

Abstract
This review analyzes the state of the art of targeted therapies for several tumors, starting from the paradigmatic example of Imatinib treatment in chronic myelogenous leukemia (CML). We discuss how rare tumors can be models for various mechanisms of receptor tyrosine kinase (RTK) activation, and provide the opportunity to develop new therapies also for more common cancer types. We discuss the activation of the downstream RTK effectors as further targets for therapies in colorectal cancer. Finally, we highlight how a novel multidimensional approach which adds an in silico dimension to the in vitro and in vivo approach, can predict clinical results.
Keywords: Targeted therapies, Imatinib treatment, Chronic myelogenous leukemia, Activation of downstream RTK effectors
1. Introduction
The last two decades have witnessed enormous progress in our knowledge and understanding of the molecular basis of neoplastic transformations. The complete sequencing of the human genome has represented one milestone on this path and, as it was anticipated at the beginning of the genome project, the completion of this enterprise brought particularly significant advancements to our understanding of cancer. By now, the view that cancer is a disease that has its origin in alterations of specific genes within the cell is a well structured theory and, thanks to high throughput technologies, many “cancer genes” have been identified and characterized. Among them, a crucial role is played by those coding for enzymatic activities, such as tyrosine kinases, particularly when they function as transmembrane receptors, the so‐called receptor tyrosine kinases (RTKs). Their activation, physiologically achieved by binding with cognate ligands, hormones, peptides or growth factors, triggers a cascade of biochemical reactions and lead to a nuclear translocation of DNA‐binding proteins which act by switching on/off sets of genes regulating fundamental processes such as cell growth, cell proliferation, cell differentiation, cell migration/invasion and apoptosis (Luo et al., 2009).
Any disturbance in any of the components of the process termed “signal transduction pathway” can potentially result in a cancerous transformation. This concept is essential from a therapeutic point of view. In fact, it has been argued that the products of altered genes coding for an enzymatic activity can become targets for compounds which inhibit their activity. As will be reported in this review, this hypothesis has been confirmed in several malignancies, where RTKs are altered by mutations (e.g. EGFR in lung carcinoma), or translocations (e.g. ALK in anaplastic large cell lymphoma). A number of these RTKs can now be targeted by small molecule inhibitors, and clinical evidence indicates that tumors carrying mutations involving these genes are particularly susceptible to such inhibitors, as the tumors are “addicted” to oncogenic signalling through the kinase pathways.
The term oncogene addiction describes a phenomenon in which a tumor becomes largely reliant on a single activated oncogene. A targeted therapy is a biologic treatment that exploits the activated oncogene as the Achille's heel of the disease and uses this molecular entity as a target for treatment.
Beside Gastrointestinal stromal tumors (GISTs), where the high expression of RTKs originates in the gain of function mutations in the protein itself (KIT or PDGFRA) (Figure 1, panel A), in sarcomas other mechanisms may result in oncogene addiction which is not mutation‐related but rearrangement‐mediated (Figure 1, panel B). Consequently, the activation in proteins that may become targets for treatment is associated with translocations involving the receptor ligand, as suggested for Platelet‐derived growth factor beta (PDGFB) in dermatofibrosarcoma protuberans (DFSP) and, more recently, Colony stimulating factor 1 (CSF1) in tenosynovial giant cell tumor/pigmented villonodular synovitis (TCGT/PVNS) (although in a minority of tumorous cells).
Figure 1.
Mechanisms of RTK activation. Gain of function mutation (A); gene translocation (B); autocrine/paracrine loop activation (C); and signalling pathway deregulation related to altered effectors of the RTK pathway (D).
Furthermore, according to recent data even autocrine/paracrine loop activation, with or without intra–inter‐family RTK cross talk not mediated via genomic mutation and/or rearrangement, may contribute to the identification of novel targets for therapies for sarcoma (Figure 1, panel C) such as aggressive fibromatosis, chordoma and alveolar sarcoma.
Interestingly, most of the tumors where molecular targets have been identified and appropriate drugs have been designed belonged to the category of the so‐called “rare tumors”. They are called “rare” due to their relative low frequency (with a prevalence lower than 50/100,000/year), but, in many instances, studies on them have provided a significant “proof of the concept” in different areas of cancer research and care so that their results have subsequently been extended to the more frequent neoplasms.
This review will analyse the state of the art of targeted therapies for several rare tumors, starting from the paradigmatic example of Imatinib treatment in chronic myelogenous leukemia (CML). The only case of a non‐rare tumor which will be examined is colorectal cancer (CRC): here the concept of a targeted therapy directed against single genetic elements has been widened so as to include the idea that, in a context of signalling pathway deregulation, an altered effector of the RTK pathway other than the receptor itself could represent a “druggable” target. A meaningful example of this situation is provided by RAS mutations in this tumor type. In this case, a mutation in RAS downstream activated or non‐activated EGFR triggers the MAP/ERK signalling to which the tumor becomes “addicted” so that this pathway yields a potential target of therapeutic agents for clinical interventions. Thus, in addition to mutational‐ or non‐mutational‐related oncogene addiction, a tumor may present a “signalling‐dependent addiction” when RTKs are considered in a context of activated signalling pathway that, containing positively or negatively regulatory elements, could be controlled at different levels of its cascade (Figure 1, panel D).
One of the weak points of this biological treatment, which by definition is not cytotoxic and can thus be chronically administered, is the development of resistance.
In a tumor where a single dominant oncogene such as the RTK is activated by mutations, acquired resistance is the consequence of secondary mutations generally affecting the kinase domain of the receptors and ultimately resulting in a modification of the kinase conformation which becomes no longer compatible with inhibitor binding. We will discuss this phenomenon with particular reference to GIST tumors, where we have also developed an original approach capable of predicting the binding affinity of drugs to mutated receptors by computational‐based molecular modeling.
It is mandatory to underline that in other tumor histotypes, characterized by a multi‐component deregulated RTK profile, other mechanisms responsible for secondary resistance have been found, mainly related to alterations in the hierarchy of dominant RTK pathways or other interconnected signalling pathways.
2. The first success story: Imatinib in CML
2.1. Chronic myelogenous leukemia (CML): historical outline
In 1960, with the advent of high‐resolution karyotyping, while studying myeloid blasts Peter Nowell and David Hungerford identified a consistent chromosomal abnormality: a small deletion at the end of chromosome 22. This would become the well‐known Philadelphia chromosome (Nowell and Hungerford, 1960).
However, many years had to pass before Janet Rowley, exploiting a new technique of chromosome banding, described this abnormality as a reciprocal translocation t(9;22)(q34;q11), in which the tip of the long arm of chromosome 9 (q34‐ter) is swapped with the tip of the long arm of chromosome 22 (q11‐ter) (Rowley, 1973). Time went by again until in 1984 Nora Heisterkamp, Jon Groffen, John Stephenson, and Gerard Grosveld, using fragments of the cloned Abelson murine leukemia virus oncogene (v‐abl) and c‐ABL genes, verified that CML translocations interrupt the c‐ABL gene on chromosome 9 by fusing its 3′ half to the 5′ half of a novel gene on chromosome 22, which they termed the BCR gene (break point cluster region) (Groffen et al., 1984). They also showed that a chimeric BCR‐ABL mRNA is present in CML cells (Stam et al., 1985) and, subsequently, Owen Witte and David Baltimore identified the protein product of the BCR‐ABL chimeric mRNA in CML as a 210‐kDa BCR‐ABL protein, larger than the 150‐kDa endogenous c‐ABL protein, and endowed with a more potent kinase activity (Konopka et al., 1984).
2.2. The development of Imatinib
The realization in the early 1980s that activated tyrosine kinases such as v‐Src, v‐Abl, and BCR‐ABL could have a causal role in cancer provoked a great interest in the development of small molecule inhibitors of oncogenic tyrosine kinases with the hope that they might be useful in cancer therapy. The key question was whether it would be possible to develop specific tyrosine kinase inhibitors considering the highly conserved ATP‐binding site among the eukaryotic protein kinases.
CIBA–Geigy (now Novartis) established a kinase inhibitor development program in 1984 (Zimmermann et al., 1996). An early target was the PDGF receptor, for which a 2‐phenylaminopyrimidine derivative (CGP53716) was synthesized as an inhibitor. This compound was found to selectively inhibit PDGF receptor signalling, and the growth of v‐sis‐transformed BALB/c 3T3 cells (Bozulic et al., 2007; Buchdunger et al., 1995).
The same series of 2‐phenylaminopyrimidines, including CGP57148B, inhibited the PDGF receptor and v‐Abl in vitro and in vivo with equal potency; importantly, BCR‐ABL was also inhibited by these molecules.
The lead compound CGP57148B was then termed signal transduction inhibitor 571 (STI571), and is currently worldwide known as Imatinib (generic name). When formulated in its mesylate salt form, Imatinib is marketed by Novartis as Gleevec in the USA, or Glivec in Europe (the dichotomy Gleevec/Glevec being exclusively due to phonetic problems).
2.3. The use of Imatinib in the treatment of CML patients …
Brian Druker is a physician scientist who has been working on CML since the early 1990s. With the view to develop a small molecule inhibitor of the BCR‐ABL tyrosine kinase activity, as this would ultimately block the growth of the transformed cells, in 1993 he started treating CML patients with his research group at the Oregon Health & Science University in Portland. In order to pursue this challenging task, he collaborated with the members of the tyrosine kinase inhibitor program at CIBA–Geigy. Working closely with Lydon, Buchdunger, and Zimmermann, Druker identified CGP57148B as a potent and relatively selective v‐Abl inhibitor. Together with Buchdunger, Druker showed that CGP57148B was able to inhibit the growth of CML cells and BCR‐ABL‐transformed cells both in cultures and in tumor‐bearing mice (Druker et al., 2001). Considering the low toxicity levels observed in the treated animals, in June 1998 Druker and Sawyers initiated a phase I/II clinical trial in chronic phase CML patients resistant to interferon therapy. This trial, and the subsequent large‐scale follow‐up phase II clinical trials, showed that Imatinib was very effective in treating chronic phase CML. These successes led to an accelerated approval process by the FDA, and Imatinib was approved for the treatment of CML on May 10, 2001. To day the predicted survival time from diagnosis for CML patients has moved from 15months before Imatinib to a projection of 15years.
2.4. … and in GIST patients
Fortuitously, Imatinib not only inhibits BCR‐ABL but is almost equally potent against platelet‐derived growth factor receptor alfa (PDGFRA), and c‐KIT receptor tyrosine kinases (Buchdunger et al., 2000). c‐KIT receptor tyrosine kinase is implicated through activating mutations in GISTs. This evidence led George Demetri to test Imatinib in the treatment of GIST patients whose tumors expressed activated c‐KIT. He found Imatinib to be not only quite effective in these patient population, but also, in some cases, resulting in a rapid tumor regression. This led to a full‐fledged clinical trial, and Imatinib was approved for the treatment of GIST in February 2002. Imatinib has also been found to be beneficial in the treatment of other proliferative premalignant hematopoietic diseases, including hypereosinophilia syndrome and chronic eosinophilia leukemia, which also express an activated form of PDGFRα.
3. RTK activation through gain of function mutation (mutation‐related oncogene addiction) (Figure 1, panel A)
3.1. Gastrointestinal stromal tumors
Gastrointestinal stromal tumors (GISTs) are the most frequent mesenchymal tumors of the gastrointestinal (GI) tract but represent <1% of all malignant GI neoplasms with an incidence on the order of 10–13 per million people per year (Goettsch et al., 2005). GIST most commonly present with abdominal pain, GI bleeding, or signs of obstruction or perforation, although as many as 20–25% are asymptomatic at time of presentation, being found for other reasons (Miettinen and Lasota, 2001; Corless and Heinrich, 2008). The most common primary sites are stomach (approximately 2/3 of cases) and small bowel (∼25% of cases), and cases arising from the esophagus, colon including rectum have been reported. Metastatic disease usually affects liver, and can also involve peritoneum. Rare sites of metastatic disease include bone, lymph nodes, and lungs.
GISTs are constituted by a proliferation of spindle‐shaped (70% of the cases), rarely epithelioid cells (20%), and 10% have mixed histology (Heinrich et al., 2002). They commonly express the KIT protein (CD117). Approximately 60–70% of GISTs are CD34+, while 30–40% are positive for SMA. Only rare GISTs are positive for desmin. Approximately 5% of GISTs are S‐100+ (Fletcher, 2002).
GIST are reported to be commonly refractory to conventional chemotherapic treatments in the “pre Imatinib era” when these entities were treated with doxorubicin, ifosfamide and dacarbazine.
3.1.1. Hereditary GISTs
Familial GISTs based on a hereditary predisposition to develop GIST owing to a germline mutation are exceedingly rare. These mutations have been identified in rare kindreds with multiple occurrence of GISTs in the relatives, and are commonly detected in KIT receptor (see below) (Li et al., 2005).
