

Abstract
Cytotoxicity of the topoisomerase II (topoII) poison etoposide has been ascribed to the persistent covalent trapping of topoII in DNA cleavage complexes that become lethal as cells replicate their DNA. However, short term etoposide treatment also leads to subsequent cell death, suggesting that the lesions that lead to cytotoxicity arise rapidly and prior to the onset DNA replication. In the present study 1h treatment with 25μM etoposide was highly toxic and initiated a double‐stranded DNA damage response as reflected by the recruitment of ATM, MDC1 and DNA‐PKcs to γH2AX foci. While most DNA breaks were rapidly repaired upon withdrawal of the etoposide treatment, the repair machinery remained engaged in foci for at least 24h following withdrawal. TopoII siRNA ablation showed the etoposide toxicity and γH2AX response to correlate with the inability of the cell to correct topoIIα‐initiated DNA damage. γH2AX induction was resistant to the inhibition of DNA replication and transcription, but was increased by pre‐treatment with the histone deacetylase inhibitor trichostatin A. These results link the lethality of etoposide to the generation of persistent topoIIα‐dependent DNA defects within topologically open chromatin domains.
Keywords: Topoisomerase II, Etoposide, Double-stranded DNA breaks, Non-homologous end joining
Abbreviations
- DSB
double‐stranded DNA breaks
- TopoII
topoisomerase II
- CC
cleavage complex
- NHEJ
non‐homologous DNA end joining
- DNA‐PKcs
DNA‐dependent protein kinase catalytic subunit
1. Introduction
Class II topoisomerases (topoIIα and topoIIβ) act to decatenate DNA through the passage of an intact DNA duplex through a transient double‐stranded DNA break in the second duplex. This decatenation is crucial to DNA metabolism, most notably during DNA synthesis, transcription and chromosome resolution during mitosis (Wang, 2002). TopoIIα and topoIIβ have distinct roles in the cell. TopoIIβ appears to have a specific role in transcription (Ju et al., 2006), while topoIIα is important in chromosome segregation and replication (McClendon and Osheroff, 2007; Wang, 2002). TopoIIα has also been implicated in nuclear matrix organization and higher order chromatin remodeling (Christensen et al., 2002; Montecucco and Biamonti, 2007).
Due to their intimate involvement in DNA metabolism, the topoisomerases have been prominent targets of chemotherapeutic strategies targeting cancer cells. One class of topoII poisons, the epipodophyllotoxins, which include etoposide (VP‐16) and VM‐26, bind reversibly to topoIIα/β to block religation of the cleaved DNA strand following passage of the intact duplex. This traps the topoII in complexes in which the DNA strands are tethered by their 5′ ends through the topoII dimer, with one end of the DNA attached to each topoII monomer (Jensen and Sehested, 1997). As the binding of etoposide with topoII is transient and non‐covalent, enzyme activity is rapidly restored upon clearance of the compound and religation of DNA is completed (D'Arpa and Liu, 1989).
Thus the prevalent hypothesis for etoposide‐mediated cell death has been that trapped topoII cleavage complexes (CC) are not directly cytotoxic, but become lethal upon the release of double‐stranded DNA breaks (DSBs) from trapped CC as the cells attempt to replicate their DNA. Supports for this hypothesis include the resistance of yeast to etoposide treatment when maintained in G1 (Nitiss et al., 1992) and acute etoposide sensitivity of cells synchronized to S phase (Gorczyca et al., 1993).
By contrast, it also has been known for some time that short term (1h) etoposide treatment of proliferating cells is sufficient to induce subsequent cell death. This suggests a rapid production of DNA lesions that lead to subsequent cell death via apoptosis despite the presumptive resealing of post‐cleavage topoII–DNA complexes upon etoposide withdrawal (Kalwinsky et al., 1983; Long et al., 1985). Recent studies have shown that etoposide elicits the rapid induction of γH2AX and ATM in foci that reflect the detection of double‐stranded damage by the cell prior to the onset of DNA replication (Foster and Downs, 2005; Kantidze et al., 2006; Rogakou et al., 2000; Sedelnikova et al., 2002). γH2AX phosphorylation has been reported to be reversed upon repair of various types of DNA lesions (Chowdhury et al., 2005; Keogh et al., 2006), although this has not been examined thoroughly in response to etoposide.
The two major mechanisms for the repair of double‐stranded DNA breaks are homologous recombination, which predominates in G2 when sister chromatids become aligned, and non‐homologous DNA end joining (NHEJ), which predominates in G1 (Shrivastav et al., 2008). NHEJ involves DNA end recognition by the Ku antigen and DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), followed by recruitment of a repair complex that includes artemis, XRCC4 and DNA ligase IV (Lieber, 2008). In response to γ irradiation and other DSB‐inducing agents, NHEJ proceeds in a biphasic manner, with rapid repair of simple breaks, followed by slower repair of complex breaks that require additional processing prior to religation (Foray et al., 1997; Goodarzi et al., 2008).
In this study we probed the underlying basis for the cytotoxicity of short term (1h) etoposide treatment. We find that the rapid appearance of γH2AX foci results from the generation of DSBs to which ATM, MDC1 and catalytically active DNA‐PKcs are recruited. These foci persist for at least 24h following withdrawal of the etoposide treatment, despite extensive repair of the DNA. Generation of the long‐lived foci was predominately dependent on topoIIα and was enhanced by co‐treatment with a histone deacetylase inhibitor. These results provide strong evidence that etoposide cytotoxicity results from the persistence DSBs or defective DSBs rejoining originating in topoIIα CC within the euchromatin of proliferating cells.
2. Experimental procedures
2.1. Reagents
Etoposide/VP‐16 (25μM or as indicated), aphidicolin (0.5μg/ml) and 5,6‐dichloro‐1‐β‐d‐ribofuranosylbenzimidazole (DRB; 250μM), camptothecin (1μM), αamanitin (100μg/ml), trichostatin A and actinomycin D (10μg/ml) were from Sigma–Aldrich (St. Louis, MO). ICRF‐193 (2μM) was from MP Biomedicals (Irvine, CA). The rabbit PARP‐1 antibody (H250), which recognizes both full‐length and caspase cleaved PARP‐1, was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit ATM‐pS1981 and ATM‐S1981 antibodies were obtained from Rockland (Gilbertsville, PA). DNA‐PKcs‐pT2609 and the MDC1 antibodies were from Abcam (Cambridge, MA). The mouse γH2AX and goat H4 antibodies were from Upstate (Chicago, IL) and the mouse ATR antibody was from Novus Biologicals (Littletown, CO). The topoIIα and the topoIIβ antibodies were from Bethyl (Montgomery, TX) and BD Biosciences (Franklin Lakes, NJ), respectively. The hnRNPU antibody was from Immunoquest (Cleveland, UK) and the rabbit H2A antibody from Cell Signaling (Danvers, MA). The mouse hsp70 antibody was obtained from Stressgen (Vancouver, BC). siRNA were obtained from Dharmacon (topoIIα, AAG ACU GUC UGU UGA AAG A; topoIIβ, GAA GUU GUC UGU UGA GAG A; control, ACU ACC GUU GUU AUA GGU G).
2.2. Cell culture and treatments
HCT116 cells were maintained in McCoy's 5A medium containing 10% fetal bovine serum, while HeLa cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum. Cells were maintained in 5% CO2 and 37°C. Etoposide treatment was for 1h. Withdrawal was initiated by washing the cells five times (three times 2min in PBS with 10% BSA, followed by two times 5min in complete medium, all at 37°C). γ irradiation was performed with a Pantak irradiator (0.9Gy/min). For gene silencing 60pmol of oligonucleotide RNA was transfected in 3.5cm dishes using oligofectamine (Invitrogen, Carlsbad, CA) for 48h. For survival assays, cells seeded as single cells (5000 for control cells, 25,000 for test cells per well in 12‐well plates) 24h prior to treatment. Survival at day 7/14 was measured by formazan formation using the CellTiter assay (Promega Madison, WI) or by counting Trypan Blue‐stained fixed colonies.
