

Abstract
Epigenetic changes can be defined as stable molecular alterations of a cellular phenotype such as the gene expression profile of a cell that are heritable during somatic cell divisions (and sometimes germ line transmissions) but do not involve changes of the DNA sequence itself. Epigenetic phenomena are mediated by several molecular mechanisms comprising histone modifications, polycomb/trithorax protein complexes, small non‐coding or antisense RNAs and DNA methylation. These different modifications are closely interconnected. Epigenetic regulation is critical in normal growth and development and closely conditions the transcriptional potential of genes. Epigenetic mechanisms convey genomic adaption to an environment thereby ultimately contributing towards given phenotype. In this review we will describe the various aspects of epigenetics and in particular DNA methylation in breast carcinogenesis and their potential application for diagnosis, prognosis and treatment decision.
Keywords: Breast cancer; DNA methylation; Histone modification, genomic instability; Basal-like; Tp53
1. Introduction
Epigenetic alterations in transformed cells involve changes in DNA methylation including global hypomethylation and locus‐specific hypermethylation, altered histone tail modifications patterns and nucleosomal remodeling. DNA methylation is an enzyme‐driven chemical change to the DNA sequence that most commonly occurs at CpG dinucleotides in mammals (Hinshelwood and Clark, 2008) (Figure 1). DNA hypomethylation can be associated with gene reactivation and chromosomal instabilities and might lead to the upregulation or overexpression of proto‐oncogenes, increased recombination and mutation rates, skewed or loss of X‐chromosomal inactivation and loss of imprinting (De Smet et al., 2004). DNA hypermethylation is frequently associated with gene repression and genomic instability (through silencing of DNA repair genes) and can lead to the suppression of tumor‐suppressor genes and compaction of chromatin. Associated with DNA methylation are post‐translational histone tail modifications, another epigenetic mechanism that can modulate chromatin structure to regulate gene expression (Baylin and Ohm, 2006; Esteller, 2007). In addition, it has been shown that some regulators that control nucleosomal remodeling are also involved in the regulation of DNA methylation and histone modification (Baylin and Ohm, 2006; Esteller, 2007; Jones and Baylin, 2007; Martin and Zhang, 2005). The understanding of all these epigenetics changes and their contribution to breast tumorigenesis is very important for further progress in the field of diagnosis, prognosis and therapy of breast cancer.
Figure 1.
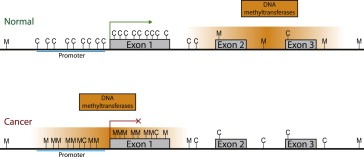
DNA methylation in normal and cancer cell. In a normal cell CpG islands (C) in actively transcribed promoter regions are not methylated allowing transcriptional activity (green arrow). CpG islands elsewhere, within genes and intergenic spaces are more often methylated (M). In a cancer cell the reverse is observed. Red line with X: repressed transcription.
2. DNA methylation, histone modification and chromatin structure
DNA methylation is a post‐replication modification, almost exclusively found on the 5th position of the pyrimidine ring of cytosines in the context of the dinucleotide sequence CpG (Bird, 2002). 5‐Methylcytosine accounts for ∼1% of all bases, varying slightly in different tissue types and the majority (75%) of CpG dinucleotides throughout mammalian genomes are methylated. More than 90% of all methylated cytosines reside within repetitive elements and transposons that together account for one third of the human genome. During evolution, the number of CpG dinucleotides in the genome has been reduced because of the inherent mutagenicity of methylated cytosines and the result is a reduced number of CpGs compared to the mathematically expected number. Methylated cytosines are more susceptible to modifications by endogenous and exogenous mutagenic processes than the other bases of the DNA and mutation rates at CpG sites have been estimated to be about 40 times higher than other transitional mutations (Pfeifer and Besaratinia, 2009). About one third of all known somatic and germ line mutations are C to T transitions at CpG dinucleotides, although there are some differences in the distribution according to the tumor type (Jones et al., 2008).
CpG islands are short stretches of DNA (200 bp to several kb in length) and are located in the 5′‐regions of 60% of all genes. Despite the general trend for CpGs throughout the genome to be methylated, CpG sites in CpG islands and especially those associated with gene promoters are usually unmethylated which correlates with their potential for active gene transcription (De Smet et al., 2004; Stearns et al., 2007). In cancer cells, CpG islands that are normally unmethylated may become methylated, which might result in silencing of important genes, such as tumor‐suppressor genes (Figure 2). At the same time, CpG dinucleotides in other regions may become unmethylated, leading to poor transcriptional repression of normally silenced genes such as oncogenes or retrotransposons. Different classes of genes are silenced by DNA methylation: tumor‐suppressor genes, genes that suppress tumor invasion, and metastasis; DNA repair genes; genes for hormone receptors; and genes that inhibit angiogenesis. DNA methylation affects gene expression because the transcriptional regulatory proteins that bind to methylated DNA differ from those that bind to unmethylated DNA. Gene silencing by hypermethylation of promoter genes is an important mechanism of carcinogenesis that has great promise for cancer prevention and therapy (“epigenetic therapy”). Many tumor‐suppressor and other cancer‐related genes have been found to be hypermethylated in human cancer cells, and primary tumors (Lo and Sukumar, 2008). Their biological function includes cell‐cycle regulation, apoptosis, DNA repair, cellular homeostasis, cell adhesion and invasion. A selection of the most frequently methylated genes in breast cancer is given in Table 1. A new dimension has been added to epigenetic cancer research with the demonstration of long‐range gene silencing by epigenetic modifications (Frigola et al., 2006). Long‐range epigenetic silencing seems to be a prevalent phenomenon during carcinogenesis as a recent survey identified 28 regions of copy‐number independent transcriptional deregulation that are potentially regulated through epigenetic mechanisms (Stransky et al., 2006). Breast cancer does not constitute an exception and the distribution of aberrantly methylated regions across the genome was found to be non‐random and tended to concentrate in relatively small genomic regions spanning up to several hundred kilobases which are frequently located at gene family clusters such as the HOXA gene cluster (Novak et al., 2006). Remodeling of the cancer genome thus occurs through a combination of hypermethylation and long‐range epigenetic silencing together with loss of heterozygosity and genomic deletions.
Figure 2.
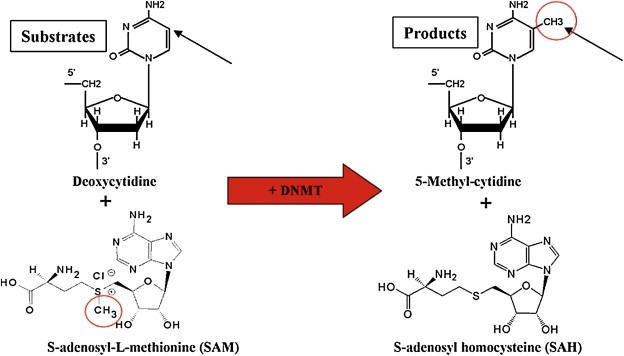
DNA methylation mediated by DNA methyltransferases (DNMTs).
Table 1.
.Hypermethylated and hypomethylated genes in human breast cancer cells.