3.1.2. Syndrome association
Cases of GISTs have been reported to arise in association with syndrome like NF1, Carney's triad and Carney–Stratakis dyad (Carney and Stratakis, 2002).
3.1.3. Molecular targets
The fundamental molecular event determining GIST development is the activation of the KIT protein and its pathway. However, in a relatively small percentage of GIST patients, an alternative activated RTK is present: PDGFRA. More in detail, KIT mutations are detected in about 75–85% of GISTs, while PDGFRA mutations amount to 5–10%. About 10–15% of these tumors lack detectable mutations in both the receptors (Lasota and Miettinen, 2008).
KIT and PDGFRA belong to the same class of receptor (class III) tyrosine kinases, the family of PDGF receptor, which is characterized by a conserved structure of the extracellular domain featuring five IgG‐like domains, and by a tyrosine kinase domain (TK), the region with enzymatic activity, which in turn is split in two parts (TK1 and TK2) connected by a flexible loop.
In physiological conditions, these receptors become activated when they are bound by their specific ligands. The binding is thought to induce a conformational change in their structure, resulting in protein dimerization, intramolecular phosphorylation, and TK activation. It was proposed that the fourth IgG‐like domain present in the extracellular portion of the receptor is responsible for KIT dimerization in response to monovalent or bivalent ligand binding (Yuzawa et al., 2007).
In pathological conditions, and in GIST tumors, the activating event for KIT or PDGFRA receptors appears to be the presence of mutations in peculiar exons, more frequently affecting their juxtamembrane regions (encoded by exons 11 and 12 in KIT and PDGFRA, respectively), their kinase domains (TK1 encoded by exons 13 in KIT and 14 in PDGFRA, and the TK2 encoded by exons 17 and 18 in KIT and PDGFRA, respectively), or the dimerization domain (exon 9 in KIT).
KIT mutations are more frequently detected in exon 11, followed by exons 9, 13 and 17. Exon 18 is the more frequently altered region in PDGFRA, followed by exon 12 and (rarely) by exon 14.
The three‐dimensional conformation of these two receptors, derived from X‐ray crystallography and molecular modeling techniques, shows how these exons adopt well defined conformations in space. Exons 13 and 17 in KIT (corresponding to TK1 and TK2, respectively), and their counterparts in PDGFRA, delimitate a binding pocket in which ATP is located during protein phosphorylation. In particular, the activation loop (exon 17), whose position acts as a “gate” for the ATP‐binding site, plays a fundamental role in the RTK activity, presiding the thermodynamic equilibrium between an “active/open” and “inactive/closed” receptor conformation (Figure 2). The RTK juxtamembrane region encoded by exon 11, takes up a harpin conformation which allows this fragment to act as an autoinhibitory domain for these kinases on one side, and to shape the ATP‐binding pocket on the other. Crystallographic coordinates available to date unfortunately do not comprise the dimerization domain of these receptors; therefore, our idea that mutations affecting exon 9 could mimic the ligand binding in the context of a completely wild‐type ATP pocket is confined to a speculative level.
Figure 2.
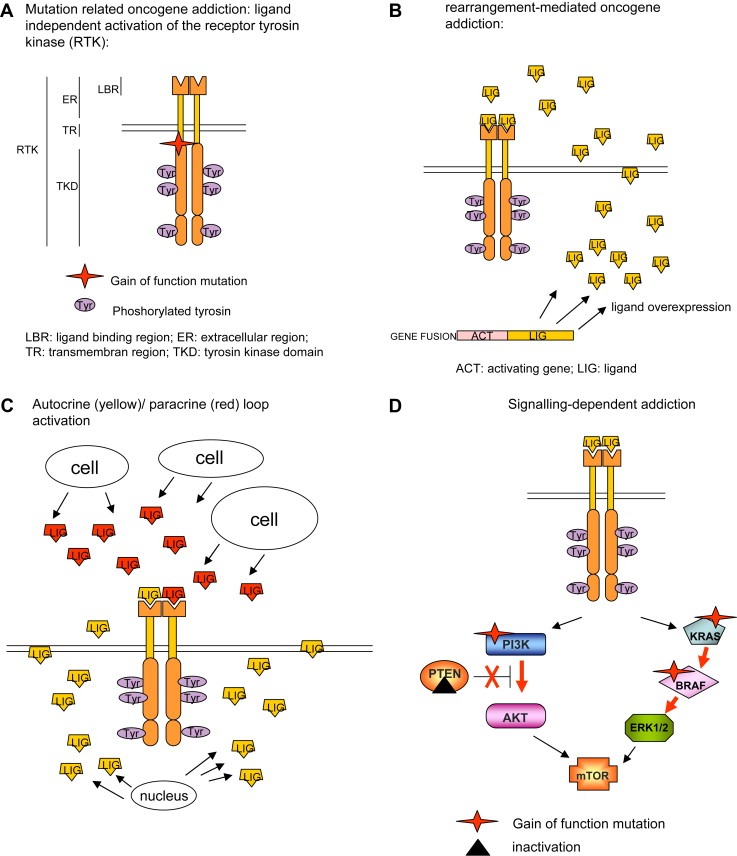
Molecular dynamics simulations. Molecular dynamics simulations snapshots of the three‐dimensional structure of (A) inactive (autoinhibited or “closed”) KIT conformation; and (B) active (“open”) KIT conformation. Secondary structure motifs are colored as follows: light gray, coils; deep sky blue, b‐sheets; orange, a‐helices. The juxtamembrane domain is depicted in plum, the P‐loop is in sienna, the A‐loop in forest green, and the control C‐helix in gold).
Globally, mutations affecting all these domains alter in a way this peculiar architecture and, hence, lead to a deregulation of the physiological RTK functions.
3.1.4. Emerging therapies
As anticipated, Imatinib has proved to be superior to any other chemotherapy for metastatic GISTs. Imatinib is a competitive inhibitor of the KIT tyrosine kinase as well as other tyrosine kinases (e.g. BCR‐ABL and PDGFR). It competes with ATP for binding to the kinases, preventing the transfer of the gamma‐phosphate group to the suitable tyrosine residues, and is able to inhibit their downstream pathways. Imatinib binds the receptor when it is in the closed conformation, and modeling studies associated with thermodynamic simulations indicate that Imatinib has more affinity for a mutated receptor than for its wild‐type counterpart. Evidence derived from the clinical experience points out that (i) tumors carrying a mutated KIT respond better than those with a wild‐type receptor; (ii) GISTs carrying mutations in exon 11 usually respond well to the drug and better than mutations in exon 9 (Corless et al., 2004); and (iii) different c‐Kit exon 11 mutation types correlate with a different response rate to Imatinib.
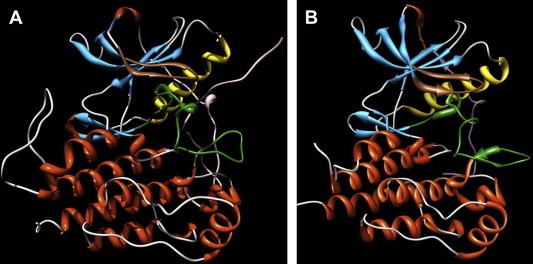
The more frequent exon 11 mutation of KIT is represented by the deletion Δ558/559 and patients carrying this mutation respond well when treated with Imatinib. Molecular modeling techniques provided a molecular rationale for this finding based on the evaluation of the inhibitor affinity for the mutant KIT. The free energy of binding ΔG bind (calculated by using the Molecular Mechanics/Poisson–Boltzmann (MM‐PBSA) method) between wild‐type KIT and Imatinib is found equal to −10.2±0.2kcal/mol, whereas that of the mutant Δ558/559 kinase and Imatinib is ΔG bind=−12.3±0.3kcal/mol. According to these results, the affinity of the mutant for Imatinib is more negative: that means that more energy is released from the system upon drug binding to the mutant KIT than to the native receptor or, in other terms, the mutant isoform is a tighter binder of Imatinib than the corresponding wild‐type counterpart (Figure 3A,B) (Tamborini et al., 2006a). The main reason for the difference in affinity can be traced to the fact that the molecular dimensions of the inhibitor are somewhat too big to result in a snug fit within the pocket formed at the N‐ and C‐lobe interface of the inactive structure of the wild‐type kinase. Accordingly, when KIT is in the closed, inactive form, the distorted juxtramembrane conformation causes a global, conformational rearrangement which involves also the ATP‐binding pocket, allowing for a larger space to accommodate the inhibitor (Figure 3B,C).
Figure 3.
Molecular dynamics simulations. Molecular dynamics simulations snapshots of the three‐dimensional structure of (A) wild‐type KIT; (B) Δ558–559 mutant Kit; and (C) V560G mutant Kit in complex with Imatinib. Secondary structure motifs are colored as follows: light green, coils; gold, b‐sheets; plum, a‐helices. The juxtamembrane domain is depicted in red. Imatinib is in stick representation (atom color code: gray, carbon; red, oxygen, blue, nitrogen), and its molecular surface is highlighted in light blue. Water molecules and hydrogen atoms are omitted for clarity.
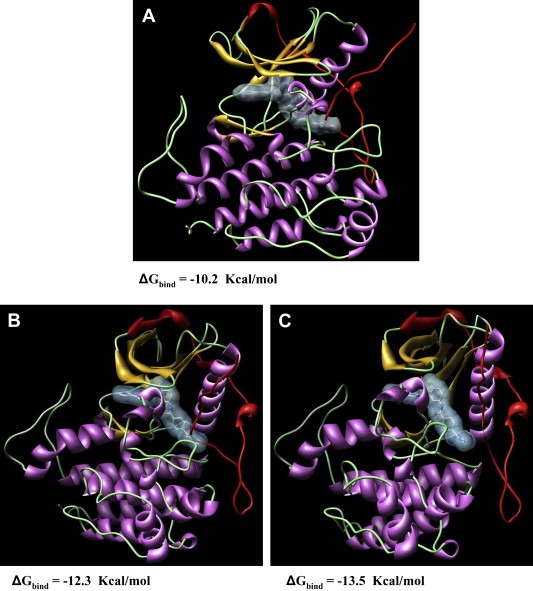
In the majority of cases in which disease progression is observed despite the targeted treatment, it is generally ascribable to the presence of secondary mutations, usually affecting the catalytic domain of KIT. Among them, the Val 654 Ala substitution, affecting the ATP‐binding pocket of the kinase, is one of the most commonly detected in Imatinib refractory GISTs. To demonstrate Imatinib insensitivity, this mutation was introduced by site‐direct mutagenesis in an expressing vector and transiently transfected into COS1 African green monkey kidney cells. Protein extracts were analyzed for KIT activation by immunoprecipitation and immunoblotting and it was demonstrated that this mutation was inhibited by 6μM of Imatinib, while another secondary mutation, the substitution Thr670Ile of KIT was insensitive to the drug at all the applied concentrations (Figure 4D). The computer modeling of the mutated receptors revealed in fact that both substitutions, although situated in the Imatinib binding site, alter the receptor in a different way: T670I substantially modifies the binding pocket whilst V654A induces only relatively confined structural changes (Figure 4B,C). Moreover, the application of molecular simulations allowed us to quantify the interactions between the mutated receptors and Imatinib, and to propose a molecular rationale for this type of drug resistance. In this light, this combined approach (biochemical and modeling analyses) yielded important information to medical oncologists suggesting the most suitable dose for escaping secondary resistance (Tamborini et al., 2006a). In fact, whereas T670I dictates a drug change, the response of patients carrying the V654A mutation is restored by increasing the dose up to 800mg/day (which roughly correspond to 6μM Imatinib).
Figure 4.
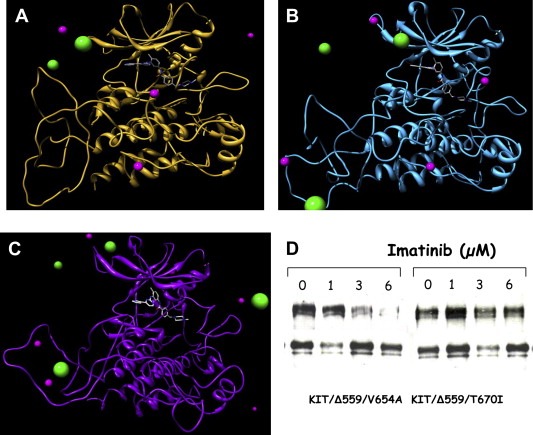
Molecular dynamics simulations. Molecular dynamics simulations snapshots of the three‐dimensional structure of (A) wild‐type KIT; (B) V654A mutant Kit; and (C) T670I mutant Kit in complex with Imatinib. Imatinib is in stick representation (atom color code: gray, carbon; red, oxygen, blue, nitrogen). Some chlorine and sodium counterions are visualized as green and magenta spheres, respectively (sphere size not in scale for graphical purposes). Water molecules and hydrogen atoms are omitted for clarity. Note how Imatinib conformation and position in its binding pocket is notably altered in the presence of the T670I mutation (C), while its structure and the whole pocket conformation is less perturbed in the V654A mutant kinase (B) with respect to the wild‐type counterpart. (D) Biochemical analysis of the two mutants showing the different sensitivity to Imatinib. The activated KIT carrying the substitution V654A is inhibited by a 6μM of Imatinib while the one carrying the T670I does not.