2.3. Indirect immunofluorescence
Following treatment, cells seeded on glass coverslips were fixed with 4% paraformaldehyde, permeabilized in TBS/0.1% Triton X‐100 and blocked in 5% goat serum in TBS/0.1% Tween. Incubations with the primary antibodies were performed for 1h in TBS/0.1% Tween. After four rinses with TBS/0.1% Tween, secondary antibodies (donkey anti‐mouse Alexa 594 and donkey anti‐rabbit Alexa 488 antibodies; Invitrogen, Carlsbad, CA) were added for another 0.5–1h. Following washes, the coverslips were mounted for microscopy in DAPI‐containing solution. Co‐localization experiments were performed on a Bio‐Rad MRC 1024 confocal microscope using a 60× oil immersion objective whereas wide field images were acquired with a fluorescence microscope equipped with a 40× objective. Quantification of indirect immunofluorescence signal intensity was performed using Image J. Signal threshold was defined as the level at which nuclear intensity exceeded the background level in three randomly chosen fields containing at least 15 cells each. Approximately 200 cells per experiment were scored.
2.4. DNA damage analysis
For alkaline single cell electrophoresis (comet assay), cells were trypsinized, centrifuged and washed in 1× PBS, resuspended in 1% low melting agarose (3×104 cells/ml) containing 0.5mg/ml proteinase K and pipetted onto CometSlides™ (Trevigen, Gaithersburg, MD). Quantification of olive tail moment was performed with TriTek CometScore freeware (TriTek Corporation, Summerduck, VA).
2.5. Analysis of cellular extracts
Cells were extracted in 50mM Tris–HCl, 0.3M NaCl, 0.5% Triton X‐100, 1mM EDTA, 1mM DTT, 0.1mM PMSF, pH 7.9 for 15min at 4°C. 20,000×g supernatants (50μg/lane) were analyzed for non‐histone proteins by 6% SDS‐PAGE. 20,000×g pellets containing the histones were resuspended in SDS sample buffer, boiled 5min, and sonicated. Following recentrifugation (20,000×g, 5min, 4°C), histones were analyzed through 15% SDS‐PAGE (20μg/lane) through Ponceau red staining of PVDF membranes or using H2A and H4 antibodies. Whole cell lysate was obtained in SDS sample buffer. Western blot and Ponceau quantification was on a Syngene2 system using GenTool.
3. Results
3.1. One hour etoposide treatment is highly toxic
The peak plasma concentration of etoposide in standard therapeutic protocols is 25–75μM and recedes within 6h (Chabner et al., 2005; Relling et al., 1998). Although lipophilic, in tissue culture the intracellular concentration of etoposide equilibrates with the extracellular concentration (Wright et al., 1990). Thus treatment of tissue culture cells with 25μM etoposide provides an approximate mimic of peak in vivo exposure and is a concentration at which physiologically relevant cellular responses may be expected to be acutely activated, with subsequent washout of the medium allowing for rapid depletion of the drug from the cells.
Increased γH2AX levels, being closely coupled to the generation of the DSBs, were selected as early indicators of the damage induced by etoposide. In HeLa cells, the response to etoposide was rapid, reaching a maximum by approximately 1h (Figure 1A; see also Figure 2C); thus, the 1h time point was chosen to study the early cellular response to etoposide. While 1h of 25μM etoposide treatment of asynchronously growing cervical carcinoma HeLa and colon carcinoma HCT116 cells was highly cytotoxic to both cell types, HCT116 cells displayed greater overall sensitivity to etoposide, with over 99% cell death over 7–14days following treatment compared with 90% death of the HeLa cells (Figure 1B). In both instances cell death occurred slowly: cell membrane integrity was preserved (as assessed by Trypan Blue staining) and no signs of chromatin compaction were visible by 24h post‐damage (data not shown).
Figure 1.
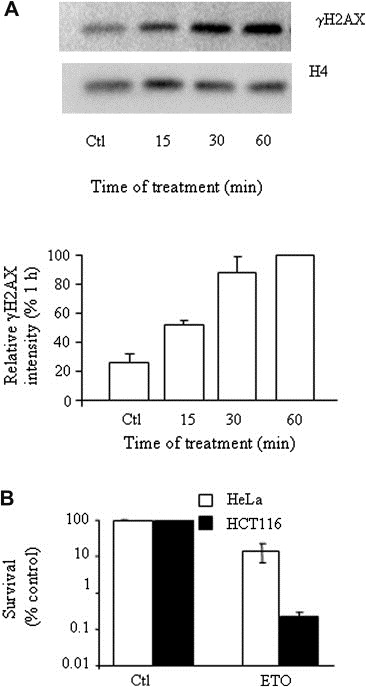
Short etoposide treatment is lethal to HeLa and HCT116 cells. (A) Time course of γH2AX induction in response to etoposide. HeLa cells were harvested at the indicated time post‐treatment with 25μM etoposide and analyzed by Western blot for the γH2AX level, corrected for histone H4 signal. Data was normalized to the 1h value, set as 100%. Analysis represents the average of duplicate determinations (±SD) and is representative of two experiments. (B) HeLa (open bars) and HCT116 (solid bars) survival at 7 and 14days, respectively, following etoposide (ETO) withdrawal measured by colony forming assay and displayed as a percentage of vehicle‐treated (Ctl) cell survival. Error reflects the mean of duplicates ±SD of two experiments.
Figure 2.
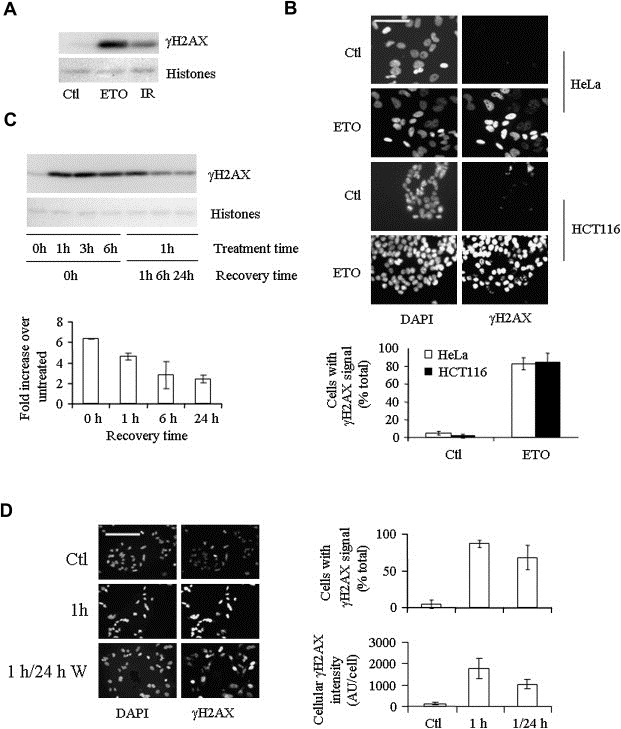
Etoposide induces a rapid, widespread and persistent γH2AX response. (A) HeLa cell γH2AX levels in response to 1h of 25μM etoposide (ETO) or 6Gy ionizing radiation (IR) followed by 1h recovery compared with vehicle‐treated (Ctl) cells. (B) Indirect immunofluorescence analysis of γH2AX induction by 1h etoposide treatment of HeLa and HCT116 cells compared with DAPI staining of nuclei. Error is expressed as SD of three experiments of a minimum of 200 cells distributed over 4–5 fields. Scale bar=50μm. (C) The left side of the Western blot shows γH2AX levels following continuous etoposide exposure of HeLa cells for 0–6h, while the right side shows γH2AX levels following1–24h withdrawal from 1h etoposide treatment. γH2AX during the withdrawal phase is quantified relative to 1h treatment. The data are representative of the results of three independent experiments performed in duplicate. (D) Indirect immunofluorescence comparison of γH2AX levels in HeLa cells following 1h of etoposide treatment and 24h of withdrawal from treatment. The percentage of cells that display a γH2AX signal after 1h treatment and 24h recovery (top right) is compared with the level of γH2AX fluorescence within each cell (bottom right) (SD of a minimum of three experiments performed in duplicate). Scale bar=100μm.
