

Abstract
Transient or long‐term quiescence, the latter referred to as dormancy are fundamental features of at least some adult stem cells. The status of dormancy is likely a critical mechanism for the observed resistance of normal HSCs and leukemic stem cells (LSCs) to anti‐proliferative chemotherapy. Recent studies have revealed cytokines such as Interferon‐alpha (IFNα) and G‐CSF as well as arsenic trioxide (As2O3) to be efficient agents for promoting cycling of dormant HSCs and LSCs. Most interestingly, such cell cycle activated stem cells become exquisitely sensitive to killing by different chemotherapeutic agents, suggesting that dormant LSCs in patients may be targeted by a sequential two‐step protocol involving an initial activation by IFNα, G‐CSF or As2O3, followed by targeted chemotherapy.
Keywords: Stem cells, Leukemia, Therapy
1.
Hematopoietic stem cells (HSCs) are the first stem cells that have been used in the clinic to correct hematopoietic deficiencies or to reconstitute the blood system after stringent chemotherapy of cancer patients (Shizuru et al., 2005). Bone marrow HSCs are probably the best understood somatic stem cells due to the availability of a number of cell surface markers that allow their prospective identification and isolation by FACS followed by assays to examine their function both in vitro and in vivo. Mouse HSCs in healthy adult bone marrow show a LinnegSca1+Kit+CD34negCD150hiCD48neg phenotype, with approximately half of these cells being capable of life‐long reconstitution of lethally irradiated mice following single cell adoptive transfer (Ema et al., 2005, 2005, 1996, 2010, 2008, 2007). Human HSCs are less well defined but represent >10% of the LinnegCD34+CD38negCD90+CD45RAneg population within cord blood (Majeti et al., 2007; McDermott et al., 2010).
Most interestingly, several hematological malignancies including acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) often contain a small population of malignant cells that show similarities in phenotype and function to normal HSCs. As these, but not their more differentiated leukemic cell progeny can transplant the disease to immuno‐compromised mice they are called leukemic stem cells (LSCs) (Wang and Dick, 2005). These studies on leukemias provoked the general cancer stem cell (CSC) concept suggesting that many tumors are hierarchically organized with these putative CSCs being at the top of the hierarchy (Cho and Clarke, 2008; Dick, 2008; Reya et al., 2001). CSCs are genetically identical to the rest of the malignant clone, but constitute the only cells with tumor propagation potential within the overall tumor population. Simultaneously, they show significant resistance to radiation and anti‐proliferative chemotherapy due to their distinctive properties which seem to be related to their stem cell‐like character. Thus, although these therapies often significantly reduce the bulk of tumor cells, resistant CSCs are often retained in the body, and are thought to be causative for the frequently observed relapse and metastasis formation after initially successful cancer therapies. Although populations of tumor cells containing CSC activity have now been identified from various solid cancers including brain, breast, colon and prostate, the search for reliable markers to identify CSCs is still ongoing (Trumpp and Wiestler, 2008). It appears that marker expression on CSCs depends on tumor type and stage as well as on the mutations present in each individual tumor. Moreover, the expression of certain markers on CSCs may not be stable within the tumor at any stage and time making it notoriously difficult to identify reliable, general markers for CSCs in vitro or in vivo. In addition, not all types of cancers are hierarchically organized, and even in those that are, the hierarchy may flatten during tumor progression towards highly metastatic “undifferentiated” cancers, in which most tumor cells show CSC properties (Eaves, 2008; Kelly et al., 2007; Passegue et al., 2009; Quintana et al., 2008). Nevertheless, the functional similarities of CSCs with normal stem cells are significant, and may provide an explanation as to why CSCs show a high degree of therapy resistance, a feature of greatest clinical relevance (Zhou et al., 2009).
A number of mechanisms have been suggested to explain the exquisite resistance of CSCs to radio‐ and chemotherapies. For example, CSCs, like normal stem cells express various ABC transporter pumps that export small molecules (drugs) out of the cell. They also have a very efficient DNA repair mechanism and are thought to be located in hypoxic extracellular matrix rich niches, which would mediate resistance to radiotherapy‐induced DNA damage and prevent drugs from reaching sufficiently high concentrations within CSCs. Finally, like many normal HSCs, CSCs may also divide only rarely, or at least are transiently in a state of deep long‐term quiescence called dormancy. Such a state would make them resistant to anti‐proliferative and other chemotherapy regimens as they are not only non‐cycling, but also require little energy or oxygen and thus may as well be rather insensitive to signaling pathway inhibitors (Trumpp and Wiestler, 2008).
1. Quiescence and dormancy in stem cells
It has long been recognized that although tissue stem cells have the highest self‐renewal potential and are individually able to produce a clone comprising millions of cells, they are themselves rather inactive in terms of cell cycle activity (Yamazaki et al., 2006). Moreover, long‐term labeling studies combined with mathematical modeling and functional transplantation assays have demonstrated that the most potent HSCs in the adult healthy mouse are in a state of dormancy (Foudi et al., 2009; Wilson et al., 2008). These dormant HSCs (dHSCs), which show the highest self‐renewal potential of all HSCs, divide only about five times per lifetime, and are almost permanently in the G0 phase of the cell cycle (van der Wath et al., 2009). They are found as individual cells in niches within the cavities of trabecular bone, and have shut down their entire DNA replication machinery, which is likely associated with a massive reduction of their general energy metabolism (Trumpp et al., 2010, 2008, 2009).
Why does the body keep such a reservoir of dormant HSCs in the adult bone marrow? It turns out that loss of hematopoietic cells by bleeding, toxic insults or chemotherapy induces a feedback loop leading to the awakening of dHSCs (Trumpp et al., 2010) (Figure 1). Although the signals that are involved remain elusive, they efficiently turn dHSCs into rapidly dividing active HSCs (aHSC), which are able to produce rapidly dividing progenitors that can then differentiate into mature cell types lost after the initial insult (Figure 1). After homeostasis is re‐installed, a‐HSCs return to their niches and become dormant again (Figure 1). Thus dHSCs most likely serve as a reservoir of highly potent but well protected HSCs essential for rapid and efficient reconstitution of the blood system in response to severe and life threatening bone marrow stress (Trumpp et al., 2010; Wilson et al., 2008). Long‐term quiescent, but activatable stem cells have also been identified outside the blood system. Examples are stem cells within the Drosophila hindgut or epidermal stem cells in the bulge region of the epidermis (Fuchs, 2009; Xie, 2009). Thus it is possible that many tissues may harbor highly potent, rare dormant stem cells as a reservoir reserved for emergencies.
Figure 1.
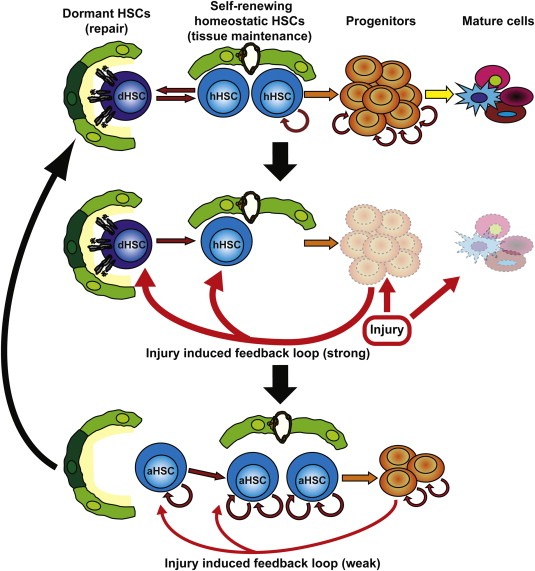
A positive feedback loop leads to the activation of dormant and homeostatic HSCs in response to hematopoietic cell loss. Dormant and homeostatic HSCs reside in distinct bone marrow niches. During homeostasis, dormant HSCs (dHSCs) remain inactive, whereas homeostatic HSCs (hHSCs) divide and self‐renew if the number of progenitors and differentiated cells drop below a homeostatic threshold level. The self‐renewal activity of hHSCs results in the constant generation of rapidly dividing progenitors, which eventually differentiate into mature blood cells. Injury to the hematopoietic system (i.e. irradiation, chemotherapy or bleeding) results in severe loss of hematopoietic cells, inducing strong positive feedback loops (red arrow). As a consequence, dHSCs and hHSCs exit their niches and undergo major self‐renewing divisions, thus replenishing first the progenitor compartment, followed by the recovery of all hematopoietic cell populations and return to homeostasis.
Most interestingly, dHSCs have been shown to be resistant to anti‐proliferative chemotherapy such as 5‐FU, which is in agreement with clinical data showing that normal tissues most affected by chemotherapy (bone marrow, hair and gastrointestinal mucosa), readily recover after drug treatment suggesting that also most normal tissue stem cells are not killed by these regimens (Essers et al., 2009; Morrison et al., 1997; Venezia et al., 2004). Interestingly, activation or priming of dHSCs using certain cytokines can convert normally resistant dHSCs into chemotherapy sensitive cells raising the possibility of designing sequential treatment regimens that may permit targeting of LSCs, a topic discussed in the remainder of this review.
2. Gaining chemotherapy sensitivity by breaking dormancy of normal and malignant stem cells
Studies on normal HSCs suggest that the status of dormancy protects them from chemotherapy‐induced killing in vivo. The reasons for this may not be limited to their non‐cycling stage, but may also be associated with their extremely low metabolism, that also renders them rather insensitive to toxic substances affecting energy metabolism or signaling pathway inhibitors. However, whether CSCs are indeed in a (transient) dormant phase in vivo has not yet been explored in patients, and requires the development of novel protocols. Nevertheless, in certain malignancies, such as breast cancer, metastatic relapse can occur more than a decade after the initial treatment. A phenomenon that can be explained by the survival and long‐term persistence of dormant CSCs (Aguirre‐Ghiso, 2007). In addition, Ishikawa and colleagues have recently provided evidence that AML stem cells are located at the endosteal region of the bone marrow and are mainly non‐cycling (Saito et al., 2010). Moreover, Tessa Holyoake and colleagues showed that cultured CD34+ stem/progenitor cells isolated from BCR‐ABL positive chronic myeloid leukemia (CML) patients also contain quiescent cells, and that these are resistant to the tyrosine kinase inhibitor Imatinib mesylate (IM) (also known as Gleevec), which blocks the constitutively active BCR‐ABL kinase produced by the Philadelphia chromosome (Goldman et al., 2009). Imatinib is the first example of targeted chemotherapy as it inhibits the causative mutation (BCR‐ABL) that initiates the disease (Goldman, 2007). Indeed, most CML patients respond very well to Imatinib, thus revolutionizing the treatment of this disease. Nevertheless, even patients showing complete molecular response (BCR‐ABL transcripts are no longer detectable by PCR) are not cured, since stopping Imatinib treatment frequently leads to relapse of the disease, likely due to a few Imatinib resistant quiescent CML stem cells retained in these patients (Goldman, 2009). Whether their quiescence stage, independency of BCR‐ABL signaling or possibly their sequestering in specific niches or a combination thereof is the reason for their resistance is still under debate. However it is likely that overcoming LSC dormancy is a critical step towards a cure for this leukemia, and possibly for other CSC‐driven cancers.
Recently, several studies have revealed agents that can activate quiescent/dormant HSCs. These include certain cytokines such as G‐CSF and IFNα, as well as arsenic trioxide (As2O3), a compound that targets PML for proteasomal degradation, all of which can efficiently activate dHSCs by inducing their cell cycle entry. Furthermore, activation of HSCs with these agents is correlated with an increased sensitivity to chemotherapy. Moreover, in mouse models for leukemia, G‐CSF and arsenic trioxide (As2O3) not only affect mouse HSCs, but also LSCs, thus opening up the idea of combining these agents with chemotherapeutic drugs to efficiently eliminate LSCs.
2.1. G‐CSF
One of the first cytokines reported to have an effect on HSCs was Granulocyte‐colony‐stimulating‐factor (G‐CSF). Treatment of mice with G‐CSF results in efficient activation of dormant HSCs, followed by the mobilization of activated HSCs into the blood stream (Jorgensen et al., 2006; Morrison et al., 1997; Wilson et al., 2008). Although the mechanism of cell cycle activation via G‐CSF remains largely unknown, mobilization is induced by the release of proteolytic enzymes such as matrix metalloproteinases (MMPs), elastase or cathepsin G by neutrophils in the bone marrow. Released enzymes will cleave and degrade stem cell anchorages between HSCs and osteoblasts of the bone marrow stem cell niche, such as SDF1/CXCR4, homotypic N‐cadherin, and VCAM‐1/VLA‐4 interactions (Heissig et al., 2002; Petit et al., 2002), between HSCs and osteoblasts in the bone marrow stem cell niche, thus resulting in the release of HSCs from their niche cells. Interestingly, G‐CSF induced activation and mobilization of HSCs in mice makes them susceptible to killing by chemotherapeutic agents like 5‐FU and cytarabine (Jorgensen et al., 2006; Morrison et al., 1997; Saito et al., 2010). These data would indicate that G‐CSF induced activation of HSCs can be combined with chemotherapy to efficiently eliminate dormant HSCs.
The first evidence for such a model was obtained by culturing CML CD34+ enriched progenitor cells in the presence of G‐CSF before exposure to IM. Indeed, intermittent exposure of these LSCs to G‐CSF in vitro, resulting in activation of quiescent LSCs, was shown to enhance the effects of IM on CML cells. Thus, these data indicated that activation of CML LSCs by G‐CSF makes them susceptible to IM treatment. Unfortunately, the first clinical pilot study has failed to confirm that combined G‐CSF and IM treatment can eliminate CML‐SCs in patients. An explanation for this might be that the exact timing of activation of CML‐SCs and IM treatment may be critical or G‐CSF may not be efficient enough to activate all stem cells. Thus further tests i.e. in xenografts need to be undertaken to improve the protocol and obtain proof‐of principle evidence for such a strategy (Drummond et al., 2009).
Therapy resistance, not only in CML, but also in AML has been suggested to be due to the relative state of quiescence in LSCs. To overcome this, G‐CSF treatment was used in a xenograft model, in which purified LSCs from AML patients were successfully engrafted into NOD‐SCID; IL2Rγ chain null (NOG) mice. Treatment of these xenografts with G‐CSF resulted in efficient activation of dormant human AML stem cells located in their endosteal bone marrow niches. Interestingly, activated AML stem cells were then eliminated by the chemotherapeutic agent cytarabine, although this was not complete as mice could not be cured from the leukemia (Saito et al., 2010). In contrast to LSCs, treatment of G‐CSF followed by cytarabine did not affect normal mouse HSCs in these animals (Saito et al., 2010). Although these data are somewhat unexpected, they indicate that human leukemic stem cells respond differently than normal HSCs to human G‐CSF, creating a window for combined treatment of G‐CSF with chemotherapeutic agents. However, the normal HSCs in this model are of mouse origin and the effects of combined G‐CSF/cytarabine on normal human HSCs still need to be determined.
Both studies suggest that G‐CSF mediated priming may significantly enhance the killing of AML and CML stem cells by cytarabine and IM respectively. Nevertheless, the efficiency needs to be further improved in order to eliminate all LSCs, a requirement towards a cure from the disease.
2.2. Arsenic trioxide
The possibility to activate quiescent HSCs and LCSc by influencing the activity of PML was recently reported by Pier Paolo Pandolfi and colleagues. The nuclear factor promyelocytic leukemia (PML) protein is localized to PML nuclear bodies and control processes such as apoptosis, cellular proliferation and senescence (Ito et al., 2009). It encodes a tumor suppressor and is highly expressed in HSCs (Ito et al., 2008). Recently, PML has been shown to play a role in HSC self‐renewal, since loss of PML enhances cycling of HSCs associated with an increase in mTOR activity (Ito et al., 2008). Arsenic trioxide (As2O3) targets the PML protein for proteosomal‐dependent degradation and this causes an increase in general RNA content as measured by Pyronin Y staining in cultured HSCs, which is indicative of HSC activation (Ito et al., 2008).
The role of PML in LSCs was also tested in a mouse model for CML. Although loss of PML induced cycling of normal HSCs the exit from quiescence was even more profound in LSCs (Ito et al., 2008), suggesting a possible therapeutic window for targeting PML in leukemic patients. Even though the effects of As2O3 on normal dormant HSCs have not yet been addressed in detail, low doses of As2O3 in these mice disrupted LSC maintenance through the inhibition of PML (Ito et al., 2008). Even more importantly, in combination with the chemotherapeutic drug Ara‐C, As2O3 increased the efficacy of this anti‐leukemic treatment for LSCs, most likely by inducing their exit from quiescence. Both human and mouse LSCs were sensitized to pro‐apoptotic stimuli (cytarabine) upon treatment with As2O3 (Ito et al., 2008). Significantly, As2O3 has successfully been used clinically to treat PML‐retinoic acid receptor‐α (RARα) induced acute promyelocytic leukemia showing that this drug can be effectively used in patients (Nasr et al., 2008, 2009, 2010). Future studies need to determine whether As2O3 can activate dormant stem cells also in other forms of human leukemia, not driven by oncogenic PML.
2.3. IFNα
IFNs play a critical role in the regulation of resistance to viral infections and enhancement of the innate and acquired immune responses via the transcriptional regulation of a whole set of IFN‐stimulated genes (ISGs) (de Veer et al., 2001). In addition to their role during host defense, IFNs were also shown to have anti‐proliferative properties on many cell types in vitro, making them highly attractive, not only as therapy for infections but also for the treatment of cancer (Borden et al., 2007).
In contrast to the previous in vitro data, we have recently shown that, treatment of mice with IFNα induces transient proliferation of progenitors and quiescent HSCs within hours of application of IFNα in vivo (Figure 2) (Essers et al., 2009). Moreover, LRC experiments revealed that even the most dormant HSCs are effectively activated by IFNα stimulation. In contrast to G‐CSF induced activation, IFNα does not induce mobilization of HSCs to the periphery, although it remains likely that IFNα alters the interaction of HSCs with their niche. This could also provide an explanation for the apparent distinct response to IFNα of HSCs in vivo compared to cultured HSCs. Upon administration, IFNα binds to its receptor on the surface of HSCs, leading to rapid phosphorylation of STAT1 and transcription of a set of IFNα target genes (Figure 2) (Essers et al., 2009). Moreover, IFNα treatment causes an increase in both transcription and cell surface presentation of the stem cell marker Sca‐1 on HSCs and progenitors (Essers et al., 2009) (Figure 2). Remarkably, Sca‐1 functionally mediates the proliferative response of HSCs to IFNα, since Sca‐1 deficient HSCs fail to respond, identifying this protein as a key player downstream of IFNα signaling in HSCs (Essers et al., 2009).
Figure 2.
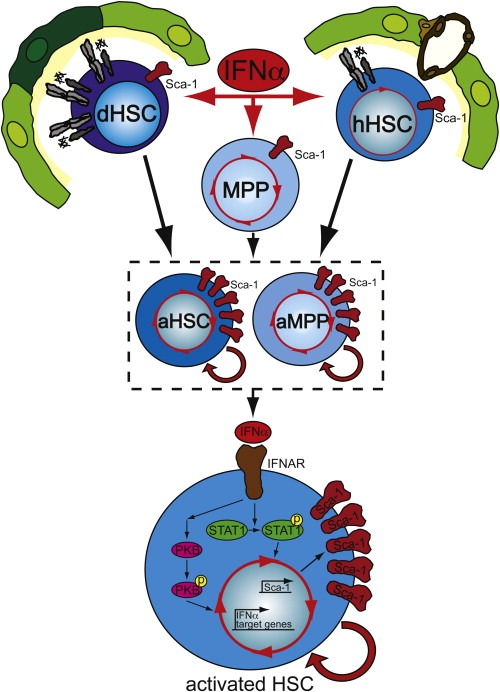
IFNα induces activation of HSCs in vivo. In vivo treatment of mice with the cytokine IFNα induces cell cycle entry of dormant and homeostatic HSCs as well as multipotent progenitors (MPPs). IFNα signaling leads to rapid phosphorylation of STAT1 and PKB, as well as activation of a set of IFNα target genes. The IFNα response is also associated with a robust up‐regulation of the stem cell marker Sca‐1 (red) on the cell surface of stimulated cells. This up‐regulation of Sca‐1 is required for the IFNα‐mediated activation and proliferation of stem/progenitors, since dHSCs lacking Sca‐1 no longer divide in response to IFNα.
Chronic IFNα treatment of HSCs, leading to permanent activation of HSCs, does not severely affect HSC function, since these HSCs are still able to efficiently reconstitute lethally irradiated mice (Essers et al., 2009). However, in a competitive setting, these chronically IFNα‐stimulated HSCs are rapidly out‐competed by HSCs engineered to be unable to respond to IFNα stimulation, due to the lack of the IFNAReceptor (Essers et al., 2009). A possible explanation for this observation is that chronically stimulated HSCs fail to compete for the dormant niche space and thus are lost when normal, dormant HSCs are also present. Similar observations have been made in mice genetically deficient for Interferon response factor‐2 (IRF2), a transcriptional repressor of type I IFN signaling (Sato et al., 2009). Although they initially proliferate more due to constitutive IFN signaling, IRF2−/− HSCs are incapable of reconstituting lethally irradiated mice. These data confirm that chronic activation of IFNα signaling in HSCs induces a competitive disadvantage in repopulation assays indicating that chronic INFα exposure has a negative impact on self‐renewal and function.
Our observation that IFNα induces activation of the dormant HSC pool, suggests that IFNα could be used as a stem cell targeting drug in a combined treatment protocol. Thus, dormant LSCs would first be activated by IFNα treatment followed by Imatinib, which should then be able to eliminate the activated CML stem cells. Priming of dormant mouse HSCs for 1–3 days using IFNα in vivo efficiently sensitized HSCs to 5‐FU, a chemotherapeutic agent killing only cycling cells (Figure 3). Interestingly, simultaneous treatment of IFNα and 5‐FU did not lead to elimination of the HSCs, strongly indicating that a two‐step model, rather than a combined protocol, is crucial to efficiently target the stem cells (Essers et al., 2009). Whether IFNα is also able to activate dormant human leukemic stem cells remains to be shown. Nevertheless, long‐term high dose IFNα therapy had a positive effect on a significant number of CML patients and was the first‐line treatment of newly diagnosed patients before IFNα was replaced by IM in 2001. However, IM must be taken life‐long as most patients, even those in complete molecular remission, frequently relapse upon IM discontinuation. However, in a French CML trial, a small group of patients experienced continued long‐term remission even after Imatinib discontinuation. Strikingly, these six patients had been treated with IFNα before they were switched to Imatinb in 2001 (Rousselot et al., 2007). This indicates that IFNα may have activated and mobilized LSCs in these patients, rendering them more sensitive to the subsequently administered IM, and thereby allowing eradication of the entire leukemic clone and hence cure. In addition, it is possible or even likely that effects of IFNα on cells of the immune system, such as cytotoxic T cells, may also have contributed to the eradication of the leukemic SCs. Although extended studies on CML patients are necessary to confirm these initial observations, they do raise potential clinical possibilities for combined therapy of activators of dormancy with targeted chemotherapy to target the dormant LSCs.
Figure 3.
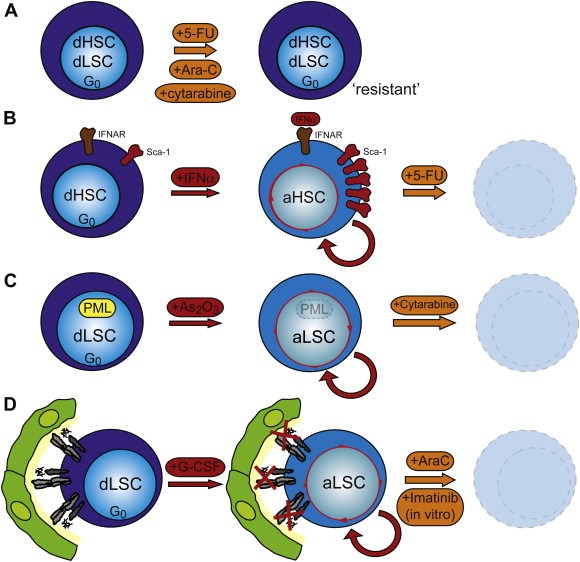
A two‐step therapy strategy to target leukemic stem cells: three examples. a. Even though leukemic stem cells (LSCs) might only be a minority within the malignant leukemic clone, they show significant resistance to anti‐proliferative chemotherapy. Their resistance is considered to be the cause of frequently observed tumor relapse. Several reasons for this resistance have been suggested amongst which is the quiescent or dormant state of LSCs. Thus, if dormancy is a main reason for LSC resistance to chemotherapy one could postulate a two‐step therapy model by which the dormant LSCs can be targeted; activators of quiescence first induce activation of dormant LSCs, followed by targeted therapy. This therapy model would not only eliminate the bulk of the leukemia, but likely also activated LSCs, thus reducing the probability of relapse of the leukemia. Three possible combinations have recently been suggested: b. IFNα efficiently activates dormant mouse HSCs in vivo via the up‐regulation of Sca‐1, thus making them susceptible to elimination by 5‐FU. c. Inhibition of PML by treatment with arsenic trioxide (As2O3) induces activation of LSCs facilitating their efficient elimination by the chemotherapeutic agent Ara‐C. d. G‐CSF induces activation and mobilization of dormant HSCs and LSCs. Combined treatment of G‐CSF and Imatinib in vitro efficiently kills dormant CML LSCs, whereas G‐CSF treatment in vivo, followed by cytarabine, leads to the elimination of AML LSCs in a xenograft model for AML.
In summary, dormancy of stem cells and their localization in dormancy inducing bone marrow niches is essential for maintenance of their self‐renewal activity. Moreover, it protects stem cells from hostile influences, like infections, mutations and chemotherapeutic drugs. In addition, the often observed resistance of leukemic stem cells to chemotherapy might at least in part be explained by their state of dormancy. If dormancy is indeed crucial for mediating drug resistance, a two‐step protocol involving activation by IFNα, G‐CSF, As2O3 or a combination thereof, followed by (targeted) chemotherapy, may target even dormant malignant stem cells. Thus it may indeed be possible to eliminate the entire leukemic clone including LSCs leading to a long‐term cure (Figure 3). Such a strategy would also be supported by mathematical modeling of CML stem cells predicting that a combined therapy could potentially eliminate all CML cells, including the CML stem cells (Roeder et al., 2006) (Figure 3). Future studies examining the molecular mechanisms of dormancy and drug resistance in normal and leukemic stem cells will be needed to fully evaluate the impact of such a two‐step therapeutic protocol for more effective future treatments of stem cell driven leukemias.
Acknowledgements
The authors would like to thank Dr. Anne Wilson for comments on the manuscript. This work was funded by the BioRN Spitzencluster “Molecular and Cell Based Medicine” funded by the German Bundesministerium für Bildung und Forschung (BMBF).
Essers Marieke Alida Gertruda, Trumpp Andreas, (2010), Targeting leukemic stem cells by breaking their dormancy, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.06.001.
References
- Aguirre-Ghiso, J.A. , 2007. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer. 7, 834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden, E.C. , Sen, G.C. , Uze, G. , Silverman, R.H. , Ransohoff, R.M. , Foster, G.R. , Stark, G.R. , 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, R.W. , Clarke, M.F. , 2008. Recent advances in cancer stem cells. Curr. Opin. Genet. Dev. 18, 48–53. [DOI] [PubMed] [Google Scholar]
- de Veer, M.J. , Holko, M. , Frevel, M. , Walker, E. , Der, S. , Paranjape, J.M. , Silverman, R.H. , Williams, B.R. , 2001. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69, 912–920. [PubMed] [Google Scholar]
- Dick, J.E. , 2008. Stem cell concepts renew cancer research. Blood. 112, 4793–4807. [DOI] [PubMed] [Google Scholar]
- Drummond, M.W. , Heaney, N. , Kaeda, J. , Nicolini, F.E. , Clark, R.E. , Wilson, G. , Shepherd, P. , Tighe, J. , McLintock, L. , Hughes, T. , 2009. A pilot study of continuous imatinib vs pulsed imatinib with or without G-CSF in CML patients who have achieved a complete cytogenetic response. Leukemia. 23, 1199–1201. [DOI] [PubMed] [Google Scholar]
- Eaves, C.J. , 2008. Cancer stem cells: here, there, everywhere?. Nature. 456, 581–582. [DOI] [PubMed] [Google Scholar]
- Ema, H. , Sudo, K. , Seita, J. , Matsubara, A. , Morita, Y. , Osawa, M. , Takatsu, K. , Takaki, S. , Nakauchi, H. , 2005. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev. Cell. 8, 907–914. [DOI] [PubMed] [Google Scholar]
- Essers, M.A. , Offner, S. , Blanco-Bose, W.E. , Waibler, Z. , Kalinke, U. , Duchosal, M.A. , Trumpp, A. , 2009. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 458, 904–908. [DOI] [PubMed] [Google Scholar]
- Foudi, A. , Hochedlinger, K. , Van Buren, D. , Schindler, J.W. , Jaenisch, R. , Carey, V. , Hock, H. , 2009. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 27, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, E. , 2009. The tortoise and the hair: slow-cycling cells in the stem cell race. Cell. 137, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, J.M. , 2007. How I treat chronic myeloid leukemia in the imatinib era. Blood. 110, 2828–2837. [DOI] [PubMed] [Google Scholar]
- Goldman, J.M. , 2009. Treatment strategies for CML. Best Pract. Res. Clin. Haematol. 22, 303–313. [DOI] [PubMed] [Google Scholar]
- Goldman, J.M. , Green, A.R. , Holyoake, T. , Jamieson, C. , Mesa, R. , Mughal, T. , Pellicano, F. , Perrotti, D. , Skoda, R. , Vannucchi, A.M. , 2009. Chronic myeloproliferative diseases with and without the Ph chromosome: some unresolved issues. Leukemia. 23, 1708–1715. [DOI] [PubMed] [Google Scholar]
- Heissig, B. , Hattori, K. , Dias, S. , Friedrich, M. , Ferris, B. , Hackett, N.R. , Crystal, R.G. , Besmer, P. , Lyden, D. , Moore, M.A. , 2002. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 109, 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, K. , Bernardi, R. , Morotti, A. , Matsuoka, S. , Saglio, G. , Ikeda, Y. , Rosenblatt, J. , Avigan, D.E. , Teruya-Feldstein, J. , Pandolfi, P.P. , 2008. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 453, 1072–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, K. , Bernardi, R. , Pandolfi, P.P. , 2009. A novel signaling network as a critical rheostat for the biology and maintenance of the normal stem cell and the cancer-initiating cell. Curr. Opin. Genet. Dev. 19, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, H.G. , Copland, M. , Allan, E.K. , Jiang, X. , Eaves, A. , Eaves, C. , Holyoake, T.L. , 2006. Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clin. Cancer Res. 12, 626–633. [DOI] [PubMed] [Google Scholar]
- Kelly, P.N. , Dakic, A. , Adams, J.M. , Nutt, S.L. , Strasser, A. , 2007. Tumor growth need not be driven by rare cancer stem cells. Science. 317, 337 [DOI] [PubMed] [Google Scholar]
- Kiel, M.J. , Yilmaz, O.H. , Iwashita, T. , Terhorst, C. , Morrison, S.J. , 2005. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Majeti, R. , Park, C.Y. , Weissman, I.L. , 2007. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 1, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott, S.P., Eppert, K., Lechman, E., Doedens, M., Dick, J.E., 2010. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. Epub ahead of print. [DOI] [PubMed]
- Morrison, S.J. , Wright, D.E. , Weissman, I.L. , 1997. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. U. S. A. 94, 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr, R. , Guillemin, M.C. , Ferhi, O. , Soilihi, H. , Peres, L. , Berthier, C. , Rousselot, P. , Robledo-Sarmiento, M. , Lallemand-Breitenbach, V. , Gourmel, B. , 2008. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 14, 1333–1342. [DOI] [PubMed] [Google Scholar]
- Nasr, R. , Lallemand-Breitenbach, V. , Zhu, J. , Guillemin, M.C. , de The, H. , 2009. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin. Cancer Res. 15, 6321–6326. [DOI] [PubMed] [Google Scholar]
- Osawa, M. , Hanada, K. , Hamada, H. , Nakauchi, H. , 1996. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 273, 242–245. [DOI] [PubMed] [Google Scholar]
- Passegue, E. , Rafii, S. , Herlyn, M. , 2009. Cancer stem cells are everywhere. Nat. Med. 15, 23 [DOI] [PubMed] [Google Scholar]
- Petit, I. , Szyper-Kravitz, M. , Nagler, A. , Lahav, M. , Peled, A. , Habler, L. , Ponomaryov, T. , Taichman, R.S. , Arenzana-Seisdedos, F. , Fujii, N. , 2002. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat. Immunol. 3, 687–694. [DOI] [PubMed] [Google Scholar]
- Quintana, E. , Shackleton, M. , Sabel, M.S. , Fullen, D.R. , Johnson, T.M. , Morrison, S.J. , 2008. Efficient tumour formation by single human melanoma cells. Nature. 456, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya, T. , Morrison, S.J. , Clarke, M.F. , Weissman, I.L. , 2001. Stem cells, cancer, and cancer stem cells. Nature. 414, 105–111. [DOI] [PubMed] [Google Scholar]
- Roeder, I. , Horn, M. , Glauche, I. , Hochhaus, A. , Mueller, M.C. , Loeffler, M. , 2006. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat. Med. 12, 1181–1184. [DOI] [PubMed] [Google Scholar]
- Rousselot, P. , Huguet, F. , Rea, D. , Legros, L. , Cayuela, J.M. , Maarek, O. , Blanchet, O. , Marit, G. , Gluckman, E. , Reiffers, J. , 2007. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 109, 58–60. [DOI] [PubMed] [Google Scholar]
- Saito, Y. , Uchida, N. , Tanaka, S. , Suzuki, N. , Tomizawa-Murasawa, M. , Sone, A. , Najima, Y. , Takagi, S. , Aoki, Y. , Wake, A. , 2010. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 28, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, T. , Onai, N. , Yoshihara, H. , Arai, F. , Suda, T. , Ohteki, T. , 2009. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med. 15, 696–700. [DOI] [PubMed] [Google Scholar]
- Shizuru, J.A. , Negrin, R.S. , Weissman, I.L. , 2005. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu. Rev. Med. 56, 509–538. [DOI] [PubMed] [Google Scholar]
- Trumpp, A. , Essers, M. , Wilson, A. , 2010. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 10, 201–209. [DOI] [PubMed] [Google Scholar]
- Trumpp, A. , Wiestler, O.D. , 2008. Mechanisms of Disease: cancer stem cells – targeting the evil twin. Nat. Clin. Pract. Oncol. 5, 337–347. [DOI] [PubMed] [Google Scholar]
- van der Wath, R.C. , Wilson, A. , Laurenti, E. , Trumpp, A. , Lio, P. , 2009. Estimating dormant and active hematopoietic stem cell kinetics through extensive modeling of bromodeoxyuridine label-retaining cell dynamics. PLoS One. 4, e6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia, T.A. , Merchant, A.A. , Ramos, C.A. , Whitehouse, N.L. , Young, A.S. , Shaw, C.A. , Goodell, M.A. , 2004. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2, e301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.C. , Dick, J.E. , 2005. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 15, 494–501. [DOI] [PubMed] [Google Scholar]
- Wilson, A. , Laurenti, E. , Oser, G. , van der Wath, R.C. , Blanco-Bose, W. , Jaworski, M. , Offner, S. , Dunant, C.F. , Eshkind, L. , Bockamp, E. , 2008. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Wilson, A. , Laurenti, E. , Trumpp, A. , 2009. Balancing dormant and self-renewing hematopoietic stem cells. Curr. Opin. Genet. Dev. 19, 461–468. [DOI] [PubMed] [Google Scholar]
- Wilson, A. , Oser, G.M. , Jaworski, M. , Blanco-Bose, W.E. , Laurenti, E. , Adolphe, C. , Essers, M.A. , Macdonald, H.R. , Trumpp, A. , 2007. Dormant and self-renewing hematopoietic stem cells and their niches. Ann. N. Y. Acad. Sci. 1106, 64–75. [DOI] [PubMed] [Google Scholar]
- Xie, T. , 2009. Stem cell in the adult Drosophila hindgut: just a sleeping beauty. Cell Stem Cell. 5, 227–228. [DOI] [PubMed] [Google Scholar]
- Yamazaki, S. , Iwama, A. , Takayanagi, S. , Morita, Y. , Eto, K. , Ema, H. , Nakauchi, H. , 2006. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 25, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.W. , Yan, X.J. , Zhou, Z.R. , Yang, F.F. , Wu, Z.Y. , Sun, H.B. , Liang, W.X. , Song, A.X. , Lallemand-Breitenbach, V. , Jeanne, M. , 2010. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 328, 240–243. [DOI] [PubMed] [Google Scholar]
- Zhou, B.B. , Zhang, H. , Damelin, M. , Geles, K.G. , Grindley, J.C. , Dirks, P.B. , 2009. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov. 8, 806–823. [DOI] [PubMed] [Google Scholar]