The already mentioned substitution Thr670Ile of KIT represents a peculiar trait which is shared by BCR‐ABL and PDGFRA in CML and idiopathic hypereosinophilic syndrome patients, respectively. The obvious question which arises is then why Thr is always replaced by Ile. To answer this question, all possible point mutations in the DNA triplet codon that could result in amino acid substitutions at Thr670 (Thr670Arg, Thr670Ile, Thr670Lys, Thr670Ala, Thr670Ser, Thr670Pro) were introduced by site‐specific mutagenesis of the complementary DNA for a constitutively active, Imatinib‐sensitive form of the KIT receptor, Δ559/KIT. The resulting mutant KIT proteins were transiently expressed in cells with and without Imatinib, and protein extracts were analyzed for KIT activation to determine autophosphorylation levels. Concomitantly, in silico experiments were conducted to estimate the relative affinities of wild‐type (Thr670) KIT and the KIT mutants for ATP and Imatinib.
Intriguingly, like the parental strain, Thr670Ala, Thr670Ser, and Thr670Lys mutants were inhibited by 5μM Imatinib, but in comparison, they were only weakly active and Thr670Pro and Thr670Arg were not active at all. Only the Thr670Ile mutant was fully active (autophosphorylated) and resistant to Imatinib. These findings were consistent with computer modeling predictions that ranked these mutants Thr∼Ile>Ala, Ser>Lys≫Pro∼Arg according to their affinity for ATP but Thr>Ala, Ser>Lys>Pro∼Arg∼Ile according to their affinity for Imatinib.
Thus, pretty unambiguously, this combination of in vitro and molecular modeling analyses revealed why, among all possible amino acid substitutions at position 670 of KIT, only Ile is naturally selected as a resistance mutant in Imatinib‐treated GIST patients (Negri et al., 2009).
A second line therapy for GIST patients who became resistant to Imatinib treatment is represented by Sunitinib, an ATP competitor that binds the receptor in its open conformation, and is thus able to bypass the major Imatinib failures (Heinrich et al., 2008a).
Figure 5 summarizes all the approaches adopted in treating GIST patients that, starting from radiological evaluations, through the molecular profiling of the tumor, led to the identification of a secondary mutation responsible for the disease progression despite Imatinib treatment. As shown, a patient under drug treatment (800mg/day), during routine radiological investigations (Figure 5A), was shown to carry liver lesions in regression whereas a peritoneal lesion in the same patient was in progression. This was confirmed also by the PET analysis (Figure 5B). After the surgical intervention the histopathological evaluation of the progressing lesions evidenced a highly cellular appearance (CD117 positive) while rare, scattered tumor cells with inconspicuous cytoplasm and stripped pyknotic nuclei embedded into an eosinophilic myxoid stroma were visible in the regressing lesion (CD117 negative) (Figure 5C). The biochemical analysis of the two lesions demonstrated a highly expressed and phosphorylated KIT receptor in the progression and a very low expressed and activated KIT in the regression (Figure 5D). Interestingly, DNA sequencing identified, besides the exon 11 activating mutation, an additional point mutation only in the progressing lesion (Figure 5E); the latter in in vitro biochemical experiments showed a total insensitivity to Imatinib (Figure 5F). The second mutation is termed “secondary resistance” mutation and is most likely the result of the selection of a preexisting cell clone which expands after drug exposure.
Figure 5.
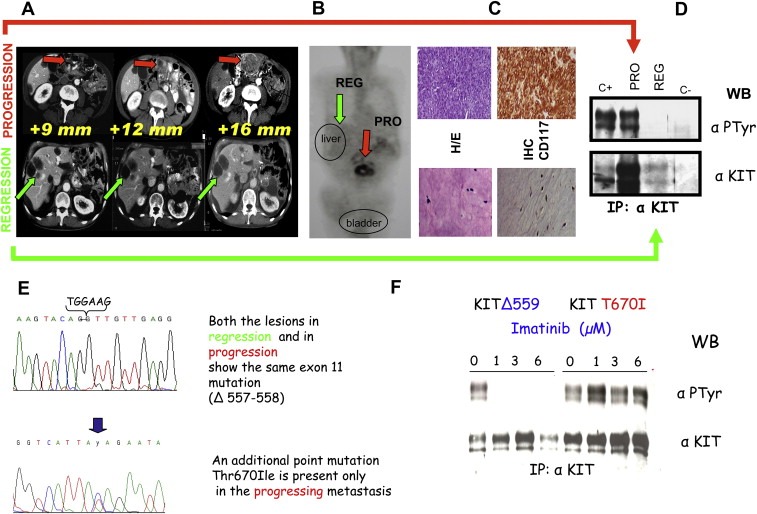
Clinical, pathological (biochemical and molecular) characterizations of GIST patients. A. CT scan of a male, 65year old patient who was operated on for a peritoneal mass (measuring 22cm) 2years previously. The diagnosis indicated a high grade GIST. He started Imatinib treatment at 800mg/day, with radiological follow‐up every 3months. In the upper lane, indicated by red arrows, the peritoneal progressing lesion is highlighted. In the lower lane, indicated by green arrows, the responding liver lesions are shown. These lesions were classified as responding due to their “hypodense” appearance. B. PET analysis. Glucose uptake, indicating a proliferating mass, is detectable only at peritoneal rather than liver site. C. Histopathological evaluation of the lesions after surgical removal. The non‐responding tumor, upper lane, nodule showed a highly cellular appearance. The tightly packed tumoral cells were arranged into large sheets of small acinus‐like clusters deposited in a myxoid stroma. The responding liver lesion, lower lane, showed marked cellular depletion. CD117 immunostaining was evidenced only in the progressing lesion. D. Biochemical analysis (immunoprecipitation of 0.5 mg of total proteins extract) of the above described lesions. A phosphorylated (active) and highly expressed KIT receptor was present in the progressing lesion and not in the responding one. E. DNA sequencing revealed in both lesions the activating exon 11 point mutation corresponding to the loss of 557–558 residues. Only in the progressing lesion an additional point mutation affecting exon 14 was present leading to the substitution T670I. F. “In vitro” analysis of COS cells, transfected with an expressing vector for KIT receptor in which was inserted the T670I substitution. Different doses of Imatinib were given and, as can be seen in the right part of the panel, T670I/KIT showed phosphorylation (activation) at all concentrations of the drug, while KIT carrying exon 11 mutation was inhibited at 1μM Imatinib.
4. RTK activation through autocrine/paracrine loop sustained by chromosomal translocation (Figure 1, panel B)
4.1. Dermatofibrosarcoma protuberans
Dermatofibrosarcoma protuberans (DFSP) represents approximately 1% of all soft tissue sarcomas; nevertheless, it is the most frequent skin sarcoma. Low‐grade classic form of DFSP is characterized by infiltrative growth in the skin and subcutaneous tissues, and by the tendency to recur locally (Lemm et al., 2009; Llombart et al., 2009). DFSP most commonly affects adults (20–50years old), and is exceedingly rare in children. Tumors clinically appear as plaque‐like or nodular lesions, and occur most commonly on the trunk (42–72% of the cases), the proximal extremities (16–30%), and head and neck (10–16%) (Lemm et al., 2009). DFSP rarely metastasizes (<5%), most often to the lung, but also to regional lymph nodes, bone, heart, and brain (Dimitropoulos, 2008). A high grade fibrosarcoma (FS‐DFSP) is present in the 10–15% of the cases, with risk to both of the local recurrence and distant metastasis (Dimitropoulos, 2008). The risk of transformation into a more malignant form is increased in repeatedly recurring tumors.
Wide excision margins (20–30mm) is the primary treatment. Mohs micrographic surgery with continuous histological margin control seems to reduce local recurrence rates. If positive margins are diagnosed after surgery, clinical practice guidelines recommend re‐resection, if necessary followed by adjuvant radiotherapy (Lemm et al., 2009). Chemotherapy produces no remarkable benefit in the treatment of DFSP (Lemm et al., 2009).
4.1.1. Molecular targets
DFSPs are cytogenetically characterized by a reciprocal translocation, t(17;22)(q22;q13) or, more frequently, supernumerary rings chromosome composed of hybrid material derived from t(17;22). These characteristic genetic rearrangements have been reported in the majority of cases (89–96%), but their true frequency is unknown, because molecular studies (RT‐PCR and/or FISH) performed to date focused on isolated cases with few large series (Llombart et al., 2009). However, in a small percentage of cases, the rearrangement is not found.
The translocated chromosome and the supernumerary rings contain the same molecular genetic rearrangement, which fuses the strongly expressed collagen type 1 alpha 1 (COL1A1) gene on chromosome 17 with the platelet‐derived growth factor B‐chain gene (PDGFB) on chromosome 22. The breakpoint localization in PDGFB is generally constant (exon 2), whereas in COL1A1 the breakpoint may occur in any of the exons in the alpha‐helical region (exons 6–49). PDGFB is thus placed under the control of the COL1A1 promoter. The COL1A1‐PDGFB fusion gene results in constitutive production of COL1A1‐PDGFB protein, which is processed into the mature PDGFB, ligand for both PDGFRB and PDGFRA. Because PDGFRB is preferentially expressed on DFSP, PDGFB is most likely to activate PDGFRB rather than PDGFRA (McArthur et al., 2005). Autocrine/paracrine stimulation of PDGFRB as the critical molecular event in the pathogenesis of DFSP led to the hypothesis that inhibition of PDGFRB by Imatinib might be effective. Accordingly, Greco et al. (Greco et al., 2001) demonstrated that the transfection of the COL1A1‐PDGFB fusion gene leads to malignant transformation of NIH‐3T3 cells, and that Imatinib reduces the growth rate of the transformed cells. Moreover, COL1A1‐PDGFB expressing cells were shown to generate tumors after subcutaneous injection into nude mice, and their growth was reduced when the mice were treated with Imatinib (Sjöblom et al., 2001).
4.1.2. Emerging therapies
Imatinib has been approved in the US and EU for the treatment of adult patients with unresectable and/or metastatic DFSP, who are not eligible for surgery, and its clinical activity has been reported in literature (McArthur et al., 2005; Lemm et al., 2008; Maki et al., 2002; Mizutani et al., 2004; Heinrich et al., 2008b).
In particular, the Imatinib Target Exploration Consortium Study B2225 performed on ten patients with locally advanced or metastatic DFSP, two of which showed the FS component, reported that Imatinib exhibits significant clinical activity in all the patients (but one who carried the FS‐DFSP) with high rates of disease regression. (McArthur et al., 2005). It is important to note that the presence of translocation, reported in the FS component of DFSP, seems to be correlated with Imatinib sensitivity (McArthur et al., 2005; Llombart et al., 2009; Mizutani et al., 2004), while the high grade FS‐DFSP lacking t(17;22) did not respond (McArthur et al., 2005). Furthermore, analysis of frozen pre‐treatment samples from patients revealed only a weak phosphorylation of both PDGFRB and PDGFRA. Accordingly, DFSP does not appear to rely on a high level of RTK activation (McArthur et al., 2005). Nevertheless, the clinical activity of Imatinib in DFSP suggests a dependence on this signalling mechanism, and histologic examination of biopsy specimen of responding patients displays marked reduction of cellularity with hyaline changes (Llombart et al., 2009; McArthur et al., 2005; Lemm et al., 2008).
Clinical trials, aimed at improving patient outcome, are needed to determine (i) Imatinib neoadjuvant use in reducing tumor burden and in facilitating surgical resection; (ii) the adjuvant use after a complete surgery; and (iii) Imatinib treatment in FS‐DFSP. Several studies are currently ongoing both in USA and Europe.
4.2. Tenosynovial giant cell tumor and pigmented villonodular synovitis
Tenosynovial giant cell tumor (TGCT), and the more aggressive pigmented villonodular synovitis (PVNS), are rare proliferative disorders affecting synovial joints and tendon sheaths characterized by localized and diffuse growth pattern, respectively (Mendenhall et al., 2006; Martin et al., 2000; WHO, 2002). The optimal treatment is surgery, although rarely local recurrence may necessitate an extensive re‐excision and radiotherapy.