3.2. Transient etoposide treatment initiates an abundant, widespread and persistent γH2AX response
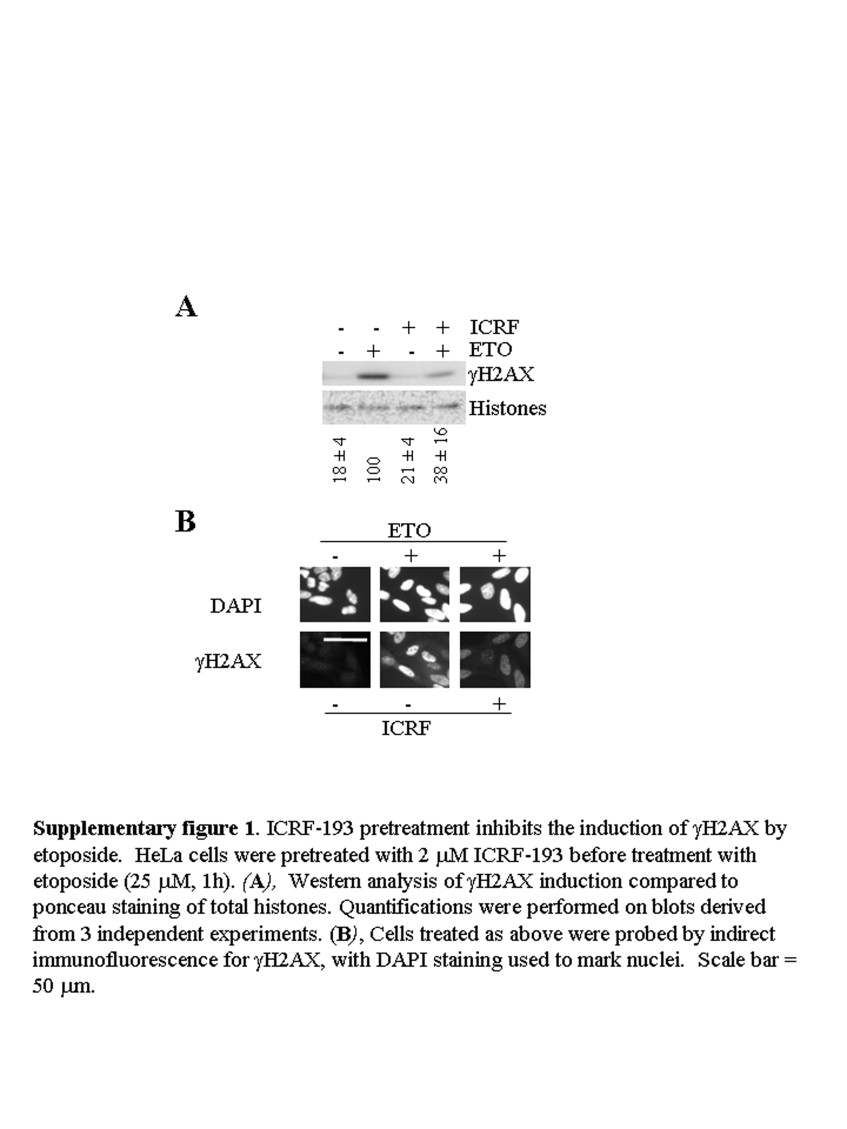
The γH2AX response to a 1h etoposide treatment was potent, exceeding that obtained following exposure of the cells to 6Gy of γ irradiation, a dose also sufficient to kill 90% of HeLa cells (Griffith and Tolmach, 1976; Hennequin et al., 1994) (Figure 2A). Indirect immunofluorescence analysis showed the induction of γH2AX in over 85% of HCT116 cells and 95% of HeLa cells (Figure 2B), far in excess of the 15–25% of cells determined by FACS analysis to be in S phase at the time of treatment (data not shown). Pre‐treatment of the cells with the topoII inhibitor bisdioxopiperazine ICRF‐193, which acts upstream of formation of the topoII CC, inhibited the γH2AX response to etoposide, verifying that the response was due to a direct effect of etoposide on topoII (Supplementary Figure 1).
Upon withdrawal of etoposide, 60% of the γH2AX signal was lost within 6h, but the remaining signal persisted thereafter for at least 24h (Figure 2C) Moreover, the signal that persisted was evenly distributed across the cell population. Thus, while the level of γH2AX signal per cell decreased, the percentage of cells exhibiting a response remained essentially the same (Figure 2D).
3.3. γH2AX response to a 1h etoposide treatment is mediated by topoIIα
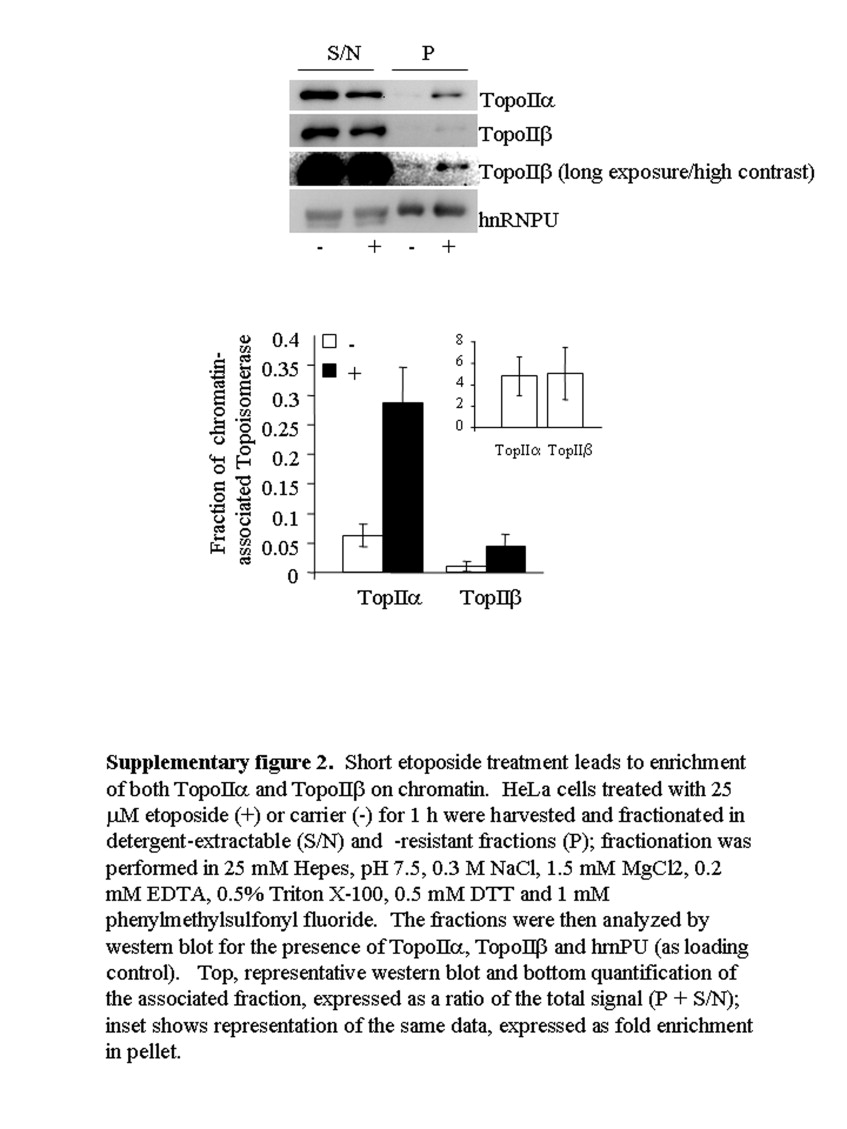
TopoIIα and topoIIβ have alternatively been implicated in the transient γH2AX response to etoposide (Houlbrook et al., 1995; Kobayashi et al., 2001; Zhang et al., 2006). Importantly, the CC of both isoforms become trapped on DNA in situ (Willmore et al., 1998). In HeLa cells, topoIIα and topoIIβ were ubiquitously expressed (Figure 3A) but with topoIIα levels varying throughout the cell population, presumably a reflection of its cell‐cycle regulation as previously reported (Woessner et al., 1991). Consistent with previously published data, a 1h etoposide treatment led to crosslinking of both isoforms to DNA (Supplementary Figure 2), suggesting that both isoforms have the potential to be involved in the response. While topoIIα was always more intimately associated with chromatin (i.e. fraction of total enzyme that is detergent‐resistant), both isoforms could be enriched to a similar extent in response to etoposide (i.e. when expressed as fold enrichment over untreated control). To assess the relative importance of topoIIα and topoIIβ to the γH2AX response, we employed a transient siRNA approach. Specific siRNA targeting reduced the levels of topoIIα and topoIIβ individually by over 90% (Figure 3A,B). Silencing of topoIIα inhibited the induction of γH2AX in response to transient etoposide exposure by over 75% (Figure 3B). By contrast, silencing of topoIIβ reduced the γH2AX response by only 15%. Coincident knockdown of topoIIα and topoIIβ together displayed little additional affect on γH2AX beyond that seen by knockdown of topoIIα alone.
Figure 3.
