplasia | 190,000 | 74 | 10.5 | 65,000 | No | 673 |
| #7 | ASM‐CMML | 2003 | 71 | m | – | – | 5780 | 0 | 8.2 | 17,000 | No | 47 |
| #7 | ASM‐AML | 2004 | 72 | m | sec. AML | AML with multi‐lineage dysplasia | 34,900 | 0 | 11.5 | 38,000 | No | 32 |
| #8 | ISM‐CMML | 2002 | 73 | m | – | – | 44,900 | 0 | 10.4 | 19,000 | No | 60 |
| #8 | ISM‐AML | 2003 | 74 | m | sec. AML | AML with multi‐lineage dysplasia | 115,000 | 49 | 7.7 | 24,000 | No | 446 |
SM indicates systemic mastocytosis; AML, acute myeloid leukemia; Pts, patients; FAB, French–American–British study group; WHO, world health organization; WBC, white blood cells; PB, peripheral blood; HGB, hemoglobin; PLT, platelets; UP, urticaria pigmentosa; MDS, myelodysplastic syndrome; ISM, indolent systemic mastocytosis; ASM, aggressive systemic mastocytosis; MCL, mast cell leukemia; RARS, refractory anemia with ringed sideroblasts; CMML, chronic myelomonocytic leukemia; f, female; m, male; sec AML, secondary AML; and ND, not determined. *Typical skin lesions of mastocytosis.
Table 2B.
Characteristics of patients with SM‐AML.
| Pts | Diagnosis | Date (Year) | Age | KIT D816V | % Blasts in BM smear | % MC in BM smear | Morphology MC** | % MC infiltrate in tryptase stain* | KIT D816V MC*** | KIT D816V CD34+*** | MC CD2 positive in BM | MC CD25 positive in BM | Karyotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | AML | 1988 | 65 | ND | 72 | <1 | ND | ND | ND | ND | ND | ||
| #1 | SM‐MDS | 1995 | 72 | ND | 8 | 5 | a I | ND | ND | ND | 46 XX | ||
| #1 | SM‐AML | 1996 | 73 | pos | 60 | 2 | a I | 5 | Pos | Pos | ND | ND. | 46 XX |
| #2 | ISM | 1993 | 55 | ND. | <5 | <1 | a I | 30 | ND | ND | ND | ||
| #2 | SM‐AML | 1995 | 57 | ND | 42 | <1 | a I | 5 | ND | ND | 46 XX | ||
| #2 | SM‐AML | 1996 | 59 | neg | 55 | <1 | a I | 5 | ND | ND | 46 XX | ||
| #3 | ASM | 2001 | 59 | pos | 1 | 4 | a I | 70 | Neg | Pos | 46 XY | ||
| #3 | MCL | 2002 | 60 | pos | 2 | 42 | a II/MB | 99 | Neg | Pos | 46 XY | ||
| #3 | ASM‐RARS | 2005 | 62 | ND | 1 | 1 | a II | 20 | Neg | Pos | ND | ||
| #3 | ASM‐AML | 2005 | 63 | pos | 25 | 1 | a II | 60 | Pos | Neg | Neg | Pos | ND |
| #4 | ASM‐CMML | 2000 | 52 | pos | 0 | 2 | a I | 50 | Neg | Pos | 46 XX | ||
| #4 | ASM‐AML | 2002 | 54 | pos | 81 | 1 | a I | 10 | Pos | Neg | Neg | ND | 46 XX |
| #4 | ASM‐CMML | 2002 | 54 | pos | 10 | 1 | a I | 20 | ND | ND. | 46 XY | ||
| #5 | ISM‐AML | 2002 | 57 | pos | 8 | <1 | a I | 5 | ND | ND | 47 XY,del(20),+22 | ||
| #6 | ASM‐CMML | 2001 | 53 | pos | ND | ND | ND | 70 | ND | ND | 46 XY | ||
| #6 | ASM‐AML | 2003 | 55 | pos | 53 | 1 | a I | <5 | Pos | Pos | ND | ND. | 46 XY,der(7),t(7;15) |
| #7 | ASM‐CMML | 2003 | 71 | pos | 1 | 2 | a I | 10 | ND | Pos | 46 XY | ||
| #7 | ASM‐AML | 2004 | 72 | pos | 53 | 1 | a I | <5 | ND | ND | 46 XY | ||
| #8 | ISM‐CMML | 2002 | 73 | ND | 5 | <1 | ND | <1 | ND. | ND. | ND | ||
| #8 | ISM‐AML | 2003 | 74 | pos | 30 | 1 | a I | 3 | Neg | Pos | ND |
SM indicates systemic mastocytosis; AML, acute myeloid leukemia; Pts, patients; BM, bone marrow; MC, mast cell; MDS, myelodysplastic syndrome; ISM, indolent systemic mastocytosis; ASM, aggressive systemic mastocytosis; MCL, mast cell leukemia; RARS, refractory anemia with ringed sideroblasts; CMML, chronic myelomonocytic leukemia; ND, not determined; pos, positive; neg, negative. Mast cell morphology (Sperr et al., 2001a,b): a I, atypical mast cell type I: spindle‐shaped hypogranulated mast cells with oval nucleus; a II, atypical mast cell type II with bilobed nuclei; and MB, metachromatic blasts. * Immunohistochemical analysis. **Predominant type of MC in bone marrow smears. ***KIT mutation analysis in microdissected MC and CD34+ cells.
2.2. Isolation of total RNA and cDNA synthesis
BM or PB mononuclear cells (MNC) were isolated by centrifugation using Ficoll. Aliquots of 1×107 cells were lysed by addition of 1.6mL RNAzol B (Biotecx, Houston, TX). RNA was extracted from MNC and from HMC‐1 cells according to a published protocol (Fritsche‐Polanz et al., 2001). One μg of total RNA was reverse transcribed into cDNA in the presence of murine leukemia virus reverse transcriptase (MMLV‐RT, Gibco, Gaitherburg, MD) and random hexamer primers (Amersham Pharmacia, Freiburg, Germany) in a final reaction volume of 20μL. cDNA synthesis was performed at 37°C (60min) followed by heat inactivation at 95°C (5min). The reaction samples were diluted to 100μL with Tris‐EDTA buffer.