4.2.1. Molecular targets
These lesions are composed of mononuclear and multinucleated cells, both overexpressing the colony‐stimulating factor receptor (CSF1R), another member of the RTK type III subgroup. By contrast, only a subset of mononuclear cells expressed high levels of colony‐stimulating factor (CSF1) mRNA encoding the ligand of CSF1R (West et al., 2006). This is in keeping with the evidence that, despite a t(1;2) translocation is found in most TGCT (87%) and PVNS (35%) where CSF1 is the gene at the chromosome 1p13 breakpoint, this translocation leads to the fusion of CSF1 and COL6A3 (2q35) resulting in high levels of CSF1 only in a small fraction of mononuclear cells. Consequently, a minority of neoplastic cells expressing the fusion protein recruit the majority of non‐neoplastic cells expressing CSF1R through a paracrine “landscape” effect.
4.2.2. Emerging therapies
The pivotal role for CSF1 in the TGCT/PVNS pathogenesis gives potential sensitivity to Imatinib. In fact, Imatinib induced complete response in relapsing TGCT/PVNS (Blay et al., 2008), offering an option when surgery is not feasible or would result in functional impairment. The effect of Imatinib seems to rely in its ability to block CSF1R activation at therapeutic concentration (Dewar et al., 2005), inhibiting the paracrine loop responsible for the growth of these tumors.
5. RTK activation through autocrine/paracrine loop up‐regulation not sustained by gene alteration (Figure 1, panel C)
5.1. Aggressive fibromatosis
Aggressive fibromatosis (AF) is a mesenchymal proliferation showing a fibroblastic to myofibroblastic differentiation that, by virtue of its clonal nature, is currently considered a true neoplasm (Lucas et al., 1997). This tumor may occur in patients with familial adenomatous polyposis (FAP) or as a sporadic form. It typically occurs in the abdomen or in the abdominal wall but can develop at other anatomic sites, most commonly in the extremities (extra‐abdominal fibromatosis). AF is characterized by an unpredictable natural history; in fact, although histologically benign, is often locally invasive, associated with a high local recurrence rate after resection and can result in pain, deformity, functional impairment and death when vital organs are involved (Mendenhall et al., 2005).
The standard treatments involve extensive surgical resection and/or radiation therapy, but there are a significant proportion of patients with local recurrence who are not amenable to surgical resection or radiotherapy. Therefore, a variety of systemic therapeutic approaches have been increasingly investigated and utilized. Responses have been reported using a variety of treatments (non‐steroidal anti‐inflammatory agents (NSAIDs), tamoxifen, interferon, chemotherapy and Imatinib). However, the optimal treatment remains to be determined, because the assessment of efficacy of the various treatments is complicated by rarity and heterogeneity of the disease (de Bree et al., 2009).
5.1.1. Molecular targets
Most AF are associated with abnormalities in the regulation of WNT pathway signalling, either because of germline/somatic inactivation of APC or for somatic beta‐catenin gain of function mutations (Li et al., 1998; Alman et al., 1997). In tumoral cells, beta‐catenin accumulates into the cytoplasm then translocates to the nucleus, where it activates the T‐cell factor. This, in turn, causes transcription of target genes such as cyclooxygenase‐2 (COX‐2). COX‐2, among its various targets, increases the expression of PDGFA and PDGFB (Dempke et al., 2001), rendering PDGFRB and/or PDGFRA activation a possible driving event in AF pathogenesis (Signoroni et al., 2008). Moreover, beta‐catenin deregulation has a potential role in AF tumorigenesis where mutations in exon 3 have been commonly identified (85%). In particular, the mutation 41A is a prognostic factor significantly associated with a higher risk of recurrence (Lazar et al., 2008).
5.1.2. Emerging therapies
Imatinib is an active agent in the treatment of advanced AF (Heinrich et al., 2006). Molecular basis for response/non‐response have been investigated by several groups (Signoroni et al., 2008; Mace et al., 2002; Liegl et al., 2006; Heinrich et al., 2006). Expression of KIT, PDGFRA and PDGFRB were analysed by immunohistochemistry with discordant results (Liegl et al., 2006; Heinrich et al., 2006). Activating mutations were not found in KIT, PDGFRA and PDGFRB genes (Signoroni et al., 2008; Liegl et al., 2006; Heinrich et al., 2006; Tamborini et al., 2006b). In particular, Heinrich et al. (2006) observed a strong expression of PDGFRB in the presence of plasma levels of PDGF ligands, while our group (Signoroni et al., 2008) has verified the activation of PDGFRA and, to a greater extent, of PDGFRB in absence of mutations, and gene number alterations in the presence of PDGFA and PDGFB transcripts. These results suggest an activation of Imatinib targets by autocrine/paracrine loop.
Response to Sunitinib has been recently reported in one case (Skubitz et al., 2009).
5.2. Alveolar soft part sarcoma
Alveolar soft part sarcoma (ASPS) is a rare tumor (less than 1% of STS) mainly affecting younger patients and occurring more frequently in the trunk and proximal extremities. ASPS pursues an indolent course when localized and resectable, but the late stage of the disease is associated with metastasis to multiple sites including lungs, bones, lymph nodes and brain. Once the tumor disseminates, it becomes resistant to conventional chemotherapy. Histologically, the tumor is characterized by organoid nests of polygonal tumor cells encompassed by dense capillary vasculature, resulting in the “alveolar” appearance.
5.2.1. Molecular targets
ASPS is characterized by the presence of a specific chromosomal translocation resulting in a fusion of the ASPSCR1 (previously known as ASPL) and TFE3 genes (chromosomes 17q25 and X11.2, respectively) (Ladanyi et al., 2001), together with a proportion of childhood RCC (Argani et al., 2001). These defects can be highlighted by IHC using a polyclonal Ab binding the C‐terminal portion of TFE3 protein retained in all known TFE3 fusion proteins (Argani et al., 2003).
Recently, two groups investigated the molecular profile of this tumor providing evidence for a role of unregulated TFE3 overexpression in the context of ASPLCR1‐TFE3 fusion protein.
The first group (Tsuda et al., 2007), after identification by expression profile analysis of a significant up‐regulation of MET in ASPS in cell lines, demonstrated through chromatin immunoprecipitation assay that ASPLCR1‐TFE3 fusion protein binds with MET promoter and activates MET leading to MET autophosphorylation and HGF co‐expression. These processes in turn trigger a strong activation of the downstream PI3K/AKT and ERK pathways (Tsuda et al., 2007). The second group (Lazar et al., 2007) identified and validated three protein products – Jag‐1, midkine and angiogenin – that were consistently up‐regulated in ASPS through expression profile analysis of three cryopreserved ASPSs. Remarkably, this quite unique angiogenic profile encompasses genes carrying putative binding sites in their promoter regions, and this strongly suggests that they may be induced by the action of ASPCR1‐TFE3 fusion transcript (Lazar et al., 2007). Several targets involved in angiogenesis were recently reconfirmed by a genome‐wide gene expression profiling approach (Stockwin et al., 2009). Finally, a study addressing RTK activation in ASPS was conducted using antibody arrays, allowing the simultaneous assessment of the phosphorylation status of 42 RTKs. These investigations, complemented by biochemical and immunohistochemical validations and downstream signalling analysis, clearly highlighted the presence of multiple RTK‐driven autocrine/paracrine loops encompassing the RTKs that are inhibited by Sunitinib malate (Stacchiotti et al., 2009a).
5.2.2. Emerging therapies
Overall, the identification of several angiogenic mediators and the evidence of MET activation provide a molecular framework supporting the use of anti‐angiogenic and MET inhibitor agents. Response to Bevacizumab has been reported in a patient (Azizi et al., 2006) and, more recently, in an in vivo model of ASPS (Vistica et al., 2009).
ARQ 197, a MET inhibitor, has been tested in a phase II trial on ASPS patients, which demonstrated stable disease (Goldberg et al., 2009).
In our institution, five patients with progressive heavily pre‐treated advanced ASPS were treated with Sunitinib malate. Among the four patients evaluable for response, two showed a partial response and one had stable disease, suggesting that this treatment could be effective in ASPS care (Stacchiotti et al., 2009a). These findings reinforce the notion that also malignancies in which RTK activation does not occur via genomic mutation and/or rearrangement (Heinrich et al., 2008b) may be associated with clinical benefit when treated with TKIs. Further, tumors showing multiple concomitantly activated RTKs may be treated with combinations of drugs against different activated RTKs or with a single drug with inhibitory action against multiple activated RTKs (Stommel et al., 2007). This is particularly true when the spectrum of TKIs applied closely matches the TK deregulation profile of the investigated tumor (Stacchiotti et al., 2009a).
5.3. Chordoma
Chordomas are rare, low‐grade bone tumors accounting for 1–4% of all malignant bone tumors (WHO, 2002). They arise mainly from the sacrum but also the skull base, the cervical vertebrae, and the thoracic–lumbar vertebrae could be affected (Mirra et al., 2002). Their peak incidence is between the fourth and sixth decades of life, although children may also be affected (5%). The gender (M:F) ratio is 2–3:1, males being affected more by sacral chordomas, whereas the frequency of chordomas at the skull base seems to be equal in the two genders (Chugh et al., 2007). Generally, chordomas are sporadic but familial cases (Miozzo et al., 2000) and tumors arising in patients with tuberous sclerosis complex (TSC) (Storm et al., 2007) have also been described.
The tumors are locally highly aggressive, although they may give rise to distant metastases (10–30% of cases or more). Surgery remains the best standard treatment for both localized and metastatic disease; radiotherapy (in particular cobalt radiation or heavy particle radiation) can control tumor growth, but the high doses required lead to significant toxicity, and standard cytotoxic chemotherapy is inefficacious (Chugh et al., 2007).
5.3.1. Molecular targets
It has been demonstrated that chordomas express an activated PDGFRB, and that it is activated by the occurrence of an autocrine/paracrine loop (Tamborini et al., 2006c). It has also been reported that a subset of chordomas express EGFR and c‐MET (Weinberger et al., 2005). The presence of activated RTKs leads to the activation of secondary transducers, belonging to the MAPK or PI3K/AKT pathways, which concur in activating mTOR (Shaw and Cantley, 2006). Downstreams of mTOR, S6K/S6, and 4E‐BP1 govern protein synthesis, and promote cell growth and proliferation. Using an RTK array, our group proved that chordomas express activated PDGFRB, FLT3 and CSF1R in the PDGFR family, as well as highly phosphorylated EGFR, HER2/neu and (to a lesser extent) HER4 in the EGFR family. This upstream analysis, followed by validation, indicates that in addition to the PDGFR also the EGFR family may be activated in chordomas. The analysis of the downstream pathways indicates that activated PI3K/AKT and RAS/MAPK cascades are both present and lead to the phosphorylation of mTOR (Tamborini et al., submitted).
5.3.2. Emerging therapies
Molecular targeted therapy has recently shown significant promise: Imatinib has induced an antitumor response in a number of patients when used alone (Casali et al., 2004), and in combination with cisplatin in patients showing secondary progression on Imatinib alone (Stacchiotti et al., 2007). Furthermore, combined treatment with Imatinib and Sirolimus has proved to be effective in patients with Imatinib‐resistant chordomas (Stacchiotti et al., in press).
Two cases responding to cetuximab (anti‐EGFR) have also been recently reported (Hof et al., 2006; Lindén et al., 2009).
5.4. Ewing sarcoma
Ewing sarcoma family tumors account for 6–8% of primary/malignant bone tumors and are the second most common sarcoma in bone and soft tissue in children; they comprise extraskeletal Ewing sarcoma, small cell tumor of the thoraco pulmonary region (Askin tumor) and the soft tissue based peripheral primitive neuroectodermal tumors (pPNET) (WHO, 2002).
Although the prognosis is better for those individuals without radiographic evidence of disseminated disease, the vast majority of people develop rapid recurrence, often to the lung, unless systemic chemotherapy is administered. For this reason, all the patients require coordinated treatments that combine both chemotherapy (neoadjuvant and adjuvant) with local control of lesions by surgery and/or radiotherapy (Subbiah et al., 2009).
A good control of the disease is generally achieved employing vincristine, actinomycin D cyclophosphamide, doxorubicin, etoposide and ifosfamide. Combinations of similar compounds are used in metastatic patients, but experimental chemotherapies are considered earlier and high doses of ifosfamide are generally used as second line therapy.
Patients with recurrences within the first 2years from the diagnosis, at 5years showed a survival around 7%, compared to 30% for recurrent disease >2years (Subbiah et al., 2009). This suggests that more tailored treatments for metastatic patients are strongly envisaged.