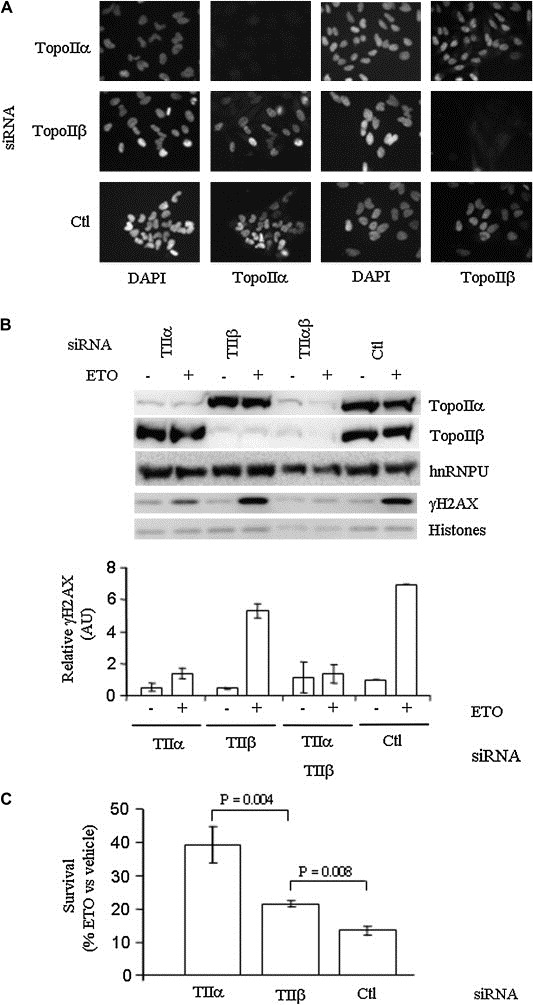
Etoposide cytotoxicity and induction of γH2AX in HeLa cells depends on topoIIα. (A) Indirect immunofluorescence of topoIIα and topoIIβ following siRNA transfection compared with DAPI nuclear staining. Ctl = siRNA directed against GFP. (B) Top, Western blot of γH2AX, topoIIα and topoIIβ subsequent to siRNA and etoposide treatment, compared with hnRNPU and total histone levels (Ponceau stain). Bottom, γH2AX levels (mean ± SD). Quantifications were normalized to either hnRNPU (topoII) or total histones (γH2AX). (C) HeLa cell survival 6 days following etoposide treatment of Ctl and topoII siRNA transfected cells analyzed by formazan formation assay. Values are ±SD of three independent experiments performed in duplicate.
The dependence of the rapid γH2AX response on topoIIα correlated directly with increased survival of topoIIα‐deficient cells subsequent to etoposide treatment (Figure 3C). Thus, while almost 90% of HeLa cells treated with control siRNA had succumbed at day 6 following etoposide treatment, siRNA ablation of 90% of topoIIα provided for an over three‐fold increase in cell survival, to 40%. By contrast, knockdown of topoIIβ, resulted in only a one‐third increase in survival. Thus γH2AX induction and the cytotoxic effects of etoposide were primarily dependent on the immobilization of topoIIα–DNA CC.
In view of the requirement for topoIIα in the induction of the γH2AX response, a potential co‐localization between γH2AX and topoIIα was investigated. Confocal microscopy revealed that topoIIα and γH2AX displayed largely distinct distribution profiles, with topoIIα being diffuse and γH2AX more punctate (Figure 4A). Permeabilization under conditions that enrich the chromatin‐associated topoIIα population in response to etoposide (same conditions as used in Supplementary Figure 2) similarly revealed thetwo protein populations to occupy largely distinct nuclear niches, although some coincidence did occur (Figure 4B).
Figure 4.
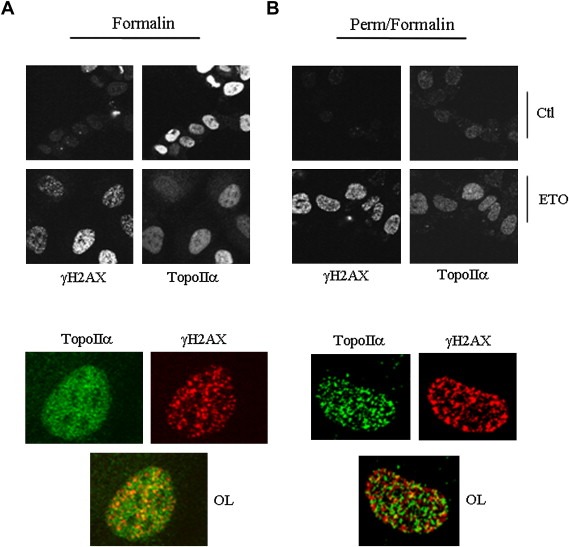
TopoIIα and γH2AX display largely distinct nuclear distributions with some signal overlap. HeLa cells were treated with 25μM etoposide for 1h, were either directly fixed in formalin. (A) or pre‐permeabilized with 25mM Hepes, pH 7.5, 0.3M NaCl, 1.5mM MgCl2, 0.2mM EDTA, 0.5% Triton X‐100, 0.5mM DTT and 1mM phenylmethylsulfonyl fluoride for 2min prior to formalin fixation. (B) Coverslips were then processed for immunocytochemistry using the indicated antibodies. Top, wide field view; bottom, magnification of cells from the 1h etoposide panel; OL, overlay of the two signals. Representative cells are shown. Similar results were obtained following 10min and 20min treatments.
3.4. TopoIIα‐dependent generation of DNA breaks in response to bleomycin
To determine whether the γH2AX response to transient etoposide treatment reflected generation of DNA breaks from processed topoII CC, we performed comet assays to monitor DNA integrity (Figure 5). Attempts at using the neutral comet assay (specific for DSBs) following 25μM etoposide indicated no meaningful difference with the mock‐treated samples, although a higher dose did reveal a significant difference (Supplementary Figure 3). By contrast, alkaline comet assay, which detects both SSBs and DSBs, revealed that a 1h etoposide treatment of HeLa cells produced widespread and abundant DNA breaks (Figure 5A,B), including DSBs as reflected by the induction of γH2AX. Again, while siRNA ablation of topoIIα dramatically reduced the number of DNA breaks generated, ablation of topoIIβ was essentially without effect. However, the topoIIα‐dependent DNA damage appeared to be efficiently repaired during the withdrawal period (Figure 5C), an observation that belied the persistence of the γH2AX signal.
Figure 5.
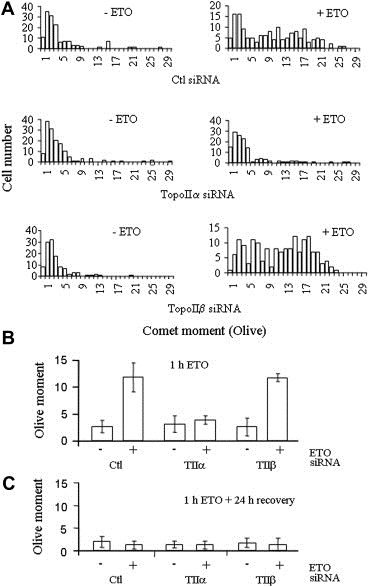
Comet analysis of etoposide‐induced HeLa cell DNA damage. Single cell electrophoresis and SYBR Green I analysis of cells treated as in Figure 3. Quantification of 100–150 cells per condition. (A) Distribution profile of the comet moments. Comets were grouped in ascending order of comet moment values (rounded up to the nearest integer). (B) Comet results expressed as the average of the moment means from two independent experiments (±SD). Wilcoxon Two Sample test indicates only the comet moment between Ctl or topoIIβ siRNA‐treated cells exposed to etoposide is significantly different from that of topoIIα‐treated cells exposed to etoposide (P<1.5×10−12). (C) Average of the moment means of cells maintained in etoposide‐free medium for 24h following 1h treatment with DMSO±25μM etoposide.
3.5. Persistent engagement of the DSB repair machinery at γH2AX foci
To probe the nature of the persistent γH2AX response, we examined recruitment of DSB repair machineries to the foci. The activation of ATM through phosphorylation on Ser‐1981 (pATM) and its recruitment to γH2AX foci, is a rapid initial event in the cellular response to DSBs (Bakkenist and Kastan, 2003). The coincident induction of pATM with γH2AX subsequent to etoposide treatment, also to a level exceeding that following 6Gy of γ‐irradiation exposure, verified that etoposide was acting through the creation of DSBs (Figure 6A,B). MDC1 is a coordinating factor that mediates the amplification of DNA damage signals from DSBs and interacts with γH2AX, ATM and Ku antigen (Lou et al., 2004). While MDC1 is localized to discrete foci in naïve cells, the size and nature of these foci change upon DNA damage induced by γ‐irradiation and etoposide treatment to largely overlap with the γH2AX foci (Figure 6C).