| Gene | Function | Methylation status |
|---|---|---|
| MGMT | DNA repair | Hyper |
| BRCA1 | Hyper | |
| MLH1 | Hyper | |
| RAD9 | Hyper | |
| LDLRAP1 (ARH1) | Cellular homeostasis | Hyper |
| GPC3 | Hyper | |
| HOXD11 | Hyper | |
| LAMA3 | Hyper | |
| LAMB3 | Hyper | |
| LAMC2 | Hyper | |
| ROBO1 | Hyper | |
| BCSG1 | Cell invasion/metastasis | Hypo |
| CDH1 | Hypo | |
| CDH3 | Hypo | |
| CDH13 | Hyper | |
| CAV1 | Hypo | |
| CST6 | Hyper | |
| NAT1 | Hypo | |
| SYK | Hyper | |
| UPA | Hypo | |
| CCND2 | Cell‐cycle regulation | Hyper |
| 14‐3‐sigma (SFN) | Hyper | |
| AK5 | Hyper | |
| CDKN1C | Hyper | |
| CDKN2A | Hyper | |
| ER | Hyper | |
| FOXA2 | Hyper | |
| PGR | Hyper | |
| RAR‐β | Hyper | |
| RASSF1A | Hyper | |
| RUNX3 | Hyper | |
| SFRP1 | Hyper | |
| WIF1 | Hyper | |
| WRN | Hyper | |
| WT1 | Hyper | |
| APC | Apoptosis | Hyper |
| HIC1 | Hyper | |
| HOXA5 | Hyper | |
| DAPK | Hyper | |
| BCL2 | Hyper | |
| DCC | Hyper | |
| TWIST | Hyper | |
| TMS1 | Hyper |
DNA is packed into chromatin, a highly organized and dynamic protein–DNA complex which forms the regulatory platform for transcription. Open (euchromatin) and closed (heterochromatin) chromatin states are controlled by histone modifications and histone composition in close crosstalk with the binding of a myriad of non‐histone proteins. The fundamental subunit of chromatin, the nucleosome, is composed of an octamer of four core histones, an H3/H4 tetramer and two H2A/H2B dimers, around which 146 bp of DNA are wrapped (Ito et al., 2002; Strahl and Allis, 2000). Local chromatin architecture is now generally recognized as an important factor in the regulation of gene expression. This architecture of chromatin is strongly influenced by post‐translational modifications of the N‐terminal tails of the histones (Figure 3). Core histones are subjected to a variety of covalent modifications including methylation, acetylation, phosphorylation, ubiquitination sumoylation, ADP ribosylation, deamination, proline isomerization (Schubeler et al., 2004; Shilatifard, 2006). The combination of different N‐terminal modifications and the incorporation of different histone variants which have distinct roles in gene regulation, have led to the proposition of a regulatory histone code which determines – at least partly – the transcriptional potential for a specific gene or a genomic region (Xu et al., 2009). During activation of gene transcription, this compact, inaccessible DNA is made available to DNA binding proteins through modification of the N‐terminal histone tails and consequently nucleosome density and positioning (Ito et al., 2002). Each histone modification is a unique mark to show the status of chromatin structure. Compared to methylation and phosphorylation, the acetylation of core histones is probably the best understood type of modification. Histone acetylation occurs at the ϵ‐amino groups of evolutionarily conserved lysine residues located at the N‐termini. Trimethylation at H3K4, H3K36, or H3K79 results in an open chromatin configuration and is often associated with active transcription. This euchromatin is also characterized by a high level of histone acetylation, which is mediated by histone acetyltransferases (HATs). Conversely, histone deacetylases (HDACs) have the ability to remove this epigenetic mark, which leads to transcriptional repression and the formation of heterochromatin. Steady‐state levels of acetylation of the core histones result from the balance between the opposing activities of histone acetyltransferases and histone deacetylases (HDACs). In general, increased levels of histone acetylation (hyperacetylation) are associated with increased transcriptional activity, whereas decreased levels of acetylation (hypoacetylation) are associated with repression of gene expression (Wade, 2001; Forsberg and Bresnick, 2001). Many non‐histone proteins have been identified to be the substrates of HDACs, such as proteins involved in transcription (p53, p73, E2F1, STAT1, STAT3, GATA1, YY1, HMGB1 and NF‐kB), hormone response (AR, ERα, GR), nuclear transport (importin‐α7), DNA repair (Ku70) WNT signaling (β‐catenin) and heat shock/chaperone response (HSP90) (Bolden et al., 2006; Kim et al., 2006).
Figure 3.
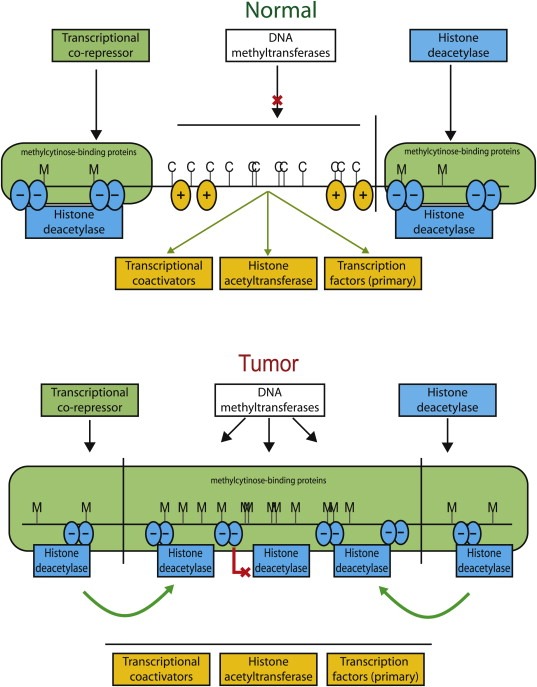
Effects of DNA methylation and chromatin structure on gene transcription in normal (upper) and tumor cells (lower panel). Blue ovals with minus signs, nucleosomes with histones having deacetylation of key histone amino acids such as lysine 9 of histone H3 and methylation of this same residue (both marks of inactive transcription); Orange ovals with plus signs, nucleosomes with acetylation of lysine 9 of histone H3 and methylation of lysine 4 of H3 (both marks of active transcription); C: CpG sites; M: hypermethylated sites; green arrow: transcriptional activity; red line with X: repressed transcription.
For a long time the presence of methylated cytosines on DNA and that of deacetylated histones have been regarded as two separate mechanisms, both capable of modulating chromatin structure and gene expression independently of each other (Turner, 2000; Roth et al., 2001). It has become evident that the activity of histone deacetylases (HDACs) is coupled to that of DNA methylation through either methyl group binding proteins such as MeCP2, which are able to recognize methylated regions on DNA and recruit HDACs to them (Nan et al., 1998; Jones et al., 1998) or through direct interaction of HDACs with DNA methyltransferases (DNMTs) (Figure 3) (Robertson et al., 2000; Bachman et al., 2001). Successively, histone methylation has appeared as an epigenetic modification connected to the two above mentioned ones in determining whether a gene will be silenced or expressed (Kouzarides, 2002; Lachner et al., 2003). For example, lysine 9 of histone H3 is acetylated in active chromatin regions and becomes methylated when the gene is silenced introducing a binding site for heterochromatin protein 1 (HP1) (Litt et al., 2001; Peters et al., 2002). Another epigenetic modification, phosphorylation, controls this event by preventing methylation of lysine 9 when serine 10 is phosphorylated (Rea et al., 2000). Another modification is poly‐ADP‐ribosylation (D'Amours et al., 1999; De Murcia and Shall, 2000). It has been suggested that poly‐ADP‐ribosylation can influence chromatin structure through two different mechanisms (Zardo et al., 2003), either covalently, building rather short chains of ADP‐ribose polymers on histone proteins or noncovalently, attracting histones to the long and branched polymers present in automodification domain of PARP‐1.
3. Epigenetics of hormonal receptors in breast cancer
Pathological estrogens have been associated with a higher risk for breast and endometrial cancer and hormone dependence of breast cancers is correlated with tumor progression and patient prognosis. Most breast cancers are initially positive for ER, and their growth can be stimulated by estrogens and inhibited by antiestrogens. DNA methylation of the ESR1 and PGR promoters has been proposed as a mechanism for the development of ER‐negative tumors in cell lines as well as primary tumors. Hypermethylation has been discussed as a possible cause of ER loss due to the findings of Weigel and deConinck (1993), who demonstrated that ER‐negative breast cancer cells are devoid of ER mRNA. Further, Ferguson et al. (1995) could reactivate ER gene expression in ER‐negative cells by inhibition of methylation. However, clinical data remains contradictory. Lapidus et al. (1996) found hypermethylation of the ER promoter region in tumors, but other groups such as Hori et al. (1999) have detected no correlation between gene methylation pattern and ER gene expression in breast tumors. In summary, current evidence suggests that there is no clear link between ESR1 methylation and ER status, while PGR methylation is significantly linked to PR expression and PGR methylation status might be a predictor for ER status (Gaudet et al., 2009). Future investigations are required to determine whether methylation actually causes loss of ER in breast cancer.
4. Epigenetics in relation to histological type, molecular profiles and clinical parameters of breast cancer
Although breast tumors are frequently hypomethylated on a genome‐wide scale the number of genes reported as hypomethylated in breast cancer is relatively small. This is probably due to the positioning of hypomethylated DNA to regions of pericentromeric DNA and gene poor regions of the genome but also to the fact that the focus on DNA methylation in cancer has been on hypermethylation of CpG islands and most techniques will only detect hypermethylated regions. Genes that are hypomethylated in primary breast tumors include the endonucleases FEN1 (Singh et al., 2008), the N‐acetyltransferase NAT1 (Kim et al., 2008) and the cadherin CDH3 (Paredes et al., 2005). Genes that have been found hypomethylated in breast cancer cell lines but where evidence for hypomethylation in primary tumors is weak include the metastasis gene PLAU (Pakneshan et al., 2004) and the breast cancer‐specific gene 1 (BCSG1) (Gupta et al., 2003). The only imprinted gene that has been reported hypomethylated in breast cancer so far is the insulin‐like growth factor II (IGF2) gene although hypomethylation has been described in cell lines also for ARH1 (Yuan et al., 2003) Recent high resolution analysis of DNA hypomethylation in breast cancer identified a large number of hypomethylated sites with around 1500 regions hypomethylated in a cancer‐specific manner (Novak et al., 2008; Shann et al., 2008). It is likely that many of these regions contain genes or regulatory sequences that play important roles in tumorigenesis.