5.4.1. Molecular targets
Karyotypic analyses have revealed a tumor‐specific chromosomal translocation t(11;22)(q24;q12) in 86% of Ewing sarcomas (ES)/pPNET. The (11;22) translocation results in the fusion of the N‐terminal region of the EWS gene rich in glutamine, serine, and tyrosine residues to the ETS‐like DNA‐binding domain of the Friend leukemia integration site 1 (FLI1) gene. EWS is an ubiquitously expressed gene, located on chromosome 22, that encodes for a RNA‐binding protein, whereas FLI1, located on chromosome 11, is a member of the ETS family of transcription factors. The oncogenic effect of the t(11;22) translocation is caused by the formation of a chimaeric protein. The protein has the potential to promote tumorigenesis by acting as an aberrant transcription factor, functionally distinct from the normal FLI1. It has been demonstrated that the FLI1 COOH‐terminal domain in addition to its DNA‐binding domain is necessary to promote cellular transformation.
Alternative fusion transcripts are present in a low percentage of Ewing sarcoma family tumors and are produced by EWS‐ERG t(21;22)(q22;q12), EWS‐ETV1 t(7;22)(p22;q12), EWS‐FEV t(2;22)(q33;q12), FUS‐ERG t(16;21)(p11;q22) translocations.
5.4.2. Emerging therapies
It has been reported that these tumors express IGF‐1R which is activated by an autocrine loop since the same malignant cells are able to produce the ligand that specifically binds the receptor present on the cell membrane. At preclinical level it has been demonstrated that the blockage of IGF‐1R mediated circuit can inhibit the in vitro growth and the motility of these tumoral cells (Scotlandi et al., 1996).
The pediatric preclinical testing program showed initial activity of IGF‐1R targeted monoclonal antibodies in a murine xonografts model of human Ewing tumor. In a phase I trial four out of nine patients appeared to have derived clinical benefits from a monoclonal based single agent therapy without significant toxicity (Subbiah et al., 2009). At present, other compounds have been reported efficacious against Ewing's cells such as NVP‐AEW541, a small inhibitory molecule of the kinase activity, even if just at the preclinical level.
Other targets could be represented by the fusion protein EWS‐FLI1 that has been downregulated in vitro by antisense oligonucleoties and RNAi and very recently by the use of a small molecule able to block the enzymatic activity of the transcription complex that involve the chimaeric protein EWS‐FLI1 and RNA helicase A (Erkizan et al., 2009).
5.5. Non‐RTK‐mediated paracrine loop in giant cell tumor of bone
According to WHO (2002) definition, giant cell tumor (GCT) of the bone is a benign locally aggressive neoplasm, mainly affecting young adult patients, made up of neoplastic ovoid mononuclear cells interspersed with osteoclast giant cells. GCT is relatively rare (4–5% of primary bone tumors) and, although classified as benign, can be aggressive. GCT recurs locally in 50% and metastasize in 5% of cases. Metastases are very slow growing (benign tumoral implants) but a small proportion are progressive and may lead to the death of the patient.
Surgery is currently the treatment of choice for resectable GCT. However, en‐bloc excision is followed by recurrence in 20% of the cases. In advanced unresectable GCTs, chemotherapy is not the standard care and local radiotherapy is used for local control with an efficacy reaching up to 80%. The role of other agents (interferon and bisphosphonates) remains unproved.
5.5.1. Molecular targets
Cross talk between osteoblast/stromal cells and osteoclasts is governed by RANK/RANKL/OPG pathway which, leading to osteoclast differentiation and their activation, provides a possible treatment of non‐tumoral and tumoral diseases characterized by excessive bone resorption.
Briefly, RANK/RANKL/OPG are members of tumor necrosis factor (TNF) receptor (TNFR)‐ligand family. Receptor activator of NF‐κB ligand (RANKL) is expressed on the cell surface of osteoblast/stromal cells and binds receptor activator of NF‐κB (RANK) expressed on the surface of osteoclast precursor cells leading to osteoclasts differentiation. OPG, which can bind to RANKL, acts as decoy receptor blocking the interaction between osteoblast/stromal cells and osteoclast precursors, inhibiting osteoclasts formation and bone resorption (Aubin and Bonnelye, 2000).
In mice, treatment with RANKL leads to severe hypercalcemia and bone loss with increasing of differentiated osteoblasts. All these effects can be blocked by OPG.
RANKL has been demonstrated to be involved in tumor cell inducing osteoclastogenesis such as in chondroblastoma as well as GCT. In the former tumor, RANKL mRNA (by RT‐PCR) and protein (by ISH and IHC) have been described as restricted to mononuclear cells (Huang et al., 2003) while, in the latter, RANKL mRNA expression (by expression profiling, confirmed by RT‐PCR, flow cytometry and IHC) seems to correlate with possible osteoclastic lineage precursors (Morgan et al., 2005). However, the genetic basis of the high expression of RANKL has not been identified (Thomas and Skubitz, 2009).
5.5.2. Emerging therapies
Denosumab is a human monoclonal antibody to RANKL, mainly investigated for the treatment of osteoporosis, myeloma and metastatic carcinomas. A recent phase II study in unresectable GCTs showed a tumor response in >85% of patients which correlated with a near complete elimination of giant cells and changes in metabolic FDG‐PET uptake or stabilization of disease (Thomas and Skubitz, 2009). The role of denosumab in the treatment of GCT is currently the subject of intense study.
OPG protein also represents a therapeutic candidate as it prevents bone lesions and inhibits associated tumor growth. However, despite this positive effect, the ability of OPG to bind TRAIL and to protect tumor cells from TRAIL‐induced apoptosis renders its use questionable in osteolysis associated with bone tumors (Lamoureux et al., 2009).
5.6. Signalling pathway deregulation and targeted therapy: the sporadic colorectal carcinoma model (Figure 1, panel D)
Colorectal cancer (CRC) is the second leading cause of cancer death in Western countries, and becomes the first when smoking‐related diseases are excluded.
When localized, CRC is often curable by surgery, but the prognosis for patients with metastatic disease remains poor. Curative‐intent resection can be performed only on 10–15% of liver metastases. In the majority of metastatic patients, the standard treatment remains palliative chemotherapy. The backbone of the treatment for metastatic CRC (mCRC) is fluorouracil, usually administrated with leucovorin. Combination of conventional chemotherapy with new anticancer drugs such as irinotecan and oxaliplatin has improved the standard chemotherapy treatment of CRC (Zuckerman and Clark, 2008). However, molecular targeted therapy seems to hold a promise for a cure of CRC.
5.6.1. Molecular targets
The identification and characterization of the genetic changes in the malignant colorectal transformation process have progressed rapidly over the last two decades, and led to the formulation of a model of CRC carcinogenesis where the temporary progression from healthy mucosa to carcinoma in situ is supported by mutations in the APC, K‐Ras, TP53, and DCC genes. This model, originally proposed for the vast majority of sporadic CRC, is also valid for familial adenomatous polyposis (FAP), characterized by an APC germline mutation. A second pathway of CRC development has been depicted in cases with a normal karyotype but featuring genetic instability at microsatellite loci. In this group of cancers (previously termed replication error (RER) positive tumors and today known as microsatellite instability‐positive (MSI‐positive) tumors), alterations in the DNA mismatch repair genes, when present in germinal cells, are responsible for the familial hereditary non‐polyposis colorectal cancer (HNPCC) and, when affecting somatic cells, may cause MSI on a subset (up to 15%) of sporadic CRC. In this second model of colorectal carcinogenesis, BRAF mutations are frequently detected (30% of cases).
5.6.2. Emerging therapies
Currently, cetuximab and panitumumab, the two monoclonal antibodies (MoAbs) targeting the extracellular portion of EGFR and approved by the US Food and Drug Administration (FDA), have been introduced into the clinical practice of patients with EGFR‐positive chemotherapy‐refractory sporadic metastatic CRC (mCRC), improving the response rate (Saltz et al., 2004; Lenz et al., 2006). The combination of these drugs with Irinotecan or best supportive care (Jonker et al., 2007) was significantly more effective than either treatments alone. However, a clinical benefit was observed in only 10–20% of the treated patients. Thus, it is important to identify biomarkers able to help clinicians in selecting patients that could potentially respond to these MoAbs. In this respect, attention was firstly focused on the EGFR analysis and, in accordance with the FDA guidelines, CRC patients are still selected on the basis of EGFR expression. However, it became soon clear that neither EGFR expression (assessed by immunohistochemistry) nor EGFR gene copy number (assessed by fluorescence in situ hybridization or comparative genomic hybridization) play a major role in predicting response to EGFR antagonists (Personeni et al., 2008; Perrone et al., 2009; Italiano et al., 2008), but, rather, that response might depend on downstream signalling elements triggered by EGFR itself. Mutations in KRAS or PI3KCA genes actually seem to constitute determinants of response to EGFR inhibitors. In fact, they lead to a permanently active state of the protein that, in turns, permits the cell to evade EGFR inhibition and apoptosis.
Many non‐randomized and randomized studies confirmed a significant correlation between KRAS codons 12 and 13 mutations and poor response to EGFR targeting MoAbs in the first‐, second‐ or third‐line settings (Jimeno et al., 2009). Thus, the European Committee for Medicinal Product for Human Use has approved the use of cetuximab and panitumumab in CRC patients harbouring wild‐type KRAS codons 12 and 13 only. Moreover, recent data showed that codon 61 and 146 mutational analysis could improve the KRAS power for predicting resistance to anti‐EGFR (Loupakis et al., 2009a). Since to date several methods are available and applied for KRAS mutation detection (Jimeno et al., 2009), the differences in sensitivity and specificity of these tests have to be taken into account as they could be critical for clinical decisions. This aspect is very important, since at present CRC patients are negatively selected for EGFR‐based therapy without biologic alternatives. To date, treatment with farnesyl transferase inhibitor R115777 failed in KRAS mutated CRC (Rao et al., 2004), and the effects of drug‐induced inhibition of targets downstream of KRAS (e.g. MEK) have still to be evaluated (Rinehart et al., 2004).
Interestingly, a wild‐type KRAS is necessary but not sufficient to derive benefit from EGFR inhibition in CRC; thus, factors others than KRAS mutation can dictate lack of efficacy for these biologic therapies. Recently, additional negative predictive factors have been identified, allowing a potential increased efficacy in treated patients. In particular, mutations of BRAF, the main downstream effector of KRAS, were described as mutually exclusive to KRAS mutations and associated with resistance to EGFR‐targeted MoAbs therapies in about 10% of KRAS wild‐type CRC patients (Di Nicolantonio et al., 2008). Accordingly, the BRAF inhibitor sorafenib restored the sensitivity to cetuximab or panitumumab of CRC cells harbouring the V600E mutation in vitro.
Moreover, as anticipated, PI3KCA mutations seem to be a further predictor factor for resistance to EGFR‐targeted MoAbs (Sartore‐Bianchi et al., 2009; Perrone et al., 2009), although this evidence was not confirmed by the study of Prenen et al. (2009). In addition, loss of PTEN expression was reported as a single marker determining resistance to cetuximab (Frattini et al., 2007; Sartore‐Bianchi et al., 2009; Loupakis et al., 2009b), as well as PTEN mutation or gene loss (Perrone et al., 2009). A comprehensive molecular analysis of KRAS, BRAF, PI3KCA, and PTEN could identify up to 70% of mCRC non‐responding patients.
All these findings highlight the ability of tumor cells to activate the EGFR signalling cascade at different EGFR downstream levels, independently from the ligand‐mediated activation of the receptor, thus not responding to EGFR drug inhibition. Consequently, in mCRC patients harbouring alterations downstream EGFR, clinical studies are required in order to verify the clinical efficacy of new inhibitors targeting the altered elements of the cascade (alone or in combination with anti‐EGFR MoAbs) or an activated effector, such as mTOR, downstream to the altered element.
Furthermore, an interaction between EGFR and TP53 pathways should also be considered. Wild‐type TP53 can suppress tumor growth acting as a “brake” for the PI3KCA transduction cascade (Kim et al., 2007); and on the other hand, TP53 inactivation could result into EGFR activation (Bheda et al., 2008). TP53 genotyping could therefore have an additional value in optimizing the selection of mCRC patients who would benefit of anti‐EGFR therapies. In accordance with this view, TP53 mutations were significantly associated with response to cetuximab, particularly in mCRC patients carrying wild‐type KRAS (Oden‐Gangloff et al., 2009).
6. Conclusions
This review started by describing the first successful story of a targeted therapy: the treatment of CML patients with the ATP antagonist Imatinib. This success triggered a series of studies that profoundly changed our understanding not only of cancer therapy but also of the classification of cancers, since the same drug, due to its intrinsic mechanism of action, beside inhibiting tyrosine kinase activity, was also found capable of inhibiting other enzymatic activities, such as those expressed by several RTKs including KIT, PDGFRA and PDGFRB. As RTKs' activities are the key pathogenetic element in different tumors, i.e. KIT and PDGFRA in GIST, PDGFRB in DFSP, PDGFRA in hypereosinophilic syndrome, the idea of “one drug for different tumor types” came to the fore.