Figure 6.
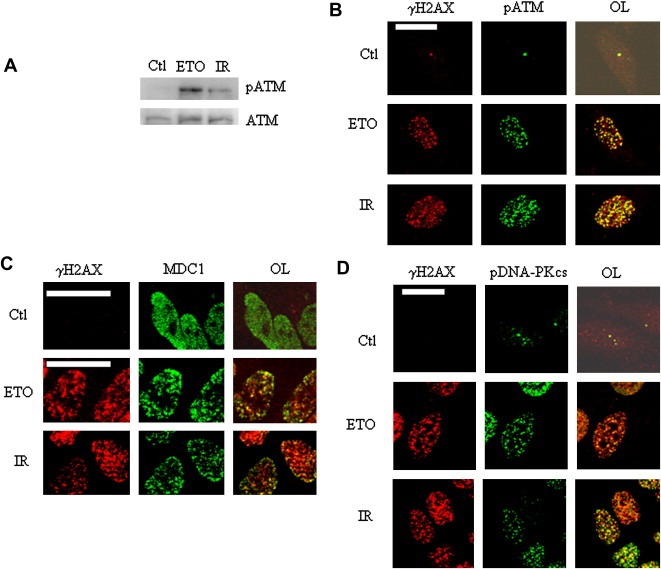
γH2AX foci mark sites of DNA repair activity. (A) ATM and pATM levels in HeLa cells treated with vehicle (Ctl), 25μM etoposide (1h) or 6Gy IR (IR). (B) Co‐localization (yellow signal in the overlay (OL)) of γH2AX (red) and pATM (green). Treatment with vehicle (Ctl), IR and etoposide (ETO) are as indicated. Scale bar=20μm. (C,D) Co‐localization of γH2AX with MDC1 (D), and pDNA‐PK (E). In B and E, the Ctl overlays were intensified in order to visualize the cells.
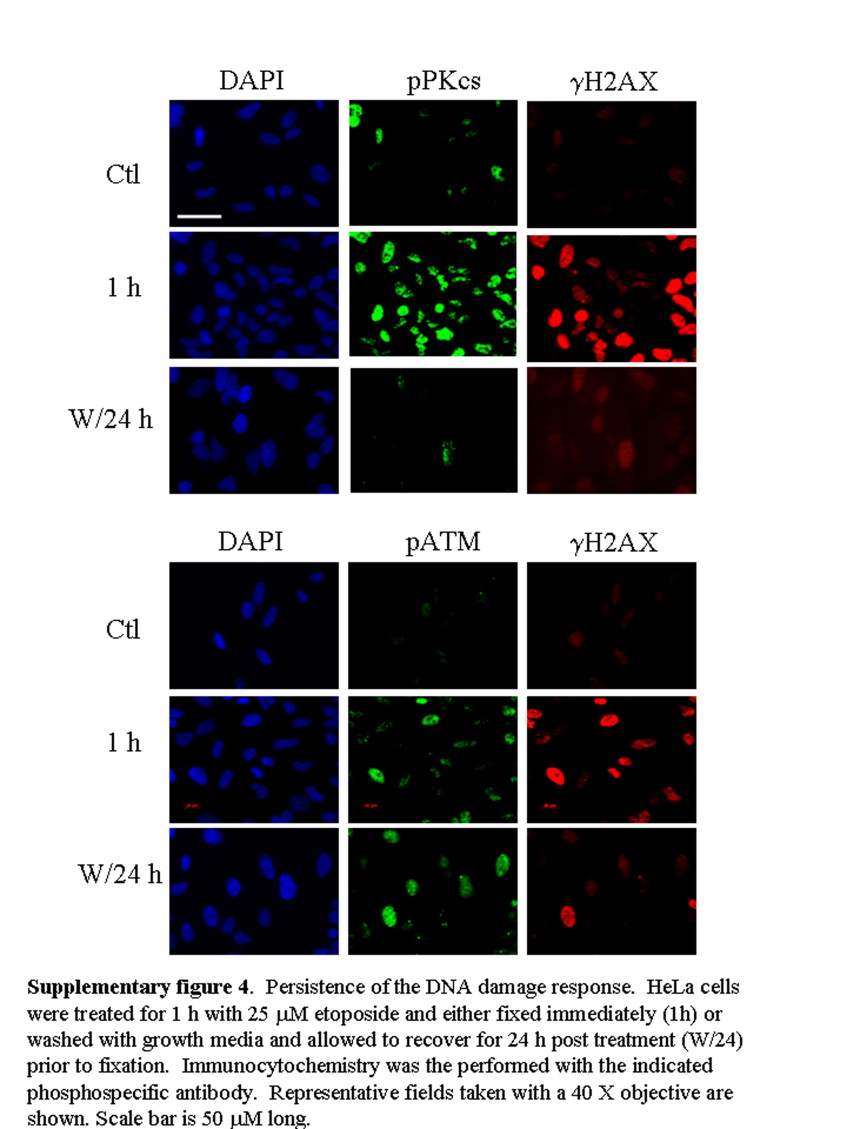
During most of the cell cycle, the predominant mechanism of DSB repair is NHEJ, with DNA‐PKcs playing a key role. Recruitment of DNA‐PKcs to DSBs leads to its phosphorylation (pDNA‐PKcs) at a number of sites including T2609 (Chan et al., 2002; Chen et al., 2007; Ding et al., 2003; Soubeyrand et al., 2003). The co‐localization of pDNA‐PKcs foci with γH2AX (Figure 6D), provided evidence of NHEJ‐associated activity at the sites of etoposide‐initiated DNA damage. Twenty‐four hours following withdrawal of etoposide treatment, γH2AX foci remained broadly distributed throughout the cell, albeit at a lower average intensity, consistent with the Western blot data; the decreased γH2AX signal was accompanied by a marked reduction of the pDNA‐PKcs signal while, surprisingly, pATM remained strong (Supplementary Figure 4). Importantly however both signals continued to co‐localize extensively with the γH2AX foci (Figure 7A,B).
Figure 7.
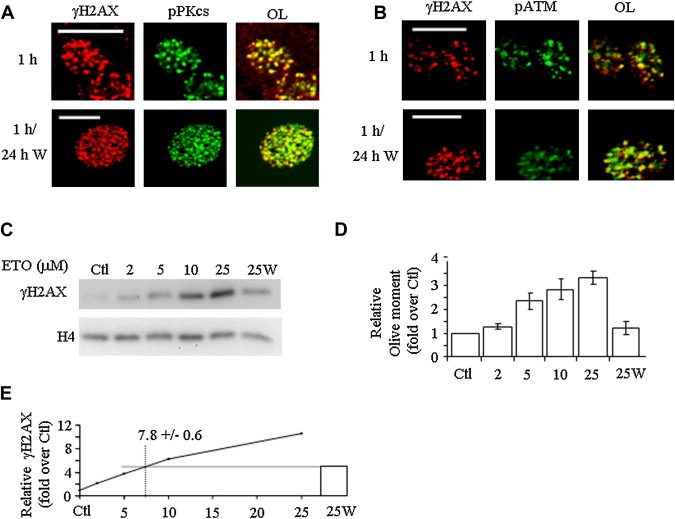
Persistence of the damage response. Co‐localization (OL, yellow) of γH2AX (red) with pDNA‐PKcs (A, green) and pATM (B, green) either 0h or 24h following withdrawal of etoposide. Scale bar=20μm. Channel intensities were optimized to facilitate overlay visualization and do not reflect absolute emission intensities (see Supplementary Figure 4 for absolute intensities). (C) Left, γH2AX levels following 1h treatment with 2–25μM etoposide (lanes 2–5) and following 24h withdrawal from 25μM treatment (25W), with H4 levels shown as loading control. (D) Average of olive moment means in comets of cells treated as in (C). (E) Quantification of γH2AX corrected for loading with the concentration of etoposide, with interpolation from a linear regression fit of the 1–10μM concentration used to predict the concentration of etoposide needed in a 1h treatment (7.8μM) to provide a γH2AX response equal to that which remained following 24h withdrawal. Error bars represent the average of two independent experiments (±SD).