More than 100 genes have been reported to be hypermethylated in breast tumors or breast cancer cell lines (Hinshelwood and Clark, 2008). Many of the genes aberrantly methylated play important roles in cell‐cycle regulation, apoptosis, tissue invasion and metastasis, angiogenesis and hormone signaling (Widschwendter and Jones, 2002). Cyclin D2 (CCND2) is an important regulator of the cell cycle and overexpression inhibits the transition between G1 and S phase. CCND2 has been found frequently methylated in breast cancer and is also methylated in DCIS (ductal carcinomas in situ) suggesting it to be an early event in tumorigenesis (Evron et al., 2001b). Another cell‐cycle regulator attracting a lot of interest is the p16ink4A/CDKN2A that is frequently methylated in many human cancers including breast cancer (Herman et al., 1995). CDKN2A inactivation associated with DNA methylation has been observed in Human Mammary Epithelial Cells (HMECs) when the cultured cells escape senescence and acquire telomere crisis and chromosomal abnormalities similar to those observed in early neoplastic lesions (Huschtscha et al., 1998; Romanov et al., 2001). CDKN2A methylation has been found in disease free breast tissue and it is speculated that this methylation originates from a subpopulation of cells in normal epithelia (Holst et al., 2003), however frequent methylation in DCIS has not been found questioning the hypothesis that CDKN2A methylation might be an early event in breast carcinogenesis (Lehmann et al., 2002). Another methylated regulator of proliferation in breast cancer is the tumor‐suppressor RAR‐β (Widschwendter et al., 2000). RAR‐β methylation is an early epigenetic event in breast cancer and is found in situ lesions from both lobular and ductal cancers (Fackler et al., 2003). RASSF1A is another tumor‐suppressor gene frequently methylated in breast cancer (Dammann et al., 2000, 2001). RASSF1A methylation is also an early epigenetic event in breast cancer and is found in DCIS and LCIS (Lehmann et al., 2002; Fackler et al., 2003). HIN1 is an inhibitor of cell growth, migration and invasion that is frequently silenced by DNA methylation in breast cancer (Krop et al., 2001, 2005). CDH3 is a cell adhesion molecule frequently silenced in breast carcinomas by DNA methylation and this silencing might be important for tumor cell invasion and metastasis (Graff et al., 1995). Given its important role in familial breast cancer and the fact that no BRCA1 mutations have been detected in sporadic breast cancers made DNA methylation induced silencing an attractive mechanism for BRCA1 silencing in these tumors. BRCA1 methylation has been found in sporadic breast cancers but it is not a frequent event (Dobrovic and Simpfendorfer, 1997; Esteller et al., 2000; Mancini et al., 1998) and it is possible that BRCA1 methylation is most common in rare subtypes of basal‐like origin (Turner et al., 2007). Sporadic and inherited breast tumors have overall similar methylation profiles but BRCA1 tumors have reduced methylation of certain non‐familial genes and have a phenotype associated with basal‐like carcinomas (Esteller et al., 2001). Although breast cancer can be divided into distinct histological subclasses there is no evidence for large differences in DNA methylation patterns between them as has been reported for ductal and lobular breast cancers (Fackler et al., 2003; Bae et al., 2004). Distinct epigenetic profiles can be identified when dividing breast tumors into groups based on hormone receptor status (Feng et al., 2007; Widschwendter et al., 2004) and candidate genes investigated are typically less methylated in ER‐negative tumors. Mutations in the TP53 gene have also been linked to differential methylation patterns in both breast cancer and colorectal cancers where tumors with TP53 mutations are hypomethylated (Feng et al., 2007; Toyota et al., 2000). Since mutations in TP53 and loss of ER expression are closely linked to the basal‐like expression subtype it is likely that tumors with a basal‐like phenotype are hypomethylated in a cancer‐specific manner.
In our own studies using quantitative methylation analysis we identified stage dependent methylation of ABCB1, FOXC1, PPP2R2B and PTEN as novel genes to be methylated as early as in DCIS. In particular, FOXC1 showed a significant increase in the methylation frequency in invasive tumors. Low FOXC1 gene expression in both methylated and unmethylated DCIS and IDCs (invasive ductal carcinoma) indicates that the loss of its expression is an early event during breast cancer progression (Figure 4) (Muggerud et al., 2010). Our studies of candidate genes suggest a non‐random distribution of methylation among breast tumors with various clinical and molecular characteristics (Figure 5A, zoom in B). This analysis has revealed new genes methylated in DCIS (ABCB1, PPP2R2B, FOXC1 and GSTP1). The role for these genes in the propagation from in situ carcinoma to invasive carcinoma needs further investigation. Associations between DNA methylation levels and clinicopathological factors were found and confirmed complex relationships between DNA methylation and TP53 status as well as Estrogen receptor status (Submitted for review). As reviewed elsewhere in this issue, gene expression analysis of breast tumors has identified different breast cancer subgroups most notably belonging to estrogen receptor negative basal‐like and the estrogen receptor positive luminal subgroups (Perou et al., 2000) with differences in outcome (Sorlie et al., 2001). Genome‐wide DNA methylation analysis using methylation specific digital karyotyping of normal breast tissue has identified cell‐type and differentiation specific DNA methylation and gene expression patterns that are also found in breast carcinoma (Bloushtain‐Qimron et al., 2008). The association between these epigenetic changes on well established prognostic factors such as tumor size, lymph node status, distant metastasis, grade, hormone receptor status, TP53 status and breast tumor expression subclasses starts to emerge.
Figure 4.
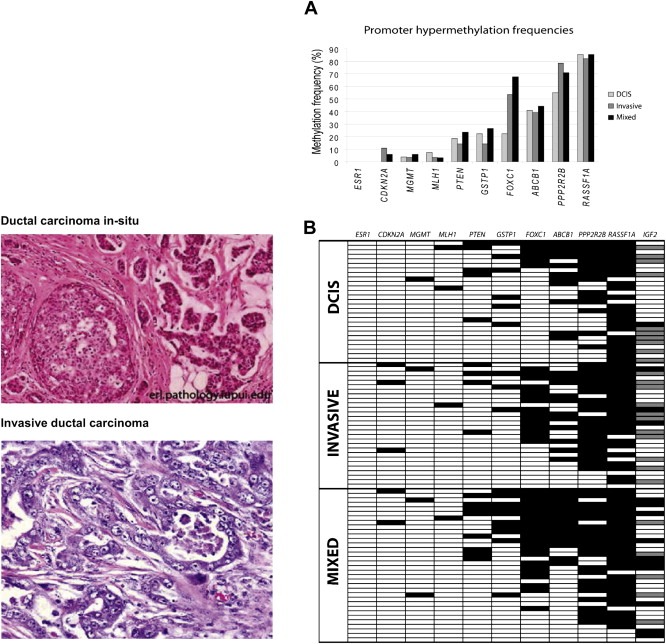
The most common histopathological types of breast cancer (left panel) and degree of methylation of selected candidate genes (right) listed with their frequency of methylation events (Panel A) and according to cancer progression (Panel B). From Muggerud et al. (2010).
Figure 5.

Hierarchical clustering of methylation levels reveal defined subgroups (Panel A). The clinical and pathological data (Panel B) for each patient was analyzed with respect to the methylation levels of the 12 selected genes (below). RASSF1A, GSTP1, ABCB1, PPP2R2B and FOXC1 were the most frequently methylated genes in the tumors and in the DCIS samples. CDKN2A, ABCB1 and FOXC1 were significantly differentially methylated between DCIS and the invasive tumors (p = 0.008, p = 0.017 and p = 0.018 respectively). PTEN, CDKN2A, MLH1 and ESR1 were methylated at a lower frequency both in the DCIS and the invasive tumors. PPP2R2B, ABCB1, FOXC1 and GSTP1 methylation levels increased in late stage breast carcinoma compared to normal breast tissue and DCIS. RASSF1A methylation levels increased in late stage BC. CDKN2A methylation levels were low and appear to increase with tumor stage but decrease with grade. Tumors with TP53 mutations had significantly lower DNA methylation levels in PTEN, PPP2R2B FOXC1 and ABCB1. RASSF1A, PTEN, PPP2R2B, GSTP1. FOXC1 and ABCB1 methylation levels were associated to hormone receptor status (submitted for review).