This new conceptualization also highlighted the need for a mechanism‐based molecular classification of tumor which became more and more significant for therapeutic interventions increasingly based on the development of targeted therapies.
Furthermore, this review emphasizes how the so‐called rare tumors became examples of different activation mechanisms of RTKs and provided the opportunity to devise therapies based on the inhibition of RTKs' deregulated enzymatic activity. In Table S1 (Supplementary material) we provide a comprehensive list of the molecular targets for which a compound has been developed, the stage of the related clinical trials and the associated tumor types. We have listed there more than 65 targeted compounds currently under clinical validations which are aimed not only at the rare tumors discussed here, but also at subgroups of basically all the most common tumor histotypes.
In this review we have also examined the case of targeted therapies in CRC. They provided a model, more inclusive approach to this kind of treatment. In fact, in this tumor model what needs to be considered in order to predict the response to the drug is not just a single element of the signal cascade or pathway, but the whole molecular profile together with the contributions of its components. The transition from a reductionist view, in which only one player in the game gets all the attention, to a more holistic perspective is the premise to a more robust approach, based on system biology, to the prediction of drug response.
Thus, in the very near future the systematic approach of molecular modeling and simulation in the selection of new drugs which is termed “virtual screening” will replace the biologically based, time‐consuming, large‐scale screening of molecules (“compound library screening”), and will become a very useful tool for the rapid ranking and selection of a smaller set of molecules to be tested in the laboratory by traditional functional assays. This new, in silico approach will undoubtedly contribute to paving the way towards truly personalized treatment in the foreseeable future, and we are confident that, through a strong interaction between in silico techniques and high throughput technologies, we will be able to design multi‐drug approaches which are effective in switching off both primary and/or expected secondary mutations.
Finally, our work on targeted therapy in GIST, particularly as regards the understanding the mechanism of the “resistance” phenomenon, has resulted in a novel multidimensional approach, which is also supported by an in silico dimension, to predict clinical results.
In fact, thanks to the availability of the crystallographic coordinates of KIT tyrosine kinase domain, the relevant 3D structure has been determined. In addition, due to the similarities between KIT and PDGFRA/B, the 3D structure of the latter could also be determined. Thanks to molecular modeling – a combination of computer‐based simulation techniques on the basis of thermodynamic parameters which are calculated on a three‐dimensional protein model derived either from crystallography or by homology techniques – we are able to estimate the affinity of a given inhibitor towards its target receptor with great accuracy. Likewise, in the case of acquired resistance, the sensitivity and affinity of these drugs for the “resistant receptor” can be predicted in the same way.
In summary, the lesson we have learnt from the analysis of targeted therapy in GIST pointed out the efficacy of a practice which leads us from bed to bench and back, i.e. the route which moves from the patient's molecular profile to the modeling of the molecular target and the examination of its data, the identification of the best available drug, the evaluation of clinic response and the possibility, even in the presence of relapse and thus of resistance to the treatment, of restarting along this virtuous circle moving from the molecular profiling this time of the reactivated tumor which developed a secondary resistance, to new modeling and data mining which in turn lead to the design of a new treatment with a new drug (Figure 6).
Figure 6.
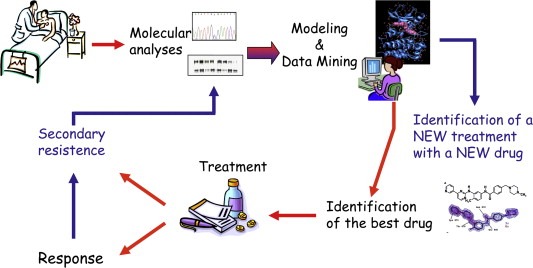
Efficacy of a personalized therapy from bed to bench and back. The route moves from the primary tumor molecular profile of the patient to the modeling of the molecular target and the examination of its data, the identification of the best available drug, the evaluation of clinic response and the possibility, even in the presence of relapse and thus of resistance to the treatment, to restart along this virtuous circle moving from the molecular profiling of the reactivated tumor which developed a secondary resistance, to new modeling and data mining which in turn lead to designing a new treatment with a new drug.
Beside this procedure that is already available, the future holds yet another momentous possibility. This experience could be used to perform a virtual screening of compounds, starting from a 3D model of the available target molecule and then adapting it according to the information drawn from the patients, from real life tumors. Our suggestion is to use such a collection of compounds to select in silico what would be the best in terms of binding energy. Only after this presumably low‐cost first step would the compounds that passed this preliminary selection undergo the usual validation testing in vitro and in vivo in order to design a truly personalized treatment (Figure 7).
Figure 7.
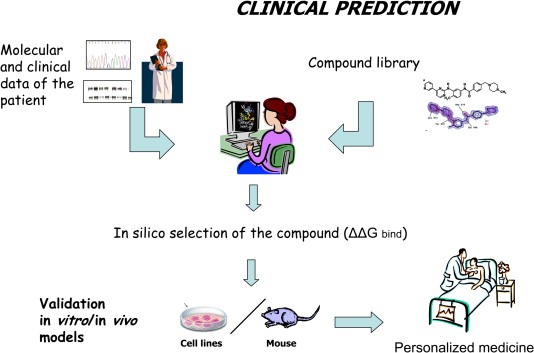
Clinical prediction. Starting from a 3D model of the target molecule (e.g. RTK) according to its molecular status detected in the tumor and patient clinical information, an “in silico” selection of the best drug in terms of binding energy could be performed using a collection of compounds (library). Only the drugs which passed this preliminary selection will undergo the usual validation “in vitro” and “in vivo” tests in order to design a truly personalised treatment.
This would make it possible to achieve an extraordinary result: the three‐dimensional model could in fact try to predict and replicate every kind of target molecule that could be generated even after the insurgence of the tumor's clones, with all the secondary mutations which would cause resistance to the originally devised treatment. Having created a model for each possible new mutation, we could then devise the drug with the greatest affinity, which would obviously be a variation of the original drug (molecular prevention by multi‐drug target).
One major objection to this dynamic therapeutic approach would be that it fails to consider that the possible variations could be innumerable. But, as we have seen before in the discussion of the emerging therapies for GIST and CML, the good news is: they are not. There are in fact specific structural and functional constraints to being resistant to the original compound, and these constrains “make sense of missense” reducing the range of possible mutations to a finite number and thus making a proactive approach to the disease entirely possible (Negri et al., 2009).
Supporting information
Supplementary data
Acknowledgments
The authors thank AIRC for its financial support. Thanks to Ms. Daniela Majerna for providing useful suggestions in editing the manuscript.
Supplemental material 1.
1.1.
Supplementary information for this manuscript can be downloaded at doi: 10.1016/j.molonc.2009.10.003.
Pierotti Marco A., Negri Tiziana, Tamborini Elena, Perrone Federica, Pricl Sabrina, Pilotti Silvana, (2010), Targeted Therapies: The Rare Cancer Paradigm, Molecular Oncology, 4, doi: 10.1016/j.molonc.2009.10.003.
References
- Alman, B.A. , Li, C. , Pajerski, M.E. , Diaz-Cano, S. , Wolfe, H.J. , 1997. Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors). Am. J. Pathol. 151, 329–334. [PMC free article] [PubMed] [Google Scholar]
- Argani, P. , Antonescu, C.R. , Illei, P.B. , Lui, M.Y. , Timmons, C.F. , Newbury, R. , Reuter, V.E. , Garvin, A.J. , Perez-Atayde, A.R. , Fletcher, J.A. , Beckwith, J.B. , Bridge, J.A. , Ladanyi, M. , 2001. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am. J. Pathol. 159, (1) 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argani, P. , Lal, P. , Hutchinson, B. , Lui, M.Y. , Reuter, V.E. , Ladanyi, M. , 2003. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am. J. Surg. Pathol. 27, (6) 750–761. [DOI] [PubMed] [Google Scholar]
- Aubin, J.E. , Bonnelye, E. , 2000 Mar. Osteoprotegerin and its ligand: a new paradigm for regulation of osteoclastogenesis and bone resorption. Medscape Womens Health. 5, (2) 5 [PubMed] [Google Scholar]
- Azizi, A.A. , Haberler, C. , Czech, T. , Gupper, A. , Prayer, D. , Breitschopf, H. , Acker, T. , Slavc, I. , 2006. Vascular-endothelial-growth-factor (VEGF) expression and possible response to angiogenesis inhibitor bevacizumab in metastatic alveolar soft part sarcoma. Lancet Oncol. 7, 533–535. [DOI] [PubMed] [Google Scholar]
- Bheda, A. , Creek, K.E. , Pirisi, L. , 2008. Loss of p53 induces epidermal growth factor receptor promoter activity in normal human keratinocytes. Oncogene. 27, 4315–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blay, J.Y. , El Sayadi, H. , Thiesse, P. , Garret, J. , Ray-Coquard, I. , 2008. Complete response to Imatinib in relapsing pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). Ann. Oncol. 19, 821–822. [DOI] [PubMed] [Google Scholar]
- Bozulic, L., Morin, P., Jr., Hunter, T., Hemmings, B.A., 2007. Meeting report: targeting the kinome — 20years of tyrosine kinase inhibitor research in Basel. Sci. STKE, pe8. [DOI] [PubMed]
- Buchdunger, E. , Zimmermann, J. , Mett, H. , Meyer, T. , Müller, M. , Regenass, U. , Lydon, N.B. , 1995. Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc. Natl. Acad. Sci. U.S.A. 92, 2558–2562. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Buchdunger, E. , Cioffi, C.L. , Law, N. , Stover, D. , Ohno-Jones, S. , Druker, B.J. , Lydon, N.B. , 2000. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 295, 139–145. [PubMed] [Google Scholar]
- Carney, J.A. , Stratakis, C.A. , 2002. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am. J. Med. Genet. 108, 132–139. [DOI] [PubMed] [Google Scholar]
- Casali, P.G. , Messina, A. , Stacchiotti, S. , Tamborini, E. , Crippa, F. , Gronchi, A. , Orlandi, R. , Ripamonti, C. , Spreafico, C. , Bertieri, R. , 2004. Imatinib mesylate in chordoma. Cancer. 101, 2086–2097. [DOI] [PubMed] [Google Scholar]
- Chugh, R. , Tawbi, H. , Lucas, D.R. , Biermann, J.S. , Schuetze, S.M. , Baker, L.H. , 2007. Chordoma: the nonsarcoma primary bone tumor. Oncologist. 12, 1344–1350. [DOI] [PubMed] [Google Scholar]
- Corless, C.L. , Heinrich, M.C. , 2008. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu. Rev. Pathol. 3, 557–586. [DOI] [PubMed] [Google Scholar]
- Corless, C.L. , Fletcher, J.A. , Heinrich, M.C. , 2004. Biology of gastrointestinal stromal tumors. J. Clin. Oncol. 22, 3813–3825. [DOI] [PubMed] [Google Scholar]
- de Bree, E. , Keus, R. , Melissas, J. , Tsiftsis, D. , van Coevorden, F. , 2009. Desmoid tumors: need for an individualized approach. Expert Rev. Anticancer Ther. 9, 525–535. [DOI] [PubMed] [Google Scholar]