3.6. High γH2AX signal to DNA break ratio 24h post‐damage
To explore the apparent dissociation between the seemingly complete DNA religation and continued engagement of the DSB repair machinery at 24h, we compared and contrasted the levels of the γH2AX and DNA damage in response to 1h of increasing amounts of etoposide treatment with those following 24h withdrawal from 25μM etoposide. Treatment of HeLa cells with increasing concentrations of etoposide resulted in a near‐linear increase in γH2AX (Figure 7C). Twenty‐four hours following etoposide withdrawal, the γH2AX signal remained high while, surprisingly, no breaks were detected (Figure 7C,D). This did not appear to be due to a sensitivity issue with the comet assay: the level of γH2AX measured is equivalent to the level expected from an 8μM etoposide treatment (Figure 7E) whereas 5μM suffice to produce substantial DNA damage (Figure 7D). Indeed the ratio of γH2AX levels to olive moment was on average 4.5×(±1.6) times higher than the mock‐treated control 24h post‐treatment. This last observation is consistent with an ongoing and disproportionably high damage signaling cascade originating from apparently rejoined break sites 24h post‐etoposide.
3.7. DNA replication and transcription play marginal roles in the rapid γH2AX response to etoposide
The action of etoposide has been linked to replication, transcription and other aspects of chromatin dynamics (Mao et al., 2001; Xiao et al., 2003; Zhang et al., 2006). To seek to define the cytotoxic target site of transient etoposide treatment, we evaluated the effects of a number of metabolic inhibitors on the rapid component of the cellular response to etoposide.
Aphidicolin (APH) blocks DNA replication by inhibiting DNA polymerase α (Wist and Prydz, 1979). Pre‐treatment with APH under conditions that abrogated 5‐bromodexoyuridine (BrdU) incorporation (Supplementary Figure 5A) modestly (17±8% inhibition, HCT116, n=2) reduced the γH2AX response (Figure 8A). In addition, APH failed to decrease the percentage of cells exhibiting a γH2AX response or improve their subsequent survival, indicating that the cytotoxic effects of etoposide were independent of DNA replication occurring during the time of etoposide exposure (Figure 8B,C). It has previously been shown that transcriptional inhibitors including DRB can protect leukemic cells from the effects of transient etoposide exposure (Kaufmann, 1991). Preliminary experiments confirmed that both DRB and amanitin prevented ongoing transcription at the dosage used (Supplementary Figure 5B). However, DRB, which promotes early termination of RNA pol II transcripts (Fraser et al., 1978), or αamanitin, which directly inhibits RNA polymerases II and III (Huang et al., 1998), inhibited γH2AX induction by only 5–15% (Figure 8D) providing an indication that the cytotoxic changes initiated by a short 25μM etoposide treatment were independent of ongoing transcription at the time of exposure and consistent with our results excluding topoIIβ.
Figure 8.
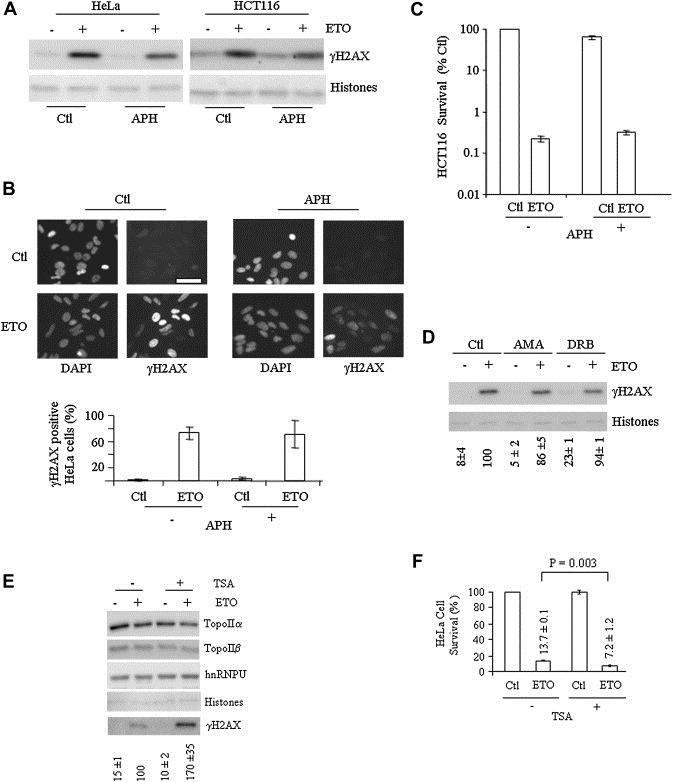
Etoposide sensitivity is independent of DNA replication and transcription but promoted by histone deacetylase inhibition. (A) Effect of aphidicolin (APH, 0.5μg/ml, begun at t=−1h) on etoposide (ETO) induction of γH2AX in HeLa and HCT116 cells compared with vehicle (Ctl). Histone levels were visualized by Ponceau staining. (B) Indirect immunofluorescence analysis of the effect of APH on γH2AX induction in HeLa cells with quantification reflecting the percentage of γH2AX positive cells. Similar results were observed with HCT116 cells (data not shown). (C) Colony formation assay at 7days following ETO and APH treatment. Quantification is relative to Ctl cells and represents the average of duplicate determinations (±SD); note log scale. Similar results were obtained with HeLa cells (data not shown). (D) γH2AX levels in vehicle, α‐amanitin (AMA) or DRB‐treated HeLa cells (4h), compared with total histone levels. (E) Effect of 4h trichostatin A (TSA, 0.2μM) pre‐treatment on the γH2AX response to 1h of etoposide (ETO) in the continuing presence of TSA, with topoIIa, topoIIb, hnRNPU and total histone levels (via Ponceau staining) also shown. Quantification of γH2AX is relative to the induction obtained by etoposide treatment alone corrected for total histone levels (mean±SD of three experiments performed in duplicate). (F) Survival of cells treated as in (E) as measured by CellTiter assay (average of two experiments performed in duplicate, ±SD); the two‐tailed P value (Student t‐test) is also shown.
3.8. Response to etoposide is enhanced by inhibition of histone deacetylase activity
Previously, histone deacetylase (HDAC) inhibitors have been shown to stabilize topoII–DNA complexes as well as increase the effectiveness of both long term and transient etoposide treatment (Kim et al., 2003; Marchion et al., 2004). Pre‐treatment of HeLa cells for 5h with 0.2μM of the HDAC inhibitor TSA had no measurable effect on the basal level of H2AX phosphorylation or topoII levels (Figure 8E). However, the same TSA treatment enhanced subsequent etoposide sensitivity, leading to an almost two‐fold increase in γH2AX levels when etoposide was added during the final hour of TSA treatment; this combined regimen also resulted in a two‐fold increase in subsequent HeLa cell mortality (Figure 8F), linking the effect of HDAC inhibitors directly to etoposide‐mediated DNA damage.
4. Discussion
We provide evidence that etoposide compromises long term cellular viability through the creation of DNA lesions of a type or structure that are unable to be correctly or completely repaired. These lesions elicit a persistent DSB response, as evidenced by the recruitment of the NHEJ machinery, that continues beyond the completion of DNA ends rejoining and reflects the long term cytotoxicity of short term etoposide exposure. These cytotoxic lesions were specifically dependent on topoIIα and were facilitated by an agent that promoted chromatin accessibility through increased histone acetylation, but were independent of transcription and replication occurring at the time of the etoposide treatment.
γH2AX induction correlates directly with the detection of DSBs by the cell and the initiation of damage response pathways (Foster and Downs, 2005). Successful DNA repair leads to loss of γH2AX and dephosphorylation of ATM (Chowdhury et al., 2005; Keogh et al., 2006). Persistence of γH2AX generally reflects the presence of DNA damage that is resistant to the DNA repair machinery and which may lead to mutagenesis or induction of cell death pathways (Cucinotta et al., 2008; Roos and Kaina, 2006). Here the induction of γH2AX by 1h of etoposide exposure reflected the introduction of widespread and substantial double‐stranded DNA damage, as assessed by the appearance of γH2AX foci, and the co‐localization of pATM, pDNA‐PKcs and MDC1. Subsequently, we observed the continuation of a long term γH2AX response in which both pDNA‐PKcs and pATM remained engaged at γH2AX foci, albeit to different extent. Persistence of etoposide in the cellular environment cannot be invoked to explain this ongoing signaling since (1) intracellular etoposide is known to equilibrate rapidly with the extracellular milieu, and is expected to be largely eliminated by the washes 1h post‐treatment; and (2) the remaining γH2AX signal would require the ongoing presence of 5–10μM etoposide, a concentration that is much higher than any possible residual etoposide.