2.3. Analysis of KIT mutations
KIT mutation analysis was performed by PCR amplification and restriction fragment length polymorphism (RFLP) analysis. The mutations investigated, the PCR primer sequences, number of PCR cycles, the restriction enzymes and the RFLP fragments generated are summarized in Table 3. PCR reactions included 3μL of cDNA in a 50μl reaction volume containing 10mM Tris–HCl (pH 8.3), 50mM KCl, 0.1 % Triton X‐100, 50pmol of each primer, 1.5mM MgCl2, 200μM of each dNTP, and 1.25 units of AmpliTaq Gold DNA Polymerase (PE Biosystems, Foster City, CA). The thermal cycling conditions were: denaturation at 94°C (1min), annealing at 55°C (2min), and extension at 72°C (1min), preceded by an initial denaturation step at 94°C for 6min, and a terminal extension step of 10min at 72°C (MultiCycler PTC 200, MJ Research, Miami, FL). PCR amplification products were analyzed on 6% polyacrylamide gels (Novex, San Diego, CA) and were stained with SYBR Green I (Molecular Probes, Eugene, Oregon) before RFLP analysis. RFLP analysis was performed using enzymes specifically cleaving KIT codons without mutation (Table 3). For detection of KIT D816V a previously described RFLP system cutting mutant KIT was applied (Fritsche‐Polanz et al., 2001). All digests were analyzed by electrophoresis through 6% polyacrylamide gels as described above for PCR products.
2.4. Determination of sensitivity of RFLP systems
The sensitivity of the RFLP systems for KIT mutation detection was evaluated using the HMC‐1 cell line (Butterfield et al., 1988) kindly provided by Dr. J.H. Butterfield (Mayo Clinic Rochester, MN). Two subclones of HMC‐1 were used: HMC‐1.1 expressing KIT G560V but not KIT D816V, and HMC‐1.2 expressing both mutations (Akin et al., 2003). HMC‐1 cells were cultured in Iscove's Modified Dulbecco's medium (IMDM; Gibco Life Technologies, Gaithersburg, MD) with 10% fetal calf serum (FCS) (PAA laboratories, Pasching, Austria). Total RNA was isolated from 1×107 cells and subjected to cDNA synthesis as described above. KIT mRNA was quantified in both subclones using pre‐designed TaqMan probes and primer sets (Applied Biosystems 7900 RT‐PCR System, Foster City, CA). Amplification of eukaryotic 18S rRNA, ACTB (beta actin), and GAPD (GAPDH), served as control. Quantification in 3 independent experiments revealed a 2‐fold higher expression of KIT mRNA in HMC‐1.2 cells compared to HMC‐1.1. Dilutions of HMC‐1.2 RNA in HMC‐1.1 RNA were performed after adjusting for the total amount of KIT mRNA. The final dilutions of KIT mRNA were 1:2, 1:5, 1:10, 1:20, 1:50, and 1:100. After cDNA synthesis, KIT transcripts were amplified with a primer system spanning codon 816 (Fritsche‐Polanz et al., 2001). PCR products were either cleaved with Hinf I (cleavage of the 287bp product in the presence of KIT D816V, mutant RFLP system) (Fritsche‐Polanz et al., 2001) or with BsmA 1 (cleavage of the PCR product without mutation, wild‐type RFLP system) (Table 3). cDNA synthesis, PCR amplification, and RFLP were performed in duplicates on ten separate days (a total of 20 results for each dilution). The diagnostic cut‐off for reliable detection of KIT D816V was 1:20 for the mutant RFLP and 1:50 for the wild‐type RFLP system. These dilutions revealed positive results in more than 95% of RFLP analyses.
2.5. Detection of KIT D816V in microdissected mast cells and AML blasts
In 4 patients with AML and associated SM (SM‐AML), neoplastic MC and AML blasts were microdissected from immunostained BM sections and subjected to KIT mutation analysis as reported (Sotlar et al., 2002, 2003). For detection of MC, antibodies against tryptase (AA‐1 from Dako, Glostrup, Denmark; or G3 from Chemicon, Temecula, CA) were used, and for detection of AML blasts, antibodies to CD34 (581 from Becton Dickinson, San Diego, CA; or QBEND10 from Biocare, Walnut, CA) were employed. Immunohistochemistry and microdissection were performed as described (Sotlar et al., 2002, 2003). In brief, single MC and blast cells were microdissected by laser pressure catapulting on a PALM Robot MicroBeam system, and pooled to a total of about 100 cells per PCR tube. From each microdissected cell type, i.e. pooled MC and pooled blast cells (in each patient), up to 8 PCR tubes were generated. Pretreatment and amplification of microdissected cells were performed as described previously (Sotlar et al., 2002, 2003).
2.6. Staging investigations and application of SM criteria in patients with KIT D816V
In patients with KIT D816V+ disease, a thorough investigation for SM criteria, including a BM biopsy was performed. Bone marrow sections were analyzed by immunohistochemistry using an antibody against tryptase as described (Horny et al., 1998). Aspirated cells were subjected to flow cytometry using fluorochrome‐conjugated antibodies against CD2, CD25, and CD117/KIT following a published protocol (Escribano et al., 2004). Bone marrow smears were carefully inspected for the presence of atypical (spindle‐shaped or immature) MC on Wright Giemsa‐stained slides following published guidelines (Sperr et al., 2001, 2001). Major and minor SM criteria were applied according to the WHO proposal (Valent et al., 2001, 2001). If at least one major (histology) and one minor or at least three minor criteria were met, the diagnosis of (an associated) SM was established.
Serum total tryptase levels were measured by a commercial fluoroimmunoenzyme assay (Amersham Pharmacia Biotech, Uppsala, Sweden). The serum tryptase level in healthy controls usually is below 20ng/mL (Schwartz et al., 1987, 1995). Tryptase levels were determined to define the subtype of AML (tryptase‐positive versus ‐negative) and to relate enzyme levels to the presence of KIT D816V and the presence of SM. Following the recommendations of the WHO proposal elevated serum tryptase levels (more than 20ng/mL) were not employed as minor diagnostic SM criterion in our AML patients (Valent et al., 2001a).
2.7. Evaluation of patients' survival and statistical analyses
To compare survival rates in AML patients with and without KIT D816V (with or without associated SM), the log rank test was applied. Cumulative survival was determined according to the method of Kaplan and Meyer. For survival measurements, two initial time points and related ‘calculation intervals’ were employed, namely the time from onset of disease (SM or AML), and the time from onset of AML (regardless of a pre‐existing SM). In particular, the probability of survival from the disease‐onset (either SM or AML – whatever was diagnosed first) and from the onset of AML were determined separately. To determine relationships between the presence of an associated SM (presence of KIT D816V) and serum tryptase levels, linear correlations were applied.
3. Results
3.1. Prevalence of KIT D816V in patients with AML
In 7 of 101 patients with AML (6.9%) the KIT mutation D816V was detected (Figure 1). Five of these patients were found to have secondary AML evolving from CMML, one patient initially had aggressive SM and later mast cell leukemia, before progressing to AML, and one patient suddenly developed AML in a completely indolent phase of SM, i.e. ISM (2A, 2B).
Figure 1.
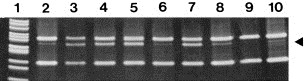
Analysis of KIT D816V in mononuclear cells of patients with AML. The presence of KIT D816V was investigated in isolated mononuclear cells by restriction fragment length polymorphism analysis of a 287bp reverse transcriptase polymerase chain reaction product. The arrow indicates the 157bp fragment generated by Hinf I restriction enzyme cleavage in the presence of KIT D816V. The mutation was only detectable in patients with AML and an associated systemic mastocytosis (lanes 2–8). In one patient with AML and associated SM, however, no KIT mutation at codon 816 was detected (lane 9). Lane 10 shows a mutation analysis performed with KIT D816V+ HMC‐1 cells diluted in KIT D816V‐cells at 1:20 (lower limit of detection – control). The Msp I‐digested plasmid pBR322 served as a molecular weight marker (lane 1).
3.2. Frequency of associated mastocytosis in patients with KIT D816V‐positive AML
In all 7 AML patients with KIT D816V, an associated SM was detected in BM sections and aspirated BM cells (major and minor SM criteria). In one AML patient with associated SM, no KIT mutation was detectable. All in all, 8 cases with SM were identified among 101 AML patients (7.9%). In 2 of these 8 patients, maculopapular lesions and histologic signs of skin involvement by SM were found (Table 2A). In all other patients, mastocytosis was only detected in the BM, but not in the skin. In 5 of the 8 patients with SM, mastocytosis in the skin had been diagnosed before AML developed. By contrast, in 3 patients with SM‐AML (all without skin lesions), AML had been diagnosed before SM was detected.
3.3. Characterization of the AML clone in patients with SM‐AML
In 7 of 8 patients with SM‐AML examined, a karyogram could be established. An abnormal karyotype was found in 2 patients: patient #5: 47XY,del(20)(q11.2q13),+22 and patient #6: 46XY,der(7);t(7;15). In the other 5 patients, no chromosomal abnormalities were found (Table 2B) which was an unexpected result. Notably, unlike reported in previous cases series with KIT D816V+ AML (Cairoli et al., 2005; Nanri et al., 2005; Goemans et al., 2005; Cammenga et al., 2005; Schnittger et al., 2006), no CBF‐related chromosome abnormalities were detected. Correspondingly, no typical AML‐related fusion transcripts (AML1/ETO, PML/RARA, CBFß/MYH11) were detected in a multiplex PCR assay. Serum tryptase levels were elevated in all patients with KIT D816V positive AML (Table 2A). A summary of clinical and laboratory parameters recorded in our AML patients with KIT D816V is shown in 2A, 2B.
3.4. Analysis of microdissected MC and AML blasts for KIT D816V
In 4 patients with KIT D816V+ SM‐AML, neoplastic tryptase+ MC and CD34+ AML blasts were microdissected from immunostained BM sections, and were analyzed for the presence of KIT D816V by melting point analysis of nested PCR products. Figure 2A–D shows typical examples of microdissection experiments using immunostained BM slides. In 2 of the 4 patients with KIT D816V+ disease, this mutation was detectable in MC, but not in isolated CD34+ blasts (Figure 2E,F; Table 2B). By contrast, in 2 patients, KIT D816V was detectable in both MC and AML blasts (Figure 2G and H; Table 2B). KIT D816V was not detected in microdissected blasts or MC in the patient with KIT D816V‐negative SM, confirming the results obtained by RFLP analysis (Table 2B).
Figure 2.
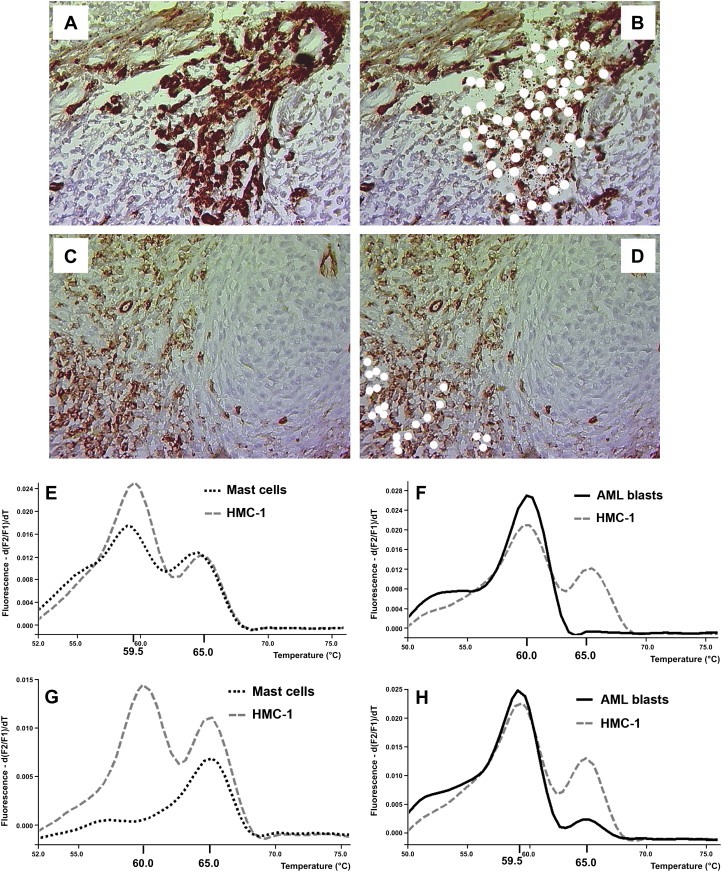
Analysis of microdissected bone marrow cells for the presence of KIT D816V. (A–D) Bone marrow sections in a patient with AML and associated systemic mastocytosis (SM) were stained with an antibody against tryptase (for mast cell detection) (A,B) and an antibody against CD34 (for detection of AML blasts) (C,D). Images were taken before (A,C) and after (B,D) microdissection by lasercapturing. Microdissected cells after successful capturing are indicated by white spots. (E–H) Melting point analysis for the presence of KIT D816V in microdissected cells in two patients with SM and associated AML (SM‐AML). In each case, HMC‐1 cells were run in parallel as an internal positive control. In one patient (E,F), CD34+ AML blast cells did not exhibit KIT D816V (E) whereas mast cells (F) clearly expressed KIT D816V. In the second patient, both the AML blasts (G) and the mast cells (H) were found to carry the KIT D816V mutant.
3.5. Investigation of other KIT mutations by RFLP
Apart from KIT D816V, we also screened for other KIT mutations in our AML patients using a wild‐type RFLP system (Figure 3). The codons analyzed were selected according to published KIT mutations known to occur in SM or AML, and included codons 52, 419, 530, 560, 820, 822 and 825 (Figure 3). None of the patients tested showed a mutation in these KIT codons.
Figure 3.
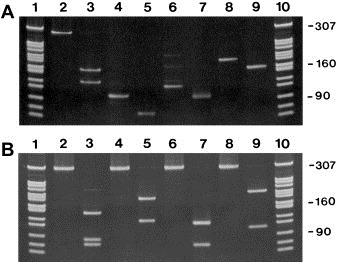
Analysis of other KIT mutations in mononuclear cells of patients with AML. The presence of other mutations in KIT was investigated in isolated mononuclear cells by restriction fragment length polymorphism (RFLP) analysis using enzymes cleaving the wild‐type sequence. A: Reverse transcriptase PCR amplification of codon 52 (280bp PCR product, lane 2), codon 419 (96bp PCR product, lane 4), codon 530 (126bp PCR product, lane 6), and codon 560 (178bp PCR product, lane 8). The PCR amplification products were cleaved with the restriction enzyme Hyp99 I (codon 52, lane 3), PflF I (codon 419, lane 5), Mae III (codon 530, lane 7), and Mse I (codon 560, lane 9) as shown in Table 3. The PCR and RFLP products were separated on 6% polyacrylamide gels. The Msp I‐digested plasmid pBR322 served as a molecular weight marker (lanes 1 and 10). B: Reverse transcriptase PCR amplification of codon 816 (lane 2), codon 820 (lane 4), codon 822 (lane 6), and codon 825 (lane 8). The PCR amplification products of 287bp were cleaved with the restriction enzyme BsmA 1 (codon 816, lane 3), Hinf I (codon 820, lane 5), Tsp509 I (codon 822, lane 7), and Mse I (codon 825, lane 9) as shown in Table 3. The PCR and RFLP products were separated on 6% polyacrylamide gels. The Msp I‐digested plasmid pBR322 served as a molecular weight marker (lanes 1 and 10).
3.6. Clinical significance of associated SM and KIT D816V in AML
To define the clinical significance of SM (displaying KIT D816V) in AML patients, survival was analyzed according to the method of Kaplan and Meyer. We found that the presence of KIT D816V (presence of concomitant SM) is associated with a significantly reduced probability of survival when survival was assessed from the onset of AML (Figure 4A). By contrast, when survival was assessed from the onset of disease (either SM or AML whatever diagnosed first), no difference in survival was found when comparing AML patients with and without concomitant SM (Figure 4B).
Figure 4.
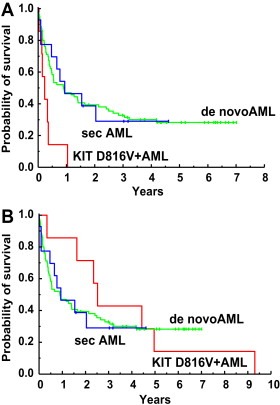
Survival in patients with AML and those with AML with associated SM (SM‐AML). Survival of patients with de novo acute myeloid leukemia (AML) (green), patients with secondary AML (blue), and patients with AML and associated systemic mastoyctosis (SM‐AML) (red) was calculated by the method of Kaplan and Meyer. Survival was calculated from the time point of first diagnosis of AML (A) and from the time point of first diagnosis of SM or AML (whatever first diagnosed) (B).
3.7. Correlation between KIT D816V and tryptase levels
In a significant number of patients with AML, an elevated serum tryptase level (more than 15ng/mL) was found. Since tryptase is produced primarily in MC, we were interested to learn whether KIT D816V (i.e. the presence of SM) correlates with serum tryptase levels. In a first step, we found that all patients with associated SM exhibit increased serum tryptase levels. However, many other patients with AML, in whom no histologic or molecular signs of SM were detected, were also found to have elevated serum tryptase levels. Thus, no significant correlation between tryptase levels and the presence of KIT D816V could be substantiated. Figure 5 shows serum tryptase levels in various groups of patients with AML.
Figure 5.
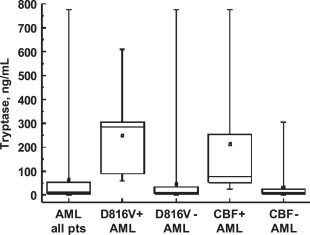
Median serum tryptase levels in patients with AML. Serum tryptase levels were determined by fluoroimmunoenzyme assay in various groups of patients with acute myeloid leukemia (AML) as indicated. The boxes represent the 25–75% percentile of serum tryptase levels in each group, the horizontal line within boxes the median, and the whiskers the range. The mean is shown as a small square within boxes. Elevated tryptase levels were found to cluster in patients with KIT D816V+ AML (D816V+) and in patients (pts) with core binding factor (CBF+) leukemias.
4. Discussion
Several reports have suggested that in a group of patients with AML, the transforming mutation KIT D816V is detectable (Cairoli et al., 2005; Nanri et al., 2005; Goemans et al., 2005; Cammenga et al., 2005; Schnittger et al., 2006). This mutation is otherwise found in neoplastic mast cells in patients with SM, but is not detectable in other myeloid neoplasms (Nagata et al., 1995; Longley et al., 1996; Fritsche‐Polanz et al., 2001; Feger et al., 2002). More recently, we have described that patients with ‘KIT D816V+ AML’ may have occult SM (SM‐AML) (Bernd et al., 2004). However, the exact frequency of occult SM in patients with ‘KIT D816V+ AML’ remains unknown. Our results suggest that the frequency of occult SM in AML with KIT D816V may be higher than has been assumed so far. In fact, all AML patients with detectable KIT D816V analyzed in this study were found to have an associated SM by histologic and/or biochemical (WHO) criteria.
According to WHO criteria, the diagnosis of SM is primarily based on the histologic demonstration of dense multifocal tryptase‐positive MC infiltrates in a representative BM biopsy section (major criterion of SM) (Valent et al., 2001, 2001). In addition, SM exhibits disease‐specific molecular features (minor criteria) including expression of CD2 and CD25 on neoplastic MC (Escribano et al., 1998, 2004), KIT D816V in affected cells (Nagata et al., 1996, 1995, 2001, 2001), and certain morphologic aspects of MC such as spindeling (Sperr et al., 2001a). In the present study, SM was detected in 8 AML patients using these criteria. Since the same markers and criteria were not employed in other studies examining patients with ‘KIT D816V+ AML’, it is difficult to relate our results to results published previously (Cairoli et al., 2005; Nanri et al., 2005; Goemans et al., 2005; Cammenga et al., 2005; Schnittger et al., 2006). However, single case report studies and smaller case series have already suggested that in several patients with AML, the associated SM had been overlooked previously (Bernd et al., 2004; Schnittger et al., 2006). In fact, AML patients with KIT D816V mutation may suffer from ‘occult’ SM. In these patients, a predominant AML clone in the BM may outnumber the smaller SM‐component of the disease, and such an occult SM may only be detected when analyzing the BM after successful cytoreductive therapy (Bernd et al., 2004). In our patients, however, co‐existing SM was readily detected in all patients, which may be explained by the thorough investigations performed, including detailed morphological studies and tryptase immunohistochemistry.
Another important question in this regard is whether an elevated tryptase level would be indicative for the presence of an occult SM. In fact, it is well known that in about 30–40% of all patients with de novo AML, an elevated serum tryptase level is found (Sperr et al., 2001b). However, in most of these patients, no associated SM is found (Sperr et al., 2001b), which could be confirmed in the present study. Correspondingly, in AML patients with elevated tryptase levels, tryptase is produced by myeloblasts (Sperr et al., 2001b). Whether tryptase+ blast cells in these AML patients aberrantly express the enzyme or represent very early mast cell‐committed progenitor cells remains unknown.
The coexistence of two distinct hematopoietic neoplasms, SM and AML, in one patient raises several questions. One question is as to whether neoplastic cells were derived from one identical clone (two subclones developed) or from two separate clones (Sperr et al., 2000). In our study, we were able to separate neoplastic MC and AML blasts by microdissection and to examine microdissected cells for the presence of KIT D816V. In these analyses, AML blasts exhibited KIT D816V in 2/4 patients with ‘KIT D816V+ AML’, whereas in the remaining 2 patients, AML blasts did not display KIT D816V. Thus, at least in a subset of patients with SM‐AML, the two disease‐components may belong to the same (sub)clone. However, our data also suggest that in several of these patients, KIT D816V is only expressed in neoplastic MC, but not in AML cells. These data are of particular interest as several AML trials using KIT D816V‐targeting drugs have been initiated. From the data generated in the current study, we recommend that (i) patients with AML should be tested for KIT D816V, and (ii) in those with detectable KIT D816V, isolated AML blasts should be examined for the presence of the mutation.
The variable expression of KIT D816V in AML blast cells in patients with SM‐AML is also of theoretical interest. The lack of the mutation in AML blasts in some of these patients (while clearly expressed in MC) may have several explanations. An attractive hypothesis would be the existence of two separate subclones derived from one stem cell (Sperr et al., 2000). In this hypothesis, one subclone acquired KIT D816V, and the other one transformed into AML (Sperr et al., 2000).
A number of recent data suggest that the presence of KIT D816V in AML is associated with a poor prognosis (Nanri et al., 2005; Schnittger et al., 2006; Care et al., 2003; Shimada et al., 2006; Cairoli et al., 2006). In our study, these data could be confirmed when comparing the outcome in these patients with patients with (de novo) AML. However, it was also found that the survival time in these patients varies depending on the definition of the checkpoint ‘disease‐onset’ in survival‐calculations. In fact, patients with SM‐AML may have SM long time before AML develops. These patients may well be regarded as secondary AML, as SM is a myeloid neoplasm – and consecutively, the survival in these patients should probably be compared to the survival of other patients with secondary AML (without SM). In the present study, we found that the survival measured from the time of AML evolution in our SM‐AML patients is poor, confirming the available literature. However, our study also demonstrates that the poor survival in these patients was similar when compared to patients with secondary AML (without co‐existing SM). The survival in our SM‐AML patients was even better than that of AML patients without SM when calculating survival times from the onset of disease, either SM or AML, whatever disease was diagnosed first. An interesting aspect in this regard is that several of these patients may not present with apparent clinical features of a BM disease or MC disease, as in these patients skin lesions often are absent – and thus, SM may be overlooked or may be occult for many years before the disease process is diagnosed (Bernd et al., 2004; Valent et al., 2007).
The frequency of KIT D816V in AML varies from study to study (Cairoli et al., 2005; Nanri et al., 2005; Goemans et al., 2005; Cammenga et al., 2005; Schnittger et al., 2006). In our patients, 6.9% of all AML patients were found to display KIT D816V. By contrast, in other studies, a smaller percentage of patients (2–5%) were reported to express the D816V‐mutated variant of KIT (Cairoli et al., 2005; Nanri et al., 2005; Goemans et al., 2005; Cammenga et al., 2005; Schnittger et al., 2006). This discrepancy may have several explanations. First, our center is a referral center for patients with mastocytosis – and all these patients have a thorough long term follow up and are also diagnosed and treated in our center when AML has developed. Second, the technique applied in our study shows a high sensitivity. In fact, KIT D816V may only be detected in SM‐AML patients when a highly sensitive technique is employed, since MC expressing KIT D816V may be outnumbered by blast cells in such patients.
Apart from KIT D816V, a number of other mutations in KIT have been described, and may occur in patients with mastocytosis (Longley et al., 1999; Garcia‐Montero et al., 2006). Some of these mutations have also been described to occur in AML. Therefore, we investigated our AML patients for additional recurrent KIT mutations in this study. However, no (KIT point) mutations in the KIT codons 52, 419, 530, 560, 816 (except for D816V), 820, 822, and 825 were detected in any of the patients examined.
In summary, our data show that expression of KIT D816V in AML is highly associated with and probably indicative of the presence of an associated (sometimes occult) SM. In several of these patients, KIT D816V may only be expressed by MC but not in AML blasts, which may have clinical and therapeutic implications.
Contributions
R.F.‐P. performed laboratory experiments, analyzed the data, and drafted the article. M. Fritz and A. H. conducted nucleic acid preparation and RFLP experiments. K. Sotlar performed microdissection experiments and analyzed microdissected cells for KIT mutations. K. Sonneck and S. F. contributed by isolating bone marrow cells and by conducting flow cytometry analysis. W. R. S. contributed patients and collected clinical data. C. M. contributed logistic support, laboratory equipment, and reagents. P. V. and M. Födinger contributed the research plan, logistics and budget, and approved the data and the final version of the manuscript.
Conflict of interest disclosure
The authors declare no competing financial interest.
Acknowledgment
This study was supported by the Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich, FWF, grant #P21173‐B13.
Fritsche-Polanz Robert, Fritz Marika, Huber Andrea, Sotlar Karl, Sperr Wolfgang R., Mannhalter Christine, Födinger Manuela, Valent Peter, (2010), High frequency of concomitant mastocytosis in patients with acute myeloid leukemia exhibiting the transforming KIT mutation D816V, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.04.008.
Supported by: Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich (FWF), grant #P21173‐B13.
References
- Akin, C. , Brockow, K. , D'Ambrosio, C. , 2003. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated forms of c-kit. Exp. Hematol. 31, 686–692. [DOI] [PubMed] [Google Scholar]
- Akin, C. , Metcalfe, D.D. , 2004. Systemic mastocytosis. Annu. Rev. Med. 55, 419–432. [DOI] [PubMed] [Google Scholar]
- Ashman, L.K. , Cambareri, A.C. , To, L.B. , Levinsky, R.J. , Juttner, C.A. , 1991. Expression of the YB5.B8 antigen (c-kit proto-oncogene product) in normal human bone marrow. Blood. 78, 30–37. [PubMed] [Google Scholar]
- Ashman, L.K. , Ferrao, P. , Cole, S.R. , Cambareri, A.C. , 2000. Effects of mutant c-kit in early myeloid cells. Leuk. Lymphoma. 37, 233–243. [DOI] [PubMed] [Google Scholar]
- Bennett, J.M. , Catovsky, D. , Daniel, M.T. , 1985. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French–American–British Cooperative Group. Ann. Intern. Med. 103, 620–625. [DOI] [PubMed] [Google Scholar]
- Bernd, H.W. , Sotlar, K. , Lorenzen, J. , 2004. Acute myeloid leukaemia with t(8;21) associated with “occult” mastocytosis. Report of an unusual case and review of the literature. J. Clin. Pathol. 57, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broudy, V.C. , Smith, F.O. , Lin, N. , Zsebo, K.M. , Egrie, J. , Bernstein, I.D. , 1992. Blasts from patients with acute myelogenous leukemia express functional receptors for stem cell factor. Blood. 80, 60–67. [PubMed] [Google Scholar]
- Brunning, R.D. , Vardiman, J. , Matutes, E. , 2001. Acute myeloid leukemia. World Health Organization (WHO) classification of tumours In Jaffe E.S., Harris N.L., Stein H., Vardiman J.W.(Eds.), Pathology & Genetics. first ed. Tumours of Haematopoietic and Lymphoid Tissues. IARC Press; Lyon: 77–105. [Google Scholar]
- Bühring, H.J. , Ullrich, A. , Schaudt, K. , Muller, C.A. , Busch, F.W. , 1991. The product of the proto-oncogene c-kit (P145c-kit) is a human bone marrow surface antigen of hemopoietic precursor cells which is expressed on a subset of acute non-lymphoblastic leukemic cells. Leukemia. 5, 854–860. [PubMed] [Google Scholar]
- Butterfield, J.H. , Weiler, D. , Dewald, G. , Gleich, G.J. , 1988. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk. Res. 12, 345–355. [DOI] [PubMed] [Google Scholar]
- Cairoli, R. , Beghini, A. , Morello, E. , 2005. Imatinib mesylate in the treatment of Core Binding Factor leukemias with KIT mutations. A report of three cases. Leuk. Res. 29, 397–400. [DOI] [PubMed] [Google Scholar]
- Cairoli, R. , Beghini, A. , Grillo, G. , 2006. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood. 107, 3463–3468. [DOI] [PubMed] [Google Scholar]
- Cammenga, J. , Horn, S. , Bergholz, U. , 2005. Extracellular KIT receptor mutants, commonly found in core binding factor AML, are constitutively active and respond to imatinib mesylate. Blood. 106, 3958–3961. [DOI] [PubMed] [Google Scholar]
- Care, R.S. , Valk, P.J. , Goodeve, A.C. , 2003. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br. J. Haematol. 121, 775–777. [DOI] [PubMed] [Google Scholar]
- Escribano, L. , Orfao, A. , Diaz-Agustin, B. , 1998. Indolent systemic mast cell disease in adults: immunophenotypic characterization of bone marrow mast cells and its diagnostic implications. Blood. 91, 2731–2736. [PubMed] [Google Scholar]
- Escribano, L. , Diaz-Agustin, B. , Lopez, A. , 2004. Spanish Network on Mastocytosis (REMA). Immunophenotypic analysis of mast cells in mastocytosis: when and how to do it. Proposals of the Spanish Network on Mastocytosis (REMA). Cytometry B. Clin. Cytom. 58, 1–8. [DOI] [PubMed] [Google Scholar]
- Estey, E. , Döhner, H. , 2006. Acute myeloid leukaemia. Lancet. 368, 1894–1907. [DOI] [PubMed] [Google Scholar]
- Feger, F. , Ribadeau Dumas, A. , Leriche, L. , Valent, P. , Arock, M. , 2002. Kit and c-kit mutations in mastocytosis: a short overview with special reference to novel molecular and diagnostic concepts. Int. Arch. Allergy Immunol. 127, 110–114. [DOI] [PubMed] [Google Scholar]
- Fritsche-Polanz, R. , Jordan, J.H. , Feix, A. , 2001. Mutation analysis of C-KIT in patients with myelodysplastic syndromes without mastocytosis and cases of systemic mastocytosis. Br. J. Haematol. 113, 357–364. [DOI] [PubMed] [Google Scholar]
- Furitsu, T. , Tsujimura, T. , Tono, T. , 1993. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of the c-kit product. J. Clin. Invest. 91, 1736–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Montero, A.C. , Jara-Acevedo, M. , Teodosio, C. , 2006. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 108, 2366–2372. [DOI] [PubMed] [Google Scholar]
- Goemans, B.F. , Zwaan, C.M. , Miller, M. , 2005. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia. 19, 1536–1542. [DOI] [PubMed] [Google Scholar]
- Horny, H.P. , Ruck, M. , Wehrmann, M. , Kaiserling, E. , 1990. Blood findings in generalized mastocytosis: evidence of frequent simultaneous occurrence of myeloproliferative disorders. Br. J. Haematol. 76, 186–193. [DOI] [PubMed] [Google Scholar]
- Horny, H.P. , Sillaber, C. , Menke, D. , 1998. Diagnostic value of immunostaining for tryptase in patients with mastocytosis. Am. J. Surg. Pathol. 22, 1132–1140. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum, A.S. , Goff, J.P. , Kessler, S.W. , Mican, J.M. , Zsebo, K.M. , Metcalfe, D.D. , 1992. Effect of IL-3 and stem cell factor on the appearance of human basophils and mast cells from CD34+ pluripotent progenitor cells. J. Immunol. 148, 772–777. [PubMed] [Google Scholar]
- Lawrence, J.B. , Friedman, B.S. , Travis, W.D. , Chinchilli, V.M. , Metcalfe, D.D. , Gralnic, H.R. , 1991. Hematologic manifestation of systemic mast cell disease: a prospective study of laboratory and morphologic features and their relation to prognosis. Am. J. Med. 91, 612–624. [DOI] [PubMed] [Google Scholar]
- Longley, B.J. , Tyrrell, L. , Lu, S.Z. , 1996. Somatic c-kit activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat. Genet. 12, 312–314. [DOI] [PubMed] [Google Scholar]
- Longley, B.J. , Metcalfe, D.D. , Tharp, M. , 1999. Activating and dominant inactivating c-kit catalytic domain mutations in distinct forms of human mastocytosis. Proc. Natl. Acad. Sci. (USA). 96, 1609–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerhofer, M. , Gleixner, K.V. , Hoelbl, A. , 2008. Unique effects of KIT D816V in BaF3 cells: induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J. Immunol. 180, 5466–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe, D.D. , 1991. Classification and diagnosis of mastocytosis: current status. J. Invest. Dermatol. 96, 2S-4S [PubMed] [Google Scholar]
- Mrozek, K. , Heerema, N.A. , Bloomfield, C.D. , 2004. Cytogenetics in acute leukemia. Blood Rev. 18, 115–136. [DOI] [PubMed] [Google Scholar]
- Nagata, H. , Worobec, A.S. , Oh, C.K. , 1995. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. (USA). 91, 10560–10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanri, T. , Matsuno, N. , Kawakita, T. , 2005. Mutations in the receptor tyrosine kinase pathway are associated with clinical outcome in patients with acute myeloblastic leukemia harboring t(8;21)(q22;q22). Leukemia. 19, 1361–1366. [DOI] [PubMed] [Google Scholar]
- Schnittger, S. , Kohl, T.M. , Haferlach, T. , 2006. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 107, 1791–1799. [DOI] [PubMed] [Google Scholar]
- Schwartz, L.B. , Metcalfe, D.D. , Miller, J.S. , Earl, H. , Sullivan, T. , 1987. Tryptase levels as an indicator of mast-cell activation in systemic anaphylaxis and mastocytosis. N. Engl. J. Med. 316, 1622–1626. [DOI] [PubMed] [Google Scholar]
- Schwartz, L.B. , Sakai, K. , Bradford, T.R. , 1995. The alpha form of human tryptase is the predominant type present in blood at baseline in normal subjects and is elevated in those with systemic mastocytosis. J. Clin. Invest. 96, 2702–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada, A. , Taki, T. , Tabuchi, K. , 2006. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t(8;21): a study of the Japanese Childhood AML Cooperative Study Group. Blood. 107, 1806–1809. [DOI] [PubMed] [Google Scholar]
- Sillaber, C. , Strobl, H. , Bevec, D. , 1991. IL-4 regulates c-kit proto-oncogene product expression in human mast and myeloid progenitor cells. J. Immunol. 147, 4224–4228. [PubMed] [Google Scholar]
- Simmons, P.J. , Aylett, G.W. , Niutta, S. , To, L.B. , Juttner, C.A. , Ashman, L.K. , 1994. c-kit is expressed by primitive human hematopoietic cells that give rise to colony-forming cells in stroma-dependent or cytokine-supplemented culture. Exp. Hematol. 22, 157–165. [PubMed] [Google Scholar]
- Sotlar, K. , Marafioti, T. , Griesser, H. , 2000. Detection of c-kit mutation Asp 816 to Val in microdissected bone marrow infiltrates in a case of systemic mastocytosis associated with chronic myelomonocytic leukaemia. Mol. Pathol. 53, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotlar, K. , Fridrich, C. , Mall, A. , 2002. Detection of c-kit point mutation Asp-816 → Val in microdissected pooled single mast cells and leukemic cells in a patient with systemic mastocytosis and concomitant chronic myelomonocytic leukemia. Leuk. Res. 26, 979–984. [DOI] [PubMed] [Google Scholar]
- Sotlar, K. , Escribano, L. , Landt, O. , 2003. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am. J. Pathol. 162, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperr, W.R. , Walchshofer, S. , Horny, H.P. , 1998. Systemic mastocytosis associated with acute myeloid leukaemia: report of two cases and detection of the c-kit mutation Asp-816 to Val. Br. J. Haematol. 103, 740–749. [DOI] [PubMed] [Google Scholar]
- Sperr, W.R. , Horny, H.P. , Lechner, K. , Valent, P. , 2000. Clinical and biologic diversity of leukemias occurring in patients with mastocytosis. Leuk. Lymphoma. 37, 473–486. [DOI] [PubMed] [Google Scholar]
- Sperr, W.R. , Jordan, J.H. , Baghestanian, M. , 2001. Expression of mast cell tryptase by myeloblasts in a group of patients with acute myeloid leukemia. Blood. 98, 2200–2209. [DOI] [PubMed] [Google Scholar]
- Sperr, W.R. , Escribano, L. , Jordan, J.H. , 2001. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk. Res. 25, 529–536. [DOI] [PubMed] [Google Scholar]
- Sperr, W.R. , Horny, H.P. , Valent, P. , 2002. Spectrum of associated clonal hematologic non-mast cell lineage disorders occurring in patients with systemic mastocytosis. Int. Arch. Allergy Immunol. 127, 140–142. [DOI] [PubMed] [Google Scholar]
- Stirewalt, D.L. , Radich, J.P. , 2003. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer. 3, 650–665. [DOI] [PubMed] [Google Scholar]
- Travis, W.D. , Li, C.Y. , Yam, L.T. , Bergstralh, E.J. , Swee, R.G. , 1988. Significance of systemic mast cell disease with associated hematologic disorders. Cancer. 62, 965–972. [DOI] [PubMed] [Google Scholar]
- Tsai, M. , Shih, L.S. , Newlands, G.F. , 1991. The rat c-kit ligand, stem cell factor, induces the development of connective tissue-type and mucosal mast cells in vivo. Analysis by anatomical distribution, histochemistry, and protease phenotype. J. Exp. Med. 174, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valent, P. , Horny, H.P. , Escribano, L. , 2001. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk. Res. 25, 603–625. [DOI] [PubMed] [Google Scholar]
- Valent, P. , Horny, H.P. , Li, C.Y. , 2001. Mastocytosis (Mast cell disease). World Health Organization (WHO) classification of tumours In Jaffe E.S., Harris N.L., Stein H., Vardiman J.W.(Eds.), Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues. vol. 1, IARC Press; Lyon: 291–302. [Google Scholar]
- Valent, P. , Akin, C. , Escribano, L. , 2007. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur. J. Clin. Invest. 37, 435–453. [DOI] [PubMed] [Google Scholar]
- Valent, P. , Spanblöchl, E. , Sperr, W.R. , 1992. Induction of differentiation of human mast cells from bone marrow and peripheral blood mononuclear cells by recombinant human stem cell factor/kit-ligand in long-term culture. Blood. 80, 2237–2245. [PubMed] [Google Scholar]
- Valent, P. , 1994. The riddle of the mast cell: kit(CD117)-ligand as the missing link?. Immunol. Today. 15, 111–114. [DOI] [PubMed] [Google Scholar]
- Valent, P. , 1996. Biology, classification and treatment of human mastocytosis. Wien. Klin. Wochenschr. 108, 385–397. [PubMed] [Google Scholar]
- Valent, P. , Akin, C. , Sperr, W.R. , 2003. Diagnosis and treatment of systemic mastocytosis: state of the art. Br. J. Haematol. 122, 695–717. [DOI] [PubMed] [Google Scholar]
- Zsebo, K.M. , Wypych, J. , McNiece, I.K. , 1990. Identification, purification, and biological characterization of hematopoietic stem cell factor from buffalo rat liver-conditioned medium. Cell. 63, 195–201. [DOI] [PubMed] [Google Scholar]