- Dempke, W. , Rie, C. , Grothey, A. , Schmoll, H.J. , 2001. Cyclooxygenase-2: a novel target for cancer chemotherapy?. J. Cancer Res. Clin. Oncol. 127, 411–417. [DOI] [PubMed] [Google Scholar]
- Dewar, A.L. , Cambareri, A.C. , Zannettino, A.C. , Miller, B.L. , Doherty, K.V. , Hughes, T.P. , Lyons, A.B. , 2005. Macrophage colony-stimulating factor receptor c-fms is a novel target of Imatinib. Blood. 105, 3127–3132. [DOI] [PubMed] [Google Scholar]
- Di Nicolantonio, F. , Martini, M. , Molinari, F. , Sartore-Bianchi, A. , Arena, S. , Saletti, P. , De Dosso, S. , Mazzucchelli, L. , Frattini, M. , Siena, S. , 2008. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 26, 5705–5712. [DOI] [PubMed] [Google Scholar]
- Dimitropoulos, V.A. , 2008. Dermatofibrosarcoma protuberans. Dermatol. Ther. 21, 428–432. [DOI] [PubMed] [Google Scholar]
- Druker, B.J. , 2001. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Erkizan, H.V. , Kong, Y. , Merchant, M. , Schlottmann, S. , Barber-Rotenberg, J.S. , Yuan, L. , Abaan, O.D. , Chou, T.H. , Dakshanamurthy, S. , Brown, M.L. , Uren, A. , Toretsky, J.A. , 2009. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat. Med. 7, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, C.D. , 2002. Clinicopathologic correlation in gastrointestinal stromal tumors. Hum. Pathol. 33, 455 [DOI] [PubMed] [Google Scholar]
- Frattini, M. , Saletti, P. , Romagnani, E. , Martin, V. , Molinari, F. , Ghisletta, M. , Camponovo, A. , Etienne, L.L. , Cavalli, F. , Mazzucchelli, L. , 2007. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer. 97, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettsch, W.G. , Bos, S.D. , Breekveldt-Postma, N. , Casparie, M. , Herings, R.M. , Hogendoorn, P.C. , 2005. Incidence of gastrointestinal stromal tumours is underestimated: results of a nation-wide study. Eur. J. Cancer. 41, 2868–2872. [DOI] [PubMed] [Google Scholar]
- Goldberg, J. , Demetri, G.D. , Choy, E. , Rosen, L. , Pappo, A. , Dubois, S. , Geller, J. , Chai, F. , Ferrari, D. , Wagner, A.J. , 2009. Preliminary results from a phase II study of ARQ 197 in patients with microphthalmia transcription factor family (Mit)-associated tumors. 2009 ASCO Annual Meeting. Abstract No: 10502. J. Clin. Oncol. 27, 15s [Google Scholar]
- Greco, A. , Roccato, E. , Miranda, C. , Cleris, L. , Formelli, F. , Pierotti, M.A. , 2001. Growth-inhibitory effect of STI571 on cells transformed by the COL1A1/PDGFB rearrangement. Int. J. Cancer. 92, 354–360. [DOI] [PubMed] [Google Scholar]
- Groffen, J. , Stephenson, J.R. , Heisterkamp, N. , de Klein, A. , Bartram, C.R. , Grosveld, G. , 1984. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 36, 93–99. [DOI] [PubMed] [Google Scholar]
- Heinrich, M.C. , Rubin, B.P. , Longley, B.J. , Fletcher, J.A. , 2002. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alteration. Hum. Pathol. 33, 484–495. [DOI] [PubMed] [Google Scholar]
- Heinrich, M.C. , McArthur, G.A. , Demetri, G.D. , Joensuu, H. , Bono, P. , Herrmann, R. , Hirte, H. , Cresta, S. , Koslin, D.B. , Corless, C.L. , 2006 Mar 1. Clinical and molecular studies of the effect of Imatinib on advanced aggressive fibromatosis (desmoid tumor). J. Clin. Oncol. 24, (7) 1195–1203. [DOI] [PubMed] [Google Scholar]
- Heinrich, M.C. , Maki, R.G. , Corless, C.L. , Antonescu, C.R. , Harlow, A. , Griffith, D. , Town, A. , McKinley, A. , Ou, W.B. , Fletcher, J.A. , Fletcher, C.D. , Huang, X. , Cohen, D.P. , Baum, C.M. , Demetri, G.D. , 2008. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in Imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 26, 5352–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich, M.C. , Joensuu, H. , Demetri, G.D. , Corless, C.L. , Apperley, J. , Fletcher, J.A. , Soulieres, D. , Dirnhofer, S. , Harlow, A. , Town, A. , 2008. Phase II, open-label study evaluating the activity of Imatinib in treating life-threatening malignancies known to be associated with Imatinib-sensitive tyrosine kinases. Imatinib Target Exploration Consortium Study B2225. Clin. Cancer Res. 14, 2717–2725. [DOI] [PubMed] [Google Scholar]
- Hof, H. , Welzel, T. , Debus, J. , 2006. Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Onkologie. 29, 572–574. [DOI] [PubMed] [Google Scholar]
- Huang, L. , Cheng, Y.Y. , Chow, L.T. , Zheng, M.H. , Kumta, S.M. , 2003. Receptor activator of NF-kappaB ligand (RANKL) is expressed in chondroblastoma: possible involvement in osteoclastic giant cell recruitment. Mol. Pathol. 56, (2) 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano, A. , Follana, P. , Caroli, F.X. , Badetti, J.L. , Benchimol, D. , Garnier, G. , Gugenheim, J. , Haudebourg, J. , Keslair, F. , Lesbats, G. , 2008. Cetuximab shows activity in colorectal cancer patients with tumors for which FISH analysis does not detect an increase in EGFR gene copy number. Ann. Surg. Oncol. 15, 649–654. [DOI] [PubMed] [Google Scholar]
- Jimeno, A. , Messersmith, W.A. , Hirsch, F.R. , Franklin, W.A. , Eckhardt, S.G. , 2009. KRAS mutations and sensitivity to epidermal growth factor receptor inhibitors in colorectal cancer: practical application of patient selection. J. Clin. Oncol. 27, 1130–1136. [DOI] [PubMed] [Google Scholar]
- Jonker, D.J. , O'Callaghan, C.J. , Karapetis, C.S. , Zalcberg, J.R. , Tu, D. , Au, H.J. , Berry, S.R. , Krahn, M. , Price, T. , Simes, R.J. , 2007. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 357, 2040–2048. [DOI] [PubMed] [Google Scholar]
- Kim, J.S. , Lee, C. , Bonifant, C.L. , Ressom, H. , Waldman, T. , 2007. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol. Cell. Biol. 27, 662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka, J.B. , Watanabe, S.M. , Witte, O.N. , 1984. An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell. 37, 1035–1042. [DOI] [PubMed] [Google Scholar]
- Ladanyi, M. , Lui, M.Y. , Antonescu, C.R. , Krause-Boehm, A. , Meindl, A. , Argani, P. , Healey, J.H. , Ueda, T. , Yoshikawa, H. , Meloni-Ehrig, A. , Sorensen, P.H. , Mertens, F. , Mandahl, N. , van den Berghe, H. , Sciot, R. , Dal Cin, P. , Bridge, J. , 2001. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 20, (1) 48–57. [DOI] [PubMed] [Google Scholar]
- Lamoureux, F. , Moriceau, G. , Picarda, G. , Rousseau, J. , Trichet, V. , Rédini, F. , 2009. Regulation of osteoprotegerin pro- or anti-tumoral activity by bone tumor microenvironment. Biochim. Biophys. Acta. Sep 2 [DOI] [PubMed] [Google Scholar]
- Lasota, J. , Miettinen, M. , 2008. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 53, 245–266. [DOI] [PubMed] [Google Scholar]
- Lazar, A.J. , Das, P. , Tuvin, D. , Korchin, B. , Zhu, Q. , Jin, Z. , Warneke, C.L. , Zhang, P.S. , Hernandez, V. , Lopez-Terrada, D. , 2007. Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin. Cancer Res. 13, 7314–7321. [DOI] [PubMed] [Google Scholar]
- Lazar, A.J. , Tuvin, D. , Hajibashi, S. , Habeeb, S. , Bolshakov, S. , Mayordomo-Aranda, E. , Warneke, C.L. , Lopez-Terrada, D. , Pollock, R.E. , Lev, D. , 2008. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am. J. Pathol. 173, 1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemm, D. , Muegge, L.O. , Hoeffken, K. , Aklan, T. , Mentzel, T. , Thorwarth, M. , Schultze-Mosgau, S. , 2008. Oral. Maxillofac. Surg. 12, 209–213. [DOI] [PubMed] [Google Scholar]
- Lemm, D. , Mügge, L.O. , Mentzel, T. , Höffken, K. , 2009. Current treatment options in dermatofibrosarcoma protuberans. J. Cancer Res. Clin. Oncol. 135, 653–665. [DOI] [PubMed] [Google Scholar]
- Lenz, H.J. , Van Cutsem, E. , Khambata-Ford, S. , Mayer, R.J. , Gold, P. , Stella, P. , Mirtsching, B. , Cohn, A.L. , Pippas, A.W. , Azarnia, N. , 2006. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J. Clin. Oncol. 24, 4914–4921. [DOI] [PubMed] [Google Scholar]
- Li, C. , Bapat, B. , Alman, B.A. , 1998. Adenomatous polyposis coli gene mutation alters proliferation through its beta-catenin-regulatory function in aggressive fibromatosis (desmoid tumor). Am. J. Pathol. 153, 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F.P. , Fletcher, J.A. , Heinrich, M.C. , Garber, J.E. , Sallan, S.E. , Curiel-Lewandrowski, C. , Duensing, A. , van de Rijn, M. , Schnipper, L.E. , Demetri, G.D. , 2005. Familial gastrointestinal stromal tumor syndrome: phenotypic and molecular features in a kindred. J. Clin. Oncol. 23, 2735–2743. [DOI] [PubMed] [Google Scholar]
- Liegl, B. , Leithner, A. , Bauernhofer, T. , Windhager, R. , Guelly, C. , Regauer, S. , Beham, A. , 2006. Immunohistochemical and mutational analysis of PDGF and PDGFR in desmoid tumours: is there a role for tyrosine kinase inhibitors in c-kit-negative desmoid tumours?. Histopathology. 49, 576–581. [DOI] [PubMed] [Google Scholar]
- Lindén, O. , Stenberg, L. , Kjellén, E. , 2009. Regression of cervical spinal cord compression in a patient with chordoma following treatment with cetuximab and gefitinib. Acta Oncol. 48, 158–159. [DOI] [PubMed] [Google Scholar]
- Llombart, B. , Sanmartín, O. , López-Guerrero, J.A. , Monteagudo, C. , Serra, C. , Requena, C. , Poveda, A. , Vistós, J.L. , Almenar, S. , Llombart-Bosch, A. , 2009. Dermatofibrosarcoma protuberans: clinical, pathological, and genetic (COL1A1-PDGFB) study with therapeutic implications. Histopathology. 54, 860–872. [DOI] [PubMed] [Google Scholar]
- Loupakis, F. , Ruzzo, A. , Cremolini, C. , Vincenzi, B. , Salvatore, L. , Santini, D. , Masi, G. , Stasi, I. , Canestrari, E. , Rulli, E. , 2009. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br. J. Cancer. 101, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loupakis, F. , Pollina, L. , Stasi, I. , Ruzzo, A. , Scartozzi, M. , Santini, D. , Masi, G. , Graziano, F. , Cremolini, C. , 2009. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J. Clin. Oncol. 27, 2622–2629. [DOI] [PubMed] [Google Scholar]
- Lucas, D.R. , Shroyer, K.R. , McCarthy, P.J. , Markham, N.E. , Fujita, M. , Enomoto, T.E. , 1997. Desmoid tumor is a clonal cellular proliferation: PCR amplification of HUMARA for analysis of patterns of X-chromosome inactivation. Am. J. Surg. Pathol. 21, (3) 306–311. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Solimini, N.L. , Elledge, S. , 2009. Principles of cancer therapies: oncogene and non oncogene addiction. Cell. 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace, J. , Sybil Biermann, J. , Sondak, V. , McGinn, C. , Hayes, C. , Thomas, D. , Baker, L. , 2002. Response of extraabdominal desmoid tumors to therapy with Imatinib mesylate. Cancer. 95, 2373–2379. [DOI] [PubMed] [Google Scholar]
- Maki, R.G. , Awan, R.A. , Dixon, R.H. , Jhanwar, S. , Antonescu, C.R. , 2002. Differential sensitivity to Imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int. J. Cancer. 100, 623–626. [DOI] [PubMed] [Google Scholar]
- Martin, R.C. , Osborne, D.L. , Edwards, M.J. , Wrightson, W. , McMasters, K.M. , 2000. Giant cell tumor of tendon sheath, tenosynovial giant cell tumor, and pigmented villonodular synovitis: defining the presentation, surgical therapy and recurrence. Oncol. Rep. 7, 413–419. [PubMed] [Google Scholar]