One possibility suggested by these results is that topoIIα trapping on DNA by etoposide leads to complex DSBs that recruit the NHEJ machinery, and perhaps other break‐repair complexes as well, but which cannot be repaired appropriately. Upon DNA end ligation, DNA‐PKcs becomes inactivated and dissociates from the DNA together with Ku antigen (Lieber et al., 2003). Notably, DNA‐PKcs kinase activity is not normally activated or maintained by B form DNA in the absence of DNA ends. However, it has previously been shown that DNA‐PK can be active from a variety of double‐stranded DNA structures that contain transitions from B form DNA that support Ku binding and by structured single‐stranded DNA (Soubeyrand et al., 2001). Thus the persistence of pDNA‐PKcs at the γH2AX foci could reflect the presence of partially or improperly joined DNA ends that yield DNA structures containing double‐ to single‐stranded DNA transitions sufficient to sustain the DNA‐PK activity.
It must be noted, however, that the NHEJ machinery seems to handle the bulk of the breaks appropriately, since the average cellular intensity of both pDNA‐PKcs and γH2AX foci decrease with time; indeed, the NHEJ repair pathway is clearly important in helping the cell recover from etoposide exposure (Adachi et al., 2004). Nevertheless, aberrant lesions that are not appropriately tackled by the cellular repair machinery likely remain as indicated by the persistence of the ATM activation.
Whereas γH2AX induction is consistent with the induction of DSBs in response to etoposide, our comet assay results reveal that a significant amount (likely the majority) of breaks generated by topoIIα consist of SSBs, since neutral comet assay, specific for DSBs, revealed no measurable breaks at 25μM. Importantly however, higher etoposide doses (200μM) which increased three‐ to five‐fold the γH2AX response, did result in significant neutral comet tails (2×background), suggesting that etoposide does indeed rapidly induce DSBs, albeit at levels below the sensitivity of the neutral comet assay when a pharmacologically relevant dose of 25μM is used. Consistent with this, Bandele and Osheroff (2008) have shown (in CEM cells) that a short 50μM etoposide can generate a neutral comet moment, although quantification was not provided. It is unclear what contributions these SSBs have on the cell survival, since they appear to be rapidly repaired (relative to DSB, i.e. γH2AX signal) as assessed by our alkaline comet data. One scenario can be envisioned in which these topoIIα‐mediated SSBs get converted to DSBs through a mechanism that has yet to be defined. These newly generated DSBs may operate in conjunction with the early‐phase DSBs and ultimately lead to cell death. While this SSB to DSB conversion process may indeed take place, it is expected to play a limited role in the whole picture in view of the fact the γH2AX levels are maximal after approximately 1h and continuously decrease thereafter, over the 24h withdrawal period.
One hour etoposide treatment did not immediately induce cell death, as cellular integrity and PARP‐1 fragmentation were delayed for over 24h following etoposide withdrawal. This suggests that the persistent lesions are tolerated by the cell for an extended period of time. Models for the cytotoxicity of continuous etoposide treatment propose that etoposide traps topoII CC that expose lethal lesions upon their encounter by DNA replication forks (Montecucco and Biamonti, 2007). Implicit to this model is that the CC half‐life is directly related to cell death, as the probability of encounter increases with persistence of the CC. Indeed, extending the CC lifetime can correlate directly with cell death (Bandele and Osheroff, 2008). However this does not suffice to explain toxicity of etoposide; for instance, transcription inhibition by DRB, which significantly stabilizes the topoII CC, offers no protection from etoposide treatment (Fan et al., 2008).
An alternative view, consistent with our data, suggests that it is not the trapped topoII–DNA complexes per se, but inappropriately repaired DNA that provides a barrier to the replication fork when the damaged cells subsequently attempt to divide. This last model appears more in line with the observation that most of the topoII–DNA crosslinks are short lived as they are largely reversed and/or processed within a few hours of etoposide removal (Bandele and Osheroff, 2008; Errington et al., 2004; Willmore et al., 1998).
In any event, the precise steps linking the formation of the CC to the downstream γH2AX induction remain unclear. In that regard the topoIIα‐γH2AX co‐localization data is intriguing. Indeed, topoIIα did not appear to particularly overlap with γH2AX foci: while there was some overlap, there was no clear enrichment in γH2AX‐rich areas as both populations appear to behave essentially independently. How then can one explain the clear causal relationship we demonstrated between the two in response to etoposide? We advance two potential explanations. For one, γH2AX foci may appear only when topoIIα is cleared from the break following processing of the CC and hence may never physically overlap at any given time. Alternatively, in view of the presence of significant SSBs, the overlap between DSBs–topoIIα lesions (γH2AX positive) may be masked by an excess of SSBs–topoIIα complexes (γH2AX negative).
DNA replication depends on topoIIα, whereas transcription shows a primary dependence on topoIIβ (Ju et al., 2006; McClendon and Osheroff, 2007; Wang, 2002). Previous studies have shown that exposure of cells to levels of etoposide 10‐fold higher (250μM) than utilized in the present study led to the induction of γH2AX foci that are dependent on the 26S proteasome‐mediated degradation of topoIIβ (Zhang et al., 2006). As we found topoIIβ to play a minor role in the γH2AX response or cell death at 25μM etoposide, our results suggest that topoIIβ only becomes a significant factor in the etoposide response at concentrations that considerably exceed those relevant to chemotherapeutic regimes. Moreover, our siRNA results are consistent with, and extend, the preponderance of the mutagenesis and knockdown/knockout data that argue for a greater role for topoIIα than topoIIβ in transmission of etoposide sensitivity (Kobayashi et al., 2001; Toyoda et al., 2008).
TopoIIα is primarily recognized for its role in DNA replication. However, the majority of the cells in this study were outside of S phase during etoposide exposure and the initiation of cytotoxic DNA damage was unaffected by inhibition of DNA replication during the etoposide treatment. DNA damage induction was similarly unaffected by transcription inhibition, despite an expected stabilization of the CC (Fan et al., 2008). Nonetheless, treatment of the cells with the HDAC inhibitor TSA, which promotes the accumulation of histone acetylation and which generally functions to facilitate gene expression through increasing the accessibility of transcriptionally active chromatin, increased the γH2AX response to etoposide and subsequent cell death. Recent work by Adimoolam et al. (2007) suggests another possible scenario, namely that TSA sensitization may reflect a direct inhibition on homologous repair inhibition via RAD51 downregulation, as was observed with PCI‐24781, another HDAC inhibitor. While it remains to be confirmed that TSA similarly interferes with homologous repair, it may offer an alternative, non‐exclusive, means through which TSA enhances etoposide sensitivity.
Together these results link the primary site of cytotoxic action of etoposide to an activity of topoIIα within euchromatin that is facilitated by histone acetylation. Indeed, the significance of the largely cell‐cycle independence of the DSB response observed in determining cellular demise is consistent with recent work from Tanaka et al. (2007), which demonstrated that a B cell lymphoblastoid cell line was sensitive to etoposide during all phases of the cell cycle.
Understanding the mechanisms through which etoposide induces cytotoxic lesions awaits further definition of the roles of topoIIα beyond DNA replication. Considerable efforts over the years have brought us closer to this goal, but much remains to be done. One interesting possibility is found in reports of a specific role for topoIIα in organization of the interaction of DNA with the nuclear matrix (Christensen et al., 2002; Danks et al., 1994). Nuclear matrix accessibility would be expected to be increased by TSA treatment and would provide for a specific site of action of topoIIα within euchromatin that would be distanced from sites of transcription. It will be important in the future to localize the specific cytotoxic target of etoposide–topoIIα trapped complex, as this could lead to the development of co‐treatment strategies aimed at increasing the effectiveness of this important class of chemotherapeutic agent.
Supporting information
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Supplementary data
{kind=link}
Acknowledgements
This work was funded by the Canadian Institutes of Health Research (MOP‐13412) and National Cancer Institute of Canada. RJGH holds a University of Ottawa Health Research Chair.
Appendix. 1.
1.1.
Supplementary data associated with this article can be found in the online version, at doi:10.1016/j.molonc.2009.9.003.
Soubeyrand Sébastien, Louise Pope, Haché Robert J.G., (2010), New specific molecular targets for radio‐chemotherapy of rectal cancer, Molecular Oncology, 4, doi: 10.1016/j.molonc.2009.11.002.
References
- Adachi, N. , Iiizumi, S. , So, S. , Koyama, H. , 2004. Genetic evidence for involvement of two distinct nonhomologous end-joining pathways in repair of topoisomerase II-mediated DNA damage. Biochem. Biophys. Res. Commun. 318, 856–861. [DOI] [PubMed] [Google Scholar]
- Adimoolam, S. , Sirisawad, M. , Chen, J. , Thiemann, P. , Ford, J.M. , Buggy, J.J. , 2007. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA. 104, 19482–19487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist, C.J. , Kastan, M.B. , 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Bandele, O.J. , Osheroff, N. , 2008. The efficacy of topoisomerase II-targeted anticancer agents reflects the persistence of drug-induced cleavage complexes in cells. Biochemistry. 47, 11900–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabner, B. , 2005. Chemotherapy of neoplastic diseases: antineoplastic agents. In Brunton L., Lazo J., Parker K.(Eds.), Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 1315–1404. [Google Scholar]
- Chan, D.W. , Chen, B.P. , Prithivirajsingh, S. , Kurimasa, A. , Story, M.D. , Qin, J. , Chen, D.J. , 2002. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 16, 2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, B.P. , 2007. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 282, 6582–6587. [DOI] [PubMed] [Google Scholar]
- Chowdhury, D. , Keogh, M.C. , Ishii, H. , Peterson, C.L. , Buratowski, S. , Lieberman, J. , 2005. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell. 20, 801–809. [DOI] [PubMed] [Google Scholar]
- Christensen, M.O. , 2002. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J. Cell Biol. 157, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucinotta, F.A. , Pluth, J.M. , Anderson, J.A. , Harper, J.V. , O’Neill, P. , 2008. Biochemical kinetics model of DSB repair and induction of gamma-H2AX foci by non-homologous end joining. Radiat. Res. 169, 214–222. [DOI] [PubMed] [Google Scholar]
- D’Arpa, P. , Liu, L.F. , 1989. Topoisomerase-targeting antitumor drugs. Biochim. Biophys. Acta. 989, 163–177. [DOI] [PubMed] [Google Scholar]
- Danks, M.K. , Qiu, J. , Catapano, C.V. , Schmidt, C.A. , Beck, W.T. , Fernandes, D.J. , 1994. Subcellular distribution of the alpha and beta topoisomerase II–DNA complexes stabilized by VM-26. Biochem. Pharmacol. 48, 1785–1795. [DOI] [PubMed] [Google Scholar]
- Ding, Q. , Reddy, Y.V. , Wang, W. , Woods, T. , Douglas, P. , Ramsden, D.A. , Lees-Miller, S.P. , Meek, K. , 2003. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 23, 5836–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington, F. , Willmore, E. , Leontiou, C. , Tilby, M.J. , Austin, C.A. , 2004. Differences in the longevity of topo IIalpha and topo IIbeta drug-stabilized cleavable complexes and the relationship to drug sensitivity. Cancer Chemother. Pharmacol. 53, 155–162. [DOI] [PubMed] [Google Scholar]
- Fan, J.R. , Peng, A.L. , Chen, H.C. , Lo, S.C. , Huang, T.H. , Li, T.K. , 2008. Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair (Amst.). 7, 452–463. [DOI] [PubMed] [Google Scholar]
- Foray, N. , Priestley, A. , Alsbeih, G. , Badie, C. , Capulas, E.P. , Arlett, C.F. , Malaise, E.P. , 1997. Hypersensitivity of ataxia telangiectasia fibroblasts to ionizing radiation is associated with a repair deficiency of DNA double-strand breaks. Int. J. Radiat. Biol. 72, 271–283. [DOI] [PubMed] [Google Scholar]
- Foster, E.R. , Downs, J.A. , 2005. Histone H2A phosphorylation in DNA double-strand break repair. FEBS J. 272, 3231–3240. [DOI] [PubMed] [Google Scholar]
- Fraser, N.W. , Sehgal, P.B. , Darnell, J.E. , 1978. DRB-induced premature termination of late adenovirus transcription. Nature. 272, 590–593. [DOI] [PubMed] [Google Scholar]
- Goodarzi, A.A. , Noon, A.T. , Deckbar, D. , Ziv, Y. , Shiloh, Y. , Lobrich, M. , Jeggo, P.A. , 2008. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 31, 167–177. [DOI] [PubMed] [Google Scholar]
- Gorczyca, W. , Gong, J. , Ardelt, B. , Traganos, F. , Darzynkiewicz, Z. , 1993. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor agents. Cancer Res. 53, 3186–3192. [PubMed] [Google Scholar]
- Griffith, T.D. , Tolmach, L.J. , 1976. Lethal response of HeLa cells to x-irradiation in the latter part of the generation cycle. Biophys. J. 16, 303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennequin, C. , Giocanti, N. , Balosso, J. , Favaudon, V. , 1994. Interaction of ionizing radiation with the topoisomerase I poison camptothecin in growing V-79 and HeLa cells. Cancer Res. 54, 1720–1728. [PubMed] [Google Scholar]
- Houlbrook, S. , Addison, C.M. , Davies, S.L. , Carmichael, J. , Stratford, I.J. , Harris, A.L. , Hickson, I.D. , 1995. Relationship between expression of topoisomerase II isoforms and intrinsic sensitivity to topoisomerase II inhibitors in breast cancer cell lines. Br. J. Cancer. 72, 1454–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. , Deerinck, T.J. , Ellisman, M.H. , Spector, D.L. , 1998. The perinucleolar compartment and transcription. J. Cell Biol. 143, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, P.B. , Sehested, M. , 1997. DNA topoisomerase II rescue by catalytic inhibitors: a new strategy to improve the antitumor selectivity of etoposide. Biochem. Pharmacol. 54, 755–759. [DOI] [PubMed] [Google Scholar]
- Ju, B.G. , Lunyak, V.V. , Perissi, V. , Garcia-Bassets, I. , Rose, D.W. , Glass, C.K. , Rosenfeld, M.G. , 2006. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 312, 1798–1802. [DOI] [PubMed] [Google Scholar]
- Kalwinsky, D.K. , Look, A.T. , Ducore, J. , Fridland, A. , 1983. Effects of the epipodophyllotoxin VP-16-213 on cell cycle traverse, DNA synthesis, and DNA strand size in cultures of human leukemic lymphoblasts. Cancer Res. 43, 1592–1597. [PubMed] [Google Scholar]
- Kantidze, O.L. , Iarovaia, O.V. , Razin, S.V. , 2006. Assembly of nuclear matrix-bound protein complexes involved in non-homologous end joining is induced by inhibition of DNA topoisomerase II.J. Cell Physiol. 207, 660–667. [DOI] [PubMed] [Google Scholar]
- Kaufmann, S.H. , 1991. Antagonism between camptothecin and topoisomerase II-directed chemotherapeutic agents in a human leukemia cell line. Cancer Res. 51, 1129–1136. [PubMed] [Google Scholar]
- Keogh, M.C. , 2006. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 439, 497–501. [DOI] [PubMed] [Google Scholar]
- Kim, M.S. , Blake, M. , Baek, J.H. , Kohlhagen, G. , Pommier, Y. , Carrier, F. , 2003. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 63, 7291–7300. [PubMed] [Google Scholar]
- Kobayashi, M. , Adachi, N. , Aratani, Y. , Kikuchi, A. , Koyama, H. , 2001. Decreased topoisomerase IIalpha expression confers increased resistance to ICRF-193 as well as VP-16 in mouse embryonic stem cells. Cancer Lett. 166, 71–77. [DOI] [PubMed] [Google Scholar]
- Lieber, M.R. , 2008. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 283, 1–5. [DOI] [PubMed] [Google Scholar]
- Lieber, M.R. , Ma, Y. , Pannicke, U. , Schwarz, K. , 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell. Biol. 4, 712–720. [DOI] [PubMed] [Google Scholar]
- Long, B.H. , Musial, S.T. , Brattain, M.G. , 1985. Single- and double-strand DNA breakage and repair in human lung adenocarcinoma cells exposed to etoposide and teniposide. Cancer Res. 45, 3106–3112. [PubMed] [Google Scholar]
- Lou, Z. , Chen, B.P. , Asaithamby, A. , Minter-Dykhouse, K. , Chen, D.J. , Chen, J. , 2004. MDC1 regulates DNA-PK autophosphorylation in response to DNA damage. J. Biol. Chem. 279, 46359–