5. Biomarkers for diagnosis, prognosis and prediction of treatment outcome in breast cancer
Tumor biomarkers are usually proteins measured either in serum, plasma or tumor tissue and can be used to identify individuals with increased predisposition to develop a cancer, screen for early malignancies and/or assist in cancer diagnosis and patient management through the stratification of patient groups and prediction of therapy response or prognosis (Duffy et al., 2009).
There is currently no ideal protein biomarker that can be measured with the desired sensitivity and specificity in plasma or serum for the early detection of breast cancer (Levenson, 2007). Gene expression signatures in peripheral blood cells have proven useful displaying a sensitivity and specificity similar to what can be obtained through mammographic screening (Aaroe et al., 2010; Sharma et al., 2005). DNA methylation changes might be useful marker for early detection of breast cancer as DNA is relatively stable compared to other sources such as mRNA and proteins and can be obtained from ductal lavage fluids, nipple aspirate fluids and cell‐free DNA in blood as well as fine needle aspirates of the primary tumor (Levenson, 2007). Using a gene panel consisting of Cyclin D2, RAR‐β and TWIST DNA methylation could be detected in ductal lavage fluids from patients with invasive breast cancer and ductal in situ carcinomas (Evron et al., 2001a). Increasing the panel size to nine genes (RASSF1A, TWIST, HIN1, Cyclin D2, RAR‐β, APC1, BRCA1, BRCA2 and p16 INk4a) the same group could show that DNA methylation doubled the sensitivity of detecting breast cancers as compared to cytology and when combined with cytology examination the accuracy reached 100% (Fackler et al., 2006). DNA methylation analysis of GSTP1, RAR‐β, p16 INk4a, p14 ARF, RASSF1A and DAP‐kinase in DCIS or stage 1 breast cancer identified identical methylation in more than 80% of the nipple aspirate fluids and DNA methylation was not detected in benign and normal breast tissue and nipple aspirate DNA from healthy women (Krassenstein et al., 2004). Cell‐free DNA from plasma was used in DNA methylation analysis of APC, GSTP1, RAR‐β, and RASSF1A for early detection (Hoque et al., 2006). This study showed that the methylation of the selected genes increased with tumor progression and that additional breast cancer‐specific genes could help in increasing the sensitivity and specificity of this approach. If the accuracy of DNA methylation analysis in plasma is increased this approach could be a valuable supplement to mammographic screening in patients with dense mammograms where mammography is less sensitive. Compared to ductal lavage and the collection of nipple aspirate fluids the methylation analysis from fine needle aspirates is a less invasive procedure that can be a supplement to histology and mammography in breast cancer detection and diagnosis. Fine needle aspirates have recently been used to detect aberrantly methylated genes in DCIS (Jeronimo et al., 2003) and to diagnose early breast cancer lesions as atypical ductal hyperplasia (ADH), DCIS and LCIS (Melnikov et al., 2008).
DNA methylation markers with independent prognostic value have been identified and can be used in tailoring treatment to patient groups that today are receiving uniform treatment regimens. Loss of P3H2 and P3H3 expression was shown to result from epigenetic silencing, and is associated with aberrant hypermethylation in the CpG islands around exon 1 of both P3H2 and P3H3 (Shah et al., 2009). Verification that P3H2 is methylated only in breast cancer would make it an attractive candidate gene in diagnosis and screening in breast cancer (Metge et al., 2008). Breast Cancer Metastasis Suppressor 1 (BMRS1) is a novel target of epigenetic silencing and aberrant methylation in the BMRS1 promoter may serve as a cause of loss of its expression (Samant et al., 2000). BMRS1 was originally identified to suppress metastasis of highly metastatic human breast cancer cell lines when injected into nude mice (Seraj et al., 2000; Samant et al., 2002; Zhang et al., 2006). Reduced expression of the BMRS1 mRNA is correlated with poor prognosis in breast cancer (Stark et al., 2005; Saunders et al., 2001). BMRS1 has been shown to play a role in cell–cell communication (Shevde et al., 2002; DeWald et al., 2005) and reduce phosphoinositide signaling (Liu et al., 2006). BMRS1 has also been reported to suppress NFk‐B signaling through blockage of IkBα (Cicek et al., 2005; Samant et al., 2007; Hurst et al., 2006). Further, regulatory mechanisms of BMRS1 have been shown to involve histone deacetylases (Meehan et al., 2004; Ki Hong et al., 1993). It has been shown that BMRS1 silencing occurs via an epigenetic mechanism in breast cancer development and progression. Treatment with 5‐aza‐2′‐deoxycytidine reversed the methylated status of the BMRS1 promoter allowing for increased expression of the BMRS1 protein. Epigenetic silencing of BMRS1 may not only be useful as a potential prognostic marker, but may also possibly be used for targeted breast cancer treatment.
The DNA methylation status of ESR1, ARH1 and CYP1B1 has been suggested as a marker for treatment response in patients receiving and not receiving Tamoxifen as hormonal treatment (Widschwendter et al., 2004) and PSAT1 has been associated with disease progression measured as time to disease progression in patients receiving Tamoxifen (Martens et al., 2005). PITX2 methylation has been identified as a prognostic marker for time to distant metastasis in untreated as well as Tamoxifen treated hormone receptor positive, node negative breast cancers (Nimmrich et al., 2008; Maier et al., 2007). Patients with methylated PITX2 are less likely to develop distant metastasis and it is possible that these patients could be spared adjuvant chemotherapy. A 4‐marker panel including PITX2, BMP4, FGF4 and C20orf55 shows an independent prognostic value for time to distant metastasis in estrogen receptor positive, node positive, HER2 negative patients who received adjuvant anthracycline‐based chemotherapy (Hartmann et al., 2009). This 4‐marker panel could delineate subgroups of patients with high overall survival suggesting that some patients within this cohort might be over treated and do not benefit from adjuvant anthracycline‐based treatment.
The presence of DNA methylation in serum has successfully been used as a marker for treatment response both to hormonal and anthracycline‐based treatment. Detection of RASSF1A methylation in serum has been used as marker for response to treatment with Tamoxifen and detection of RASSF1A methylation in sera 1 year after primary surgery was an independent prognostic factor for the risk of relapse and death (Fiegl et al., 2005). The presence of RASSF1A and APC methylation in serum of breast cancer are independent markers for poor prognosis in patients not receiving adjuvant treatment (Muller et al., 2003). The presence of methylation at the NEUROD1 promoter in serum after treatment with the anthracyclines has been shown to be an indicator of poor prognosis (Fiegl et al., 2008).
The resistance of cancer cells to chemotherapeutic agents is major challenge in clinical practice and understanding the mechanisms behind this resistance will provide valuable information that hopefully can be used to predict therapy resistance and help in tailoring treatment to the individual patient. Experiments performed in cell lines have shown that hypomethylation of drug resistance genes such as ABCB1, GSTP1, UPa and MGMT might be responsible for Doxorubicin resistance in MCF‐7 breast cancer cells (Chekhun et al., 2006). MCF‐7 cell lines resistant to either Tamoxifen or Fulvestrant have different epigenetic profiles compared to sensitive MCF‐7 cells suggesting that epigenetic mechanisms might be involved in chemoresistance (Fan et al., 2006). Activation of growth promoting genes by promoter hypomethylation was frequently observed in antiestrogen resistant cells. Also in doxorubicin and cisplatin resistant MCF‐7 cell lines frequent hypomethylation has been observed suggesting that hypomethylation might be a prevalent mechanism for drug resistance development (Chekhun et al., 2007). In a recent study analyzing quantitatively the methylation patterns in the promoter regions of 14 genes in 75 well‐described pre‐treatment samples from locally advanced breast cancer treated with the anthracycline doxorubicin, we identified the absence of methylation at the ABCB1 promoter to be correlated with progressive disease during doxorubicin treatment (Dejeux et al., in press). Further, the DNA methylation status at the promoters of GSTP1, FOXC1 and ABCB1 correlated with survival, whereby the combination of methylated genes improved the subdivision with respect to the survival of the patients. In multivariate analysis GSTP1 and FOXC1 methylation status proved to be independent prognostic markers associated with survival.
6. DNA methylation and histone modification as therapeutic targets in breast cancer
6.1. DNA methyltransferase inhibitors
The best‐established inhibitors of (cytosine‐5) DNMTs are the substrate analogues 5‐azacytidine and 5‐aza‐deoxycytidine (decitabine). These compounds act by their incorporation into DNA in the place of the natural base, cytosine, during DNA replication leading to covalent trapping of DNMTs. This causes the depletion of active DNMT enzymes and demethylation of genomic DNA through cell division. There are a few disadvantages of these compounds and the main disadvantage is that 5‐Azacytidine and 5‐aza‐deoxycytidine are highly unstable in neutral aqueous solutions, complicating the treatment regimens. The development of zebularine, a more stable derivative, might provide an alternative. In one study it has been shown that zebularine is an effective DNMT inhibitor and demethylating agent in human breast cancer cell lines. Treatment of two cancer cell lines, MDA‐MB‐231 and MCF‐7 with zebularine showed that cell growth was inhibited in a dose and time dependent manner. This was associated with increased expression of p21, decreased expression of cyclin‐D, and induction of S‐phase arrest. At high doses zebularine induced changes in apoptotic proteins manifested by alteration in caspase‐3, Bax, Bcl2 and PARP cleavage (Billam et al., 2009). The toxicity of 5‐azacytosine nucleotides has been shown to be significantly reduced in DNMT1‐deficient cells and animals, which provided a direct link between the inhibitory activity and the toxicity of the compounds. This inherent toxicity has initiated the search for alternative ways to inhibit DNMT activity in human cells.
Recently, several additional compounds have been described that inhibit DNMTs in cultured human cells. The local anaesthetic procaine and its derivative procainamide, an approved antiarrhythmic drug, have been shown to have demethylating activity in cancer cells, leading to the reexpression of silenced tumor‐suppressor genes (Brueckner and Lyko, 2004). Both compounds are derivatives of 4‐aminobenzoic acid that do not become incorporated into DNA but bind to CpG‐rich sequences. This might disturb the interactions between DNMTs and their target sequences. Another molecule with demethylating activity is (−)‐epigallocatechin‐3‐gallate (EGCG), the main polyphenol compound in green tea. Cancer cells treated with micromolar concentrations of EGCG showed reduced DNA methylation and elevated transcription of tumor‐suppressor genes. EGCG is currently being tested in phase I trials but its effects on DNA methylation in cancer patients have not been reported (Chekhun et al., 2007).
6.2. Histone deacetylase inhibitors
Recent research has focused on the role of epigenetic gene silencing, important in pathogenesis of breast cancer, in which acetylation or deacetylation of DNA modifies the expression of tumor‐suppressing genes. HDAC inhibitors could potentially represent new treatment options for breast cancer. In one small study (Serena, 2009) a combination of the HDAC inhibitor vorinostat with paclitaxel and bevacizumab induced a partial or complete response in more than 50% of patients with metastatic breast cancer. It has also been shown that HDAC inhibitors have different role in ER+ and ER− breast cancer cells. Thomas and Munster (2009) have shown in their study that HDAC inhibitors are associated with a transcriptional down‐regulation in ER positive cells while in ER‐negative cells HDAC inhibitors re‐establish expression. Hurtubise and Momparler (2006) have studied the combination of the HDAC inhibitor (LAQ824) and a DNMT inhibitor (decitabine) since LAQ824 shows antineoplastic activity and can activate genes that produce cell‐cycle arrest. A combination of LAQ824 with decitabine might therefore result in the synergistic (re‐)activation of silenced tumor‐suppressor genes. Histone deacetylase inhibitors (HDACi) can also sensitise cancer cells to topoisomerase inhibitors by increasing their access and binding to DNA. A phase I trial confirmed an acceptable toxicity profile, tolerability, and recommended phase II dose of escalating doses of the HDACi vorinostat with weekly doxorubicin (Munster et al., 2009). These studies demonstrate the potential of epigenetic therapies in combination with conventional chemotherapy to improve breast cancer patient management.
7. Outlook
A large number of breast tumor samples are currently comprehensively analyzed by the International Cancer Genome Consortium (ICGC, http://www.icgc.org). The ICGC aims at studying all major types and subtypes of cancer in adults and children. Systematic studies of over 25,000 cancer genomes at the genomic, epigenomic, and transcriptomic levels are undertaken to reveal the repertoire of oncogenic mutations, define clinically‐relevant subtypes for prognosis and therapeutic management, and enable the development of new cancer therapies. Currently three breast cancer projects within the framework of the ICGC have been announced that will analyze together at least 1500 breast tumors at the genetic, transcriptomic and epigenetic (DNA methylation) level using massively parallel sequencing technologies. The French National Cancer Institute (INCa) has agreed to fund the integrative analysis of 500 HER2 amplified breast cancer with an amplification of the HER2 gene, the Wellcome trust and the Breakthrough Foundation provide the funding for the analysis of triple negative and lobular breast cancers and a large consortium (BASIS) led by the Sanger Institute has been selected through the FP7 programme of the European Union to analyze at least 500 estrogen positive (HER2 negative) breast cancer samples. The results will yield an immense amount of data to decipher the epigenetic (and other molecular) alterations implicated in breast carcinogenesis and will permit to identify correlations between tumor‐specific epigenetic changes with clinical and histopathological data including prognosis, prediction of therapy response and tumor classification schemes for diagnosis and might eventually lead to the development of novel specific therapies.
Acknowledgements
JJ is a fellow of the University of Oslo, the International Quota Programme, JAR is a fellow at the Faculty Division the Norwegian Radium Hospital, University of Oslo. This work was supported by grant D‐03067 from The Norwegian Cancer Society (VNK), grant 152004/150 from The Functional Genomics Program (FUGE) of the Norwegian Research Council (NFR) (VNK) and the French–Norwegian cooperation program Aurora grant 15842WE (JAR, JT, VNK). We thank Daniel Nebdal for the graphical design of 1, 3, 5.
Jovanovic Jovana, Rønneberg Jo Anders, Tost Jörg, Kristensen Vessela, (2010), The epigenetics of breast cancer, Molecular Oncology, 4, doi: 10.1016/j.molonc.2010.04.002.
References
- Aaroe, J. , Lindahl, T. , Dumeaux, V. , Saebo, S. , Tobin, D. , Hagen, N. , 2010. Gene expression profiling of peripheral blood cells for early detection of breast cancer. Breast Cancer Res. 12, R7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman, K.E. , Rountree, M.R. , Baylin, S.B. , 2001. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem. 276, 32282–32287. [DOI] [PubMed] [Google Scholar]
- Bae, Y.K. , Brown, A. , Garrett, E. , Bornman, D. , Fackler, M.J. , Sukumar, S. , 2004. Hypermethylation in histologically distinct classes of breast cancer. Clin. Cancer Res. 10, 5998–6005. [DOI] [PubMed] [Google Scholar]
- Baylin, S.B. , Ohm, J.E. , 2006. Epigenetic gene silencing in cancer-mechanism for early oncogenic pathway addiction?. Nat. Rev. Cancer. 6, 107–116. [DOI] [PubMed] [Google Scholar]
- Billam, M. , Sobolewski, M.D. , Davidson, N.E. , 2009. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast Cancer Res. Treat. 120, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, A. , 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Bloushtain-Qimron, N. , Yao, J. , Snyder, E.L. , Shipitsin, M. , Campbell, L.L. , Mani, S.A. , Hu, M. , Chen, H. , Ustyansky, V. , Antosiewicz, J.E. , Argani, P. , Halushka, M.K. , Thomson, J.A. , Pharoah, P. , Porgador, A. , Sukumar, S. , Parsons, R. , Richardson, A.L. , Stampfer, M.R. , Gelman, R.S. , Nikolskaya, T. , Nikolsky, Y. , Polyak, K. , 2008. Cell type-specific DNA methylation patterns in the human breast. Proc. Natl. Acad. Sci. U.S.A. 105, 14076–14081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden, J.E. , Peart, M.J. , Johnstone, R.W. , 2006. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5, 769–784. [DOI] [PubMed] [Google Scholar]
- Brueckner, B. , Lyko, F. , 2004. DNA methyltransferase inhibitors: old and new drugs for epigenetic cancer therapy. Trends Pharmacol. Sci. 25, 551–554. [DOI] [PubMed] [Google Scholar]
- Chekhun, V.F. , Kulik, G.I. , Yurchenko, O.V. , Tryndyak, V.P. , Todor, I.N. , Luniv, L.S. , 2006. Role of DNA hypomethylation in the development of the resistance to doxorubicin in human MCF-7 breast adenocarcinoma cells. Cancer Lett. 231, 87–93. [DOI] [PubMed] [Google Scholar]
- Chekhun, V.F. , Lukyanova, N.Y. , Kovalchuk, O. , Tryndyak, V.P. , Pogribny, I.P. , 2007. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethylated targets. Mol. Cancer Ther. 6, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Cicek, M. , Fukuyama, R. , Welch, D.R. , Sizemore, N. , Casey, G. , 2005. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res. 65, 3586–3595. [DOI] [PubMed] [Google Scholar]
- Dammann, R. , Li, C. , Yoon, J.H. , Chin, P.L. , Bates, S. , Pfeifer, G.P. , 2000. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 25, 315–319. [DOI] [PubMed] [Google Scholar]
- Dammann, R. , Yang, G. , Pfeifer, G.P. , 2001. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 61, 3105–3109. [PubMed] [Google Scholar]
- D'Amours, D. , Desnoyers, S. , D'Silva, I. , 1999. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342, 242–268. [PMC free article] [PubMed] [Google Scholar]
- De Murcia, G. , Shall, S. , 2000. From DNA Damage and Stress Signaling to Cell Death: Poly ADP-ribosylation Reactions Oxford University Press; [Google Scholar]
- De Smet, C. , Loriot, A. , Boon, T. , 2004. Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells. Mol. Cell. Biol. 24, 4781–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejeux, E. , Rønneberg, J.A. , Solvang, H. , Bukholm, I. , Geisler, S. , Aas, T. , Gut, I.G. , Børresen-Dale, A.L. , Lønning, P.E. , Kristensen, V.N. , Tost, J. , 2010. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol. Cancer. 9, 68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWald, D.B. , Torabinejad, J. , Samant, R.S. , Johnston, D. , Erin, N. , Shope, J.C. , 2005. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 65, 713–717. [PubMed] [Google Scholar]
- Dobrovic, A. , Simpfendorfer, D. , 1997. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 57, 3347–3350. [PubMed] [Google Scholar]
- Duffy, M.J. , Napieralski, R. , Martens, J.W. , Span, P.N. , Spyratos, F. , Sweep, F.C. , 2009. Methylated genes as new cancer biomarkers. Eur. J. Cancer. 45, 335–346. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , Silva, J.M. , Dominguez, G. , Bonilla, F. , Matias-Guiu, X. , Lerma, E. , 2000. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 92, 564–569. [DOI] [PubMed] [Google Scholar]
- Esteller, M., Fraga, M.F., Guo, M., Garcia-Foncillas, J., Hedenfalk, I., Godwin, A.K., et al., 2001. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. 10, 3001–3007. [DOI] [PubMed]
- Esteller, M. , 2007. Cancer epigenomics: DNA methylomes and histone modification maps. Nat. Rev. Genet. 8, 286–298. [DOI] [PubMed] [Google Scholar]
- Evron, E. , Dooley, W.C. , Umbricht, C.B. , Rosenthal, D. , Sacchi, N. , Gabrielson, E. , 2001. Detection of breast cancer cells in ductal lavage fluid by methylation-specific PCR. Lancet. 357, 1335–1336. [DOI] [PubMed] [Google Scholar]
- Evron, E. , Umbricht, C.B. , Korz, D. , Raman, V. , Loeb, D.M. , Niranjan, B. , 2001. Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res. 61, 2782–2787. [PubMed] [Google Scholar]
- Fackler, M.J. , McVeigh, M. , Evron, E. , Garrett, E. , Mehrotra, J. , Polyak, K. , 2003. DNA methylation of RASSF1A, HIN-1, RAR-beta, Cyclin D2 and Twist in in situ and invasive lobular breast carcinoma. Cancer. 107, 970–975. [DOI] [PubMed] [Google Scholar]
- Fackler, M.J. , Malone, K. , Zhang, Z. , Schilling, E. , Garrett-Mayer, E. , Swift-Scanlan, T. , 2006. Quantitative multiplex methylation-specific PCR analysis doubles detection of tumor cells in breast ductal fluid. Clin. Cancer Res. 12, 3306–3310. [DOI] [PubMed] [Google Scholar]
- Fan, M. , Yan, P.S. , Hartman-Frey, C. , Chen, L. , Paik, H. , Oyer, S.L. , 2006. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 66, 11954–11966. [DOI] [PubMed] [Google Scholar]
- Feng, W. , Shen, L. , Wen, S. , Rosen, D.G. , Jelinek, J. , Hu, X. , 2007. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 9, R57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson, A.T. , Lapidus, R.G. , Baylin, S.B. , Davidson, N.E. , 1995. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 55, 2279–2283. [PubMed] [Google Scholar]
- Fiegl, H. , Millinger, S. , Mueller-Holzner, E. , Marth, C. , Ensinger, C. , Berger, A. , 2005. Circulating tumor-specific DNA: a marker for monitoring efficacy of adjuvant therapy in cancer patients. Cancer Res. 65, 1141–1145. [DOI] [PubMed] [Google Scholar]
- Fiegl, H. , Jones, A. , Hauser-Kronberger, C. , Hutarew, G. , Reitsamer, R. , Jones, R.L. , 2008. Methylated NEUROD1 promoter is a marker for chemosensitivity in breast cancer. Clin. Cancer Res. 14, 3494–3502. [DOI] [PubMed] [Google Scholar]
- Forsberg, E.C. , Bresnick, E.H. , 2001. Histone acetylation beyond promoters: long-range acetylation patterns in the chromatin world. Bioessays. 23, 820–830. [DOI] [PubMed] [Google Scholar]
- Frigola, J. , Song, J. , Stirzaker, C. , Hinshelwood, R.A. , Peinado, M.A. , Clark, S.J. , 2006. Epigenetic remodeling in colorectal cancer results in coordinate gene suppression across entire chromosome band. Nat. Genet. 38, 540–549. [DOI] [PubMed] [Google Scholar]
- Gaudet, M.M. , Campan, M. , Figueroa, J.D. , Yang, X.R. , Lissowska, J. , Peplonska, B. , 2009. DNA hypermethylation of ESR1 and PGR in breast cancer: pathologic and epidemiologic associations. Cancer Epidemiol. Biomarkers Prev. 18, 3036–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff, J.R. , Herman, J.G. , Lapidus, R.G. , Chopra, H. , Xu, R. , Jarrard, D.F. , 1995. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 55, 5195–5199. [PubMed] [Google Scholar]
- Gupta, A. , Godwin, A.K. , Vanderveer, L. , Lu, A. , Liu, J. , 2003. Hypomethylation of the synuclein gamma gene CpG island promotes its aberrant expression in breast carcinoma and ovarian carcinoma. Cancer Res. 63, 664–673. [PubMed] [Google Scholar]
- Hartmann, O. , Spyratos, F. , Harbeck, N. , Dietrich, D. , Fassbender, A. , Schmitt, M. , 2009. DNA methylation markers predict outcome in node-positive, estrogen receptor-positive breast cancer with adjuvant anthracycline-based chemotherapy. Clin. Cancer Res. 15, 315–323. [DOI] [PubMed] [Google Scholar]
- Herman, J.G. , Merlo, A. , Mao, L. , Lapidus, R.G. , Issa, J.P. , Davidson, N.E. , 1995. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 55, 4525–4530. [PubMed] [Google Scholar]
- Hinshelwood, R.A. , Clark, S.J. , 2008. Breast cancer epigenetics: normal human mammary epithelial cells as a model system. J. Mol. Med. 86, 1315–1328. [DOI] [PubMed] [Google Scholar]
- Holst, C.R. , Nuovo, G.J. , Esteller, M. , Chew, K. , Baylin, S.B. , Herman, J.G. , 2003. Methylation of p16(INK4a) promoters occurs in vivo in histologically normal human mammary epithelia. Cancer Res. 63, 1596–1601. [PubMed] [Google Scholar]
- Hoque, M.O. , Feng, Q. , Toure, P. , Dem, A. , Critchlow, C.W. , Hawes, S.E. , 2006. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J. Clin. Oncol. 24, 4262–4269. [DOI] [PubMed] [Google Scholar]
- Hori, M. , 1999. Hypermethylation of the estrogen receptor alpha gene is not related to lack of receptor protein in human breast cancer. Breast Cancer. 6, 79–86. [DOI] [PubMed] [Google Scholar]
- Hurst, D.R. , Mehta, A. , Moore, B.P. , Phadke, P.A. , Meehan, W.J. , Accavitti, M.A. , 2006. Breast cancer metastasis suppressor 1 (BRMS1) is stabilized by the Hsp90 chaperone. Biochem. Biophys. Res. Commun. 348, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtubise, A. , Momparler, R.L. , 2006. Effect of histone deacetylase inhibitor LAQ824 on antineoplastic action of 5-Aza-2′ deoxycytidine (decitabine) on human breast carcinoma cells. Cancer Chemother. Pharmacol. 58, 618–625. [DOI] [PubMed] [Google Scholar]
- Huschtscha, L.I. , Noble, J.R. , Neumann, A.A. , Moy, E.L. , Barry, P. , Melki, J.R. , 1998. Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res. 58, 3508–3512. [PubMed] [Google Scholar]
- Ito, K. , Barnes, P.J. , Adcock, I.M. , 2002. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 20, 6891–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo, C. , Costa, I. , Martins, M.C. , Monteiro, P. , Lisboa, S. , Palmeira, C. , 2003. Detection of gene promoter hypermethylation in fine needle washings from breast lesions. Clin. Cancer Res. 9, 3413–3417. [PubMed] [Google Scholar]
- Jones, P.A. , Baylin, S.B. , 2007. The epigenomics of cancer. Cell. 128, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.L. , Veenstra, G.J.C. , Wade, P.A. , 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191. [DOI] [PubMed] [Google Scholar]
- Jones, S. , Zhang, X. , Parsons, D.W. , Lin, J.C. , Leary, R.J. , Angenendt, P. , Mankoo, P. , Carter, H. , Kamiyama, H. , Jimeno, A. , Hong, S.M. , Fu, B. , Lin, M.T. , Calhoun, E.S. , Kamiyama, M. , Walter, K. , Nikolskaya, T. , Nikolsky, Y. , Hartigan, J. , Smith, D.R. , Hidalgo, M. , Leach, S.D. , Klein, A.P. , Jaffee, E.M. , Goggins, M. , Maitra, A. , Iacobuzio-Donahue, C. , Eshleman, J.R. , Kern, S.E. , Hruban, R.H. , Karchin, R. , Papadopoulos, N. , Parmigiani, G. , Vogelstein, B. , Velculescu, V.E. , Kinzler, K.W. , 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki Hong, W. , Lippman, S.M. , Hittelmann, W.N. , Lotan, R. , 1993. Retinoid chemoprevention of aerodigestive cancer: from basic research to the clinic. Clin. Oncol. 11, 1216–1222. [PubMed] [Google Scholar]
- Kim, T.Y. , Bang, Y.J. , Robertson, K.D. , 2006. Histone deacetylase inhibitors for cancer therapy. Epigenetics. 1, 14–23. [DOI] [PubMed] [Google Scholar]
- Kim, S.J. , Kang, H.S. , Chang, H.L. , Jung, Y.C. , Sim, H.B. , Lee, K.S. , 2008. Promoter hypomethylation of the N-acetyltransferase 1 gene in breast cancer. Oncol. Rep. 19, 663–668. [PubMed] [Google Scholar]
- Kouzarides, T. , 2002. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 12, 198–209. [DOI] [PubMed] [Google Scholar]
- Krassenstein, R. , Sauter, E. , Dulaimi, E. , Battagli, C. , Ehya, H. , Klein-Szanto, A. , 2004. Detection of breast cancer in nipple aspirate fluid by CpG island hypermethylation. Clin. Cancer Res. 10, 28–32. [DOI] [PubMed] [Google Scholar]
- Krop, I.E. , Sgroi, D. , Porter, D.A. , Lunetta, K.L. , LeVangie, R. , Seth, P. , 2001. HIN-1, a putative cytokine highly expressed in normal but not cancerous mammary epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 98, 9796–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krop, I. , Parker, M.T. , Bloushtain-Qimron, N. , Porter, D. , Gelman, R. , Sasaki, H. , 2005. HIN-1, an inhibitor of cell growth, invasion, and AKT activation. Cancer Res. 65, 9659–9669. [DOI] [PubMed] [Google Scholar]
- Lachner, M. , Roderick, J. , O'Sullivan, 2003. An epigenetic road map for histone lysine methylation. J. Cell. Sci. 116, 2117–2124. [DOI] [PubMed] [Google Scholar]
- Lapidus, R. , 1996. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin. Cancer Res. 2, 805–810. [PubMed] [Google Scholar]
- Lehmann, U. , Langer, F. , Feist, H. , Glockner, S. , Hasemeier, B. , Kreipe, H. , 2002. Quantitative assessment of promoter hypermethylation during breast cancer development. Am. J. Pathol. 160, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson, V.V. , 2007. Biomarkers for early detection of breast cancer: what, when, and where?. Biochim. Biophys. Acta. 1770, 847–856. [DOI] [PubMed] [Google Scholar]
- Litt, M.D. , Simpson, M. , Gaszner, M. , Allis, C.D. , Felsenfeld, G. , 2001. Correlation between histone lysine 9 methylation and developmental changes at the chicken beta-globin locus. Science. 293, 2453–2455. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Smith, P.W. , Jones, D.R. , 2006. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. 26, 8683–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, P.K. , Sukumar, S. , 2008. Epigenomics and breast cancer. Pharmacogenomics. 12, 1879–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, S. , Nimmrich, I. , Koenig, T. , Eppenberger-Castori, S. , Bohlmann, I. , Paradiso, A. , 2007. DNA-methylation of the homeodomain transcription factor PITX2 reliably predicts risk of distant disease recurrence in tamoxifen-treated, node-negative breast cancer patients – technical and clinical validation in a multi-centre setting in collaboration with the European Organisation for Research and Treatment of Cancer (EORTC) PathoBiology group. Eur. J. Cancer. 43, 1679–1686. [DOI] [PubMed] [Google Scholar]
- Mancini, D.N. , Rodenhiser, D.I. , Ainsworth, P.J. , O'Malley, F.P. , Singh, S.M. , Xing, W. , 1998. CpG methylation within the 5′ regulatory region of the BRCA1 gene is tumor specific and includes a putative CREB binding site. Oncogene. 16, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Martens, J.W. , Nimmrich, I. , Koenig, T. , Look, M.P. , Harbeck, N. , Model, F. , 2005. Association of DNA methylation of phosphoserine aminotransferase with response to endocrine therapy in patients with recurrent breast cancer. Cancer Res. 65, 4101–4117. [DOI] [PubMed] [Google Scholar]
- Martin, C. , Zhang, Y. , 2005. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell. Biol. 6, 838–849. [DOI] [PubMed] [Google Scholar]
- Meehan, W.J. , Samant, R.S. , Hopper, J.E. , Carrozza, M.J. , Shevde, L.A. , Workman, J.L. , 2004. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J. Biol. Chem. 279, 1562–1569. [DOI] [PubMed] [Google Scholar]
- Melnikov, A.A. , Scholtens, D.M. , Wiley, E.L. , Khan, S.A. , Levenson, V.V. , 2008. Array-based multiplex analysis of DNA methylation in breast cancer tissues. J. Mol. Diagn. 10, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metge, B.J. , Frost, A.R. , King, J.A. , Dyess, D.L. , Welch, D.R. , Samant, R.S. , Shevde, L.A. , 2008. Epigenetic silencing contributes to the loss of BRMS1 expression in breast cancer. Clin. Exp. Metastasis. 25, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muggerud, A.A. , Ronneberg, J.A. , Warnberg, F. , Botling, J. , Busato, F. , Jovanovic, J. , Solvang, H. , Bukholm, I. , Borresen-Dale, A.L. , Kristensen, V.N. , Sorlie, T. , Tost, J. , 2010. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. 12, R3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H.M. , Widschwendter, A. , Fiegl, H. , Ivarsson, L. , Goebel, G. , Perkmann, E. , 2003. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. 63, 7641–7645. [PubMed] [Google Scholar]
- Munster, P.N. , Marchion, D. , Thomas, S. , Egorin, M. , Minton, S. , Springett, G. , Lee, J.H. , Simon, G. , Chiappori, A. , Sullivan, D. , Daud, A. , 2009. 2.Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br. J. Cancer. 101, 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan, X. , Ng, H.H. , Johnson, C.A. , 1998. Transcriptional repression by the methyl-CpG binding protein MeCP2 involves a histone deacetylase complex. Nature. 393, 386–389. [DOI] [PubMed] [Google Scholar]
- Nimmrich, I. , Sieuwerts, A.M. , Meijer-van Gelder, M.E. , Schwope, I. , Bolt-de, V.J. , Harbeck, N. , 2008. DNA hypermethylation of PITX2 is a marker of poor prognosis in untreated lymph node-negative hormone receptor-positive breast cancer patients. Breast Cancer Res. Treat. 111, 429–437. [DOI] [PubMed] [Google Scholar]
- Novak, P. , Jensen, T. , Oshiro, M.M. , Wozniak, R.J. , Nouzova, M. , Watts, G.S. , Klimecki, W.T. , Kim, C. , Futscher, B.W. , 2006. Epigenetic inactivation of the HOX gene cluster in breast cancer. Cancer Res. 66, 10664–10670. [DOI] [PubMed] [Google Scholar]
- Novak, P. , Jensen, T. , Oshiro, M.M. , Watts, G.S. , Kim, C.J. , Futscher, B.W. , 2008. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res. 68, 8616–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakneshan, P. , Szyf, M. , Farias-Eisner, R. , Rabbani, S.A. , 2004. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J. Biol. Chem. 279, 31735–31744. [DOI] [PubMed] [Google Scholar]
- Paredes, J. , Albergaria, A. , Oliveira, J.T. , Jeronimo, C. , Milanezi, F. , Schmitt, F.C. , 2005. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin. Cancer Res. 11, 5869–5877. [DOI] [PubMed] [Google Scholar]
- Perou, C.M. , Sorlie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Rees, C.A. , Pollack, J.R. , Ross, D.T. , Johnsen, H. , Akslen, L.A. , 2000. Molecular portraits of human breast tumours. Nature. 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Peters, A.H. , Mermoud, J.E. , Carroll, D.O. , 2002. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat. Genet. 30, 77–80. [DOI] [PubMed] [Google Scholar]
- Pfeifer, G.P. , Besaratinia, A. , 2009. Mutational spectra of human cancer. Hum. Genet. 125, 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea, S. , Eisenhaber, F. , O'Carroll, D. , 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 406, 579–580. [DOI] [PubMed] [Google Scholar]
- Robertson, K.D. , Ait-Si-Ali, S. , Yokochi, T. , 2000. DNMT1 forms a complex with Rb, E2F1, and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 25, 338–342. [DOI] [PubMed] [Google Scholar]
- Romanov, S.R. , Kozakiewicz, B.K. , Holst, C.R. , Stampfer, M.R. , Haupt, L.M. , Tlsty, T.D. , 2001. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature. 409, 633–637. [DOI] [PubMed] [Google Scholar]
- Roth, S.Y. , Denu, J.M. , Allis, C.D. , 2001. Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120. [DOI] [PubMed] [Google Scholar]
- Samant, R.S. , Seraj, M.J. , Saunders, M.M. , Sakamaki, T.S. , Shevde, L.A. , Harms, J.F. , 2000. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin. Exp. Metastasis. 18, 683–693. [DOI] [PubMed] [Google Scholar]
- Samant, R.S. , Debies, M.T. , Shevde, L.A. , Verderame, M.F. , Welch, D.R. , 2002. Identification and characterization of the murine ortholog (brms1) of breast-cancer metastasis suppressor 1 (BRMS1). Int. J. Cancer. 97, 15–20. [DOI] [PubMed] [Google Scholar]
- Samant, R.S. , Clark, D.W. , Fillmore, R.A. , Cicek, M. , Metge, B.J. , Chandramouli, K.H. , 2007. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NFkappaB activation. Mol. Cancer. 6, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, M.M. , Seraj, M.J. , Li, Z. , Zhou, Z. , Winter, C.R. , Welch, D.R. , 2001. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 61, 1765–1767. [PubMed] [Google Scholar]
- Schubeler, D. , macAlpine, D.M. , Scalzo, D. , 2004. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of higher eukaryote. Genes Dev. 18, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seraj, M.J. , Samant, R.S. , Verderame, M.F. , Welch, D.R. , 2000. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 60, 2764–2769. [PubMed] [Google Scholar]
- Serena, T. , 2009. Emerging treatment combinations: integrating therapy into clinical practice. Am. J. Health Syst. Pharm. 66, 9–14. [DOI] [PubMed] [Google Scholar]
- Shah, R. , Smith, P. , Quinlan, C. , Baker, L. , Aman, P. , Thompson, A.M. , Crook, T. , 2009. The prolyl 3-hydroxylases P3H2 and P3H3 are novel targets for epigenetic silencing in breast cancer. Br. J. Cancer. 100, 1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shann, Y.J. , Cheng, C. , Chiao, C.H. , Chen, D.T. , Li, P.H. , Hsu, M.T. , 2008. Genome-wide mapping and characterization of hypomethylated sites in human tissues and breast cancer cell lines. Genome Res. 18, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, P. , Sahni, N.S. , Tibshirani, R. , Skaane, P. , Urdal, P. , Berghagen, H. , 2005. Early detection of breast cancer based on gene-expression patterns in peripheral blood cells. Breast Cancer Res. 7, R634–R644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevde, L.A. , Samant, R.S. , Goldberg, S.F. , Sikaneta, T. , Alessandrini, A. , Donahue, H.J. , 2002. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp. Cell Res. 273, 229–239. [DOI] [PubMed] [Google Scholar]
- Shilatifard, A. , 2006. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75, 243–269. [DOI] [PubMed] [Google Scholar]
- Singh, P. , Yang, M. , Dai, H. , Yu, D. , Huang, Q. , Tan, W. , 2008. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol. Cancer Res. 6, 1710–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U.S.A. 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, A.M. , Tongers, K. , Maass, N. , Mehdorn, H.M. , Held-Feindt, J. , 2005. Reduced metastasis-suppressor gene mRNA-expression in breast cancer brain metastases. J. Cancer Res. Clin. Oncol. 131, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns, V. , Zhou, Q. , Davidson, N.E. , 2007. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest. 25, 659–665. [DOI] [PubMed] [Google Scholar]
- Strahl, B.D. , Allis, C.D. , 2000. The language of covalent histone modifications. Nature. 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Stransky, N. , Vallot, C. , Reyal, F. , Bernard-Pierrot, I. , de Medina, S.G. , Segraves, R. , de Rycke, Y. , Elvin, P. , Cassidy, A. , Spraggon, C. , Graham, A. , Southgate, J. , Asselain, B. , Allory, Y. , Abbou, C.C. , Albertson, D.G. , Thiery, J.P. , Chopin, D.K. , Pinkel, D. , Radvanyi, F. , 2006. Regional copy number-independent deregulation of transcription in cancer. Nat. Genet. 38, 1386–1396. [DOI] [PubMed] [Google Scholar]
- Thomas, S. , Munster, P.N. , 2009. Histone deacetylase inhibitor induced modulation of anti-estrogen therapy. Cancer Lett. 280, 184–191. [DOI] [PubMed] [Google Scholar]
- Toyota, M. , Ohe-Toyota, M. , Ahuja, N. , Issa, J.P. , 2000. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc. Natl. Acad. Sci. U.S.A. 97, 7 [DOI] [PMC free article] [PubMed] [Google Scholar]; Furumai, R. , Komatsu, Y. , Nishino, N. , Khochbin, S. , Yoshida, M. , Horinouchi, S. , 2001. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. U.S.A. 98, 710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, N.C. , Reis-Filho, J.S. , Russell, A.M. , Springall, R.J. , Ryder, K. , Steele, D. , 2007. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 26, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Turner, B.M. , 2000. Histone acetylation and an epigenetic code. Bioessays. 22, 836–845. [DOI] [PubMed] [Google Scholar]
- Wade, P.A. , 2001. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum. Mol. Genet. 10, 693–698. [DOI] [PubMed] [Google Scholar]
- Weigel, R.J. , deConinck, E.C. , 1993. Transcriptional control of estrogen receptor in estrogen receptor-negative breast carcinoma. Cancer Res. 53, 3472–3474. [PubMed] [Google Scholar]
- Widschwendter, M. , Jones, P.A. , 2002. DNA methylation and breast carcinogenesis. Oncogene. 21, 5462–5482. [DOI] [PubMed] [Google Scholar]
- Widschwendter, M. , Berger, J. , Hermann, M. , Muller, H.M. , Amberger, A. , Zeschnigk, M. , 2000. Methylation and silencing of the retinoic acid receptor-beta2 gene in breast cancer. J. Natl. Cancer Inst. 92, 826–832. [DOI] [PubMed] [Google Scholar]
- Widschwendter, M. , Siegmund, K.D. , Muller, H.M. , Fiegl, H. , Marth, C. , Muller-Holzner, E. , 2004. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 64, 3807–3813. [DOI] [PubMed] [Google Scholar]
- Xu, D. , Bai, J. , Duan, Q. , Costa, M. , Dai, W. , 2009. Covalent modifications of histones during mitosis and meiosis. Cell Cycle. 8, 3688–3694. [DOI] [PubMed] [Google Scholar]
- Yuan, J. , Luo, R.Z. , Fujii, S. , Wang, L. , Hu, W. , Andreeff, M. , 2003. Aberrant methylation and silencing of ARHI, an imprinted tumor suppressor gene in which the function is lost in breast cancers. Cancer Res. 63, 4174–4180. [PubMed] [Google Scholar]
- Zardo, G. , Reale, A. , De Matteis, G. , 2003. A role for poly(ADP-ribosyl)ation in DNA methylation. Biochem. Cell. Biol. 81, 197–208. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Yamashita, H. , Toyama, T. , Yamamoto, Y. , Kawasoe, T. , Iwase, H. , 2006. Reduced expression of the breast cancer metastasis suppressor 1 mRNA is correlated with poor progress in breast cancer. Clin. Cancer Res. 12, 6410–6414. [DOI] [PubMed] [Google Scholar]