- McArthur, G.A. , Demetri, G.D. , van Oosterom, A. , Heinrich, M.C. , Debiec-Rychter, M. , Corless, C.L. , Nikolova, Z. , Dimitrijevic, S. , Fletcher, J.A. , 2005. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with Imatinib: Imatinib Target Exploration Consortium Study B2225. J. Clin. Oncol. 23, 866–873. [DOI] [PubMed] [Google Scholar]
- Mendenhall, W.M. , Zlotecki, R.A. , Morris, C.G. , Hochwald, S.N. , Scarborough, M.T. , 2005. Aggressive fibromatosis. Am. J. Clin. Oncol. 28, 211–215. [DOI] [PubMed] [Google Scholar]
- Mendenhall, W.M. , Mendenhall, C.M. , Reith, J.D. , Scarborough, M.T. , Gibbs, C.P. , Mendenhall, N.P. , 2006. Pigmented villonodular synovitis. Am. J. Clin. Oncol. 29, 548–550. [DOI] [PubMed] [Google Scholar]
- Miettinen, M. , Lasota, J. , 2001. Gastrointestinal stromal tumors—definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 438, 1–12. [DOI] [PubMed] [Google Scholar]
- Miozzo, M. , Dalprà, L. , Riva, P. , Volontà, M. , Macciardi, F. , Pericotti, S. , Tibiletti, M.G. , Cerati, M. , Rohde, K. , Larizza, L. , 2000. A tumor suppressor locus in familial and sporadic chordoma maps to 1p36. Int. J. Cancer. 87, 68–72. [PubMed] [Google Scholar]
- Mirra, J.M. , Nelson, S.D. , Della Rocca, C. , Mertens, F. , 2002. Chordoma. In Fletcher C.D.M., Unni K.K., Mertens F.(Eds.), Pathology and Genetics of Tumors of Soft Tissue and Bone. World Health Organization Classification of Tumors. IARC Press; Lyon: 316–317. [Google Scholar]
- Mizutani, K. , Tamada, Y. , Hara, K. , Tsuzuki, T. , Saeki, H. , Tamaki, K. , Matsumoto, Y. , 2004. Imatinib mesylate inhibits the growth of metastatic lung lesions in a patient with dermatofibrosarcoma protuberans. Br. J. Dermatol. 151, 235–237. [DOI] [PubMed] [Google Scholar]
- Morgan, T. , Atkins, G.J. , Trivett, M.K. , Johnson, S.A. , Kansara, M. , Schlicht, S.L. , Slavin, J.L. , Simmons, P. , Dickinson, I. , Powell, G. , Choong, P.F. , Holloway, A.J. , Thomas, D.M. , 2005. Molecular profiling of giant cell tumor of bone and the osteoclastic localization of ligand for receptor activator of nuclear factor kappaB. Am. J. Pathol. 167, (1) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri, T. , Pavan, G.M. , Virdis, E. , Greco, A. , Fermeglia, M. , Sandri, M. , Pricl, S. , Pierotti, M.A. , Pilotti, S. , Tamborini, E. , 2009. T670X KIT mutations in gastrointestinal stromal tumors: making sense of missense. J. Natl. Cancer Inst. 101, 194–204. [DOI] [PubMed] [Google Scholar]
- Nowell, P.C. , Hungerford, D.A. , 1960. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 25, 85–109. [PubMed] [Google Scholar]
- Oden-Gangloff, A. , Di Fiore, F. , Bibeau, F. , Lamy, A. , Bougeard, G. , Charbonnier, F. , Blanchard, F. , Tougeron, D. , Ychou, M. , Boissière, F. , 2009. TP53 mutations predict disease control in metastatic colorectal cancer treated with cetuximab-based chemotherapy. Br. J. Cancer. 100, 1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone, F. , Lampis, A. , Orsenigo, M. , Di Bartolomeo, M. , Gevorgyan, A. , Losa, M. , Frattini, M. , Riva, C. , Andreola, S. , Bajetta, E. , 2009. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann. Oncol. 20, 84–90. [DOI] [PubMed] [Google Scholar]
- Personeni, N. , Fieuws, S. , Piessevaux, H. , De Hertogh, G. , De Schutter, J. , Biesmans, B. , De Roock, W. , Capoen, A. , Debiec-Rychter, M. , Van Laethem, J.L. , 2008. Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin. Cancer Res. 14, 5869–5876. [DOI] [PubMed] [Google Scholar]
- Prenen, H. , De Schutter, J. , Jacobs, B. , De Roock, W. , Biesmans, B. , Claes, B. , Lambrechts, D. , Van Cutsem, E. , Tejpar, S. , 2009. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin. Cancer Res. 15, 3184–3188. [DOI] [PubMed] [Google Scholar]
- Rao, S. , Cunningham, D. , de Gramont, A. , Scheithauer, W. , Smakal, M. , Humblet, Y. , Kourteva, G. , Iveson, T. , Andre, T. , Dostalova, J. , 2004. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J. Clin. Oncol. 22, 3950–3957. [DOI] [PubMed] [Google Scholar]
- Rinehart, J. , Adjei, A.A. , Lorusso, P.M. , Waterhouse, D. , Hecht, J.R. , Natale, R.B. , Hamid, O. , Varterasian, M. , Asbury, P. , Kaldjian, E.P. , 2004. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 22, 4456–4462. [DOI] [PubMed] [Google Scholar]
- Rowley, J.D. , 1973. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 243, 290–293. [DOI] [PubMed] [Google Scholar]
- Saltz, L.B. , Meropol, N.J. , Loehrer, P.J. , SrNeedle, M.N. , Kopit, J. , Mayer, R.J. , 2004. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J. Clin. Oncol. 22, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Sartore-Bianchi, A. , Martini, M. , Molinari, F. , Veronese, S. , Nichelatti, M. , Artale, S. , Di Nicolantonio, F. , Saletti, P. , De Dosso, S. , Mazzucchelli, L. , 2009. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 69, 1851–1857. [DOI] [PubMed] [Google Scholar]
- Scotlandi, K. , Benini, S. , Sarti, M. , Serra, M. , Lollini, P.L. , Maurici, D. , Picci, P. , Manara, M.C. , Baldini, N. , 1996. Insulin-like growth factor I receptor-mediated circuit in Ewing's sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 56, 4570–4574. [PubMed] [Google Scholar]
- Shaw, R.J. , Cantley, L.C. , 2006. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 441, 424–430. [DOI] [PubMed] [Google Scholar]
- Signoroni, S. , Frattini, M. , Negri, T. , Pastore, E. , Tamborini, E. , Casieri, P. , Orsenigo, M. , Da Riva, L. , Radice, P. , Sala, P. , 2008. Cyclooxygenase-2 and platelet-derived growth factor receptors as potential targets in treating aggressive fibromatosis. Clin. Cancer Res. 13, 5034–5040. Erratum in: Clin. Cancer Res. 2008;14(13):4354 [DOI] [PubMed] [Google Scholar]
- Sjöblom, T. , Shimizu, A. , O'Brien, K.P. , Pietras, K. , Dal Cin, P. , Buchdunger, E. , Dumanski, J.P. , Ostman, A. , Heldin, C.H. , 2001. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 61, 5778–5783. [PubMed] [Google Scholar]
- Skubitz, K.M. , Manivel, J.C. , Clohisy, D.R. , Frolich, J.W. , 2009. Response of Imatinib-resistant extra-abdominal aggressive fibromatosis to sunitinib: case report and review of the literature on response to tyrosine kinase inhibitors. Cancer Chemother. Pharmacol. 64, 635–640. [DOI] [PubMed] [Google Scholar]
- Stacchiotti, S., Bertulli, R., Grosso, F., Dileo, P., Badalamenti, G., Messina, A., Gronchi, A., Tamborini, E., Pilotti, S., Bidoli, P., et al., 2007. Antitumor activity of Imatinib plus cisplatin in three patients with advanced chordoma. Proc. 12th Ann Connective Tissue. Oncology Society Meet., Abs. # 725, p. 58.
- Stacchiotti, S. , Tamborini, E. , Marrari, A. , Brich, S. , Rota, S.A. , Orsenigo, M. , Crippa, F. , Morosi, C. , Gronchi, A. , Pierotti, M.A. , 2009. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin. Cancer Res. 15, 1096–1104. [DOI] [PubMed] [Google Scholar]
- Stacchiotti, S. , Marrari, A. , Tamborini, E. , Palassini, E. , Virdis, E. , Messina, A. , Crippa, F. , Morosi, C. , Gronchi, A. , Pilotti, S. , 2009. Response to Imatinib plus sirolimus in advanced chordoma. Ann. Oncol. 20, 1886–1894. [DOI] [PubMed] [Google Scholar]
- Stam, K. , 1985. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N. Engl. J. Med. 313, 1429–1433. [DOI] [PubMed] [Google Scholar]
- Stockwin, L.H. , Vistica, D.T. , Kenney, S. , Schrump, D.S. , Butcher, D.O. , Raffeld, M. , Shoemaker, R.H. , 2009. Gene expression profiling of alveolar soft-part sarcoma (ASPS). BMC Cancer. 15, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel, J.M. , Kimmelman, A.C. , Ying, H. , Nabioullin, R. , Ponugoti, A.H. , Wiedemeyer, R. , Stegh, A.H. , Bradner, J.E. , Ligon, K.L. , Brennan, C. , Chin, L. , DePinho, R.A. , 2007. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 318, (5848) 287–290. [DOI] [PubMed] [Google Scholar]
- Storm, P.B. , Magge, S.N. , Kazahaya, K. , Sutton, L.N. , 2007. Cervical chordoma in a patient with tuberous sclerosis presenting with shoulder pain. Pediatr. Neurosurg. 43, 167–169. [DOI] [PubMed] [Google Scholar]
- Subbiah, V. , Anderson, P. , Lazar, A.J. , Burdett, E. , Raymond, K. , Ludwig, J.A. , 2009. Ewing's sarcoma: standard and experimental treatment options. Curr. Treat. Options Oncol. 1–2, 126–140. [DOI] [PubMed] [Google Scholar]
- Tamborini, E. , Pricl, S. , Negri, T. , Lagonigro, M.S. , Miselli, F. , Greco, A. , Gronchi, A. , Casali, P.G. , Ferrone, M. , Fermeglia, M. , Carbone, A. , Pierotti, M.A. , Pilotti, S. , 2006. Functional analyses and molecular modeling of two c-Kit mutations responsible for Imatinib secondary resistance in GIST patients. Oncogene. 25, 6140–6146. [DOI] [PubMed] [Google Scholar]
- Tamborini, E. , Negri, T. , Miselli, F. , Lagonigro, M.S. , Pricl, S. , Pilotti, S. , 2006. Re: response of a KIT-positive extra-abdominal fibromatosis to Imatinib mesylate and KIT genetic analysis. J. Natl. Cancer Inst. 98, 1583–1584. [DOI] [PubMed] [Google Scholar]
- Tamborini, E. , Miselli, F. , Negri, T. , Lagonigro, M.S. , Staurengo, S. , Dagrada, G.P. , Stacchiotti, S. , Pastore, E. , Gronchi, A. , Perrone, F. , 2006. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin. Cancer Res. 12, 6920–6928. [DOI] [PubMed] [Google Scholar]
- Thomas, D.M. , Skubitz, K.M. , 2009. Giant cell tumour of bone. Curr. Opin. Oncol. 21, (4) 338–344. [DOI] [PubMed] [Google Scholar]
- Tsuda, M. , Davis, I.J. , Argani, P. , Shukla, N. , McGill, G.G. , Nagai, M. , Saito, T. , Laé, M. , Fisher, D.E. , Ladanyi, M. , 2007. TFE3 fusions activate MET signaling by transcriptional up-regulation, defining another class of tumors as candidates for therapeutic MET inhibition. Cancer Res. 67, 919–929. [DOI] [PubMed] [Google Scholar]
- Vistica, D.T. , Hollingshead, M. , Borgel, S.D. , Kenney, S. , Stockwin, L.H. , Raffeld, M. , Schrump, D.S. , Burkett, S. , Stone, G. , Butcher, D.O. , 2009. Therapeutic vulnerability of an in vivo model of alveolar soft part sarcoma (ASPS) to antiangiogenic therapy. J. Pediatr. Hematol. Oncol. 31, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger, P.M. , Yu, Z. , Kowalski, D. , Joe, J. , Manger, P. , Psyrri, A. , Sasaki, C.T. , 2005. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch. Otolaryngol. Head Neck Surg. 131, 707–711. [DOI] [PubMed] [Google Scholar]
- West, R.B. , Rubin, B.P. , Miller, M.A. , Subramanian, S. , Kaygusuz, G. , Montgomery, K. , Zhu, S. , Marinelli, R.J. , De Luca, A. , Downs-Kelly, E. , 2006. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc. Natl. Acad. Sci. U.S.A. 103, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization Classification of Tumours, 2002. Pathology and Genetics. Tumours of soft Tissue and bone IARC Press; Lyon: [Google Scholar]
- Yuzawa, S. , Opatowsky, Y. , Zhang, Z. , Mandiyan, V. , Lax, I. , Schlessinger, J. , 2007. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 130, 323–334. [DOI] [PubMed] [Google Scholar]
- Zimmermann, J. , Caravatti, G. , Mett, H. , Meyer, T. , Müller, M. , Lydon, N.B. , Fabbro, D. , 1996. Phenylamino-pyrimidine (PAP) derivatives: a new class of potent and selective inhibitors of protein kinase C (PKC). Arch. Pharm. (Weinheim). 329, 371–376. [DOI] [PubMed] [Google Scholar]
- Zuckerman, D.S. , Clark, W. , 2008. Systemic therapy for metastatic colorectal cancer. Cancer. 112, 1879–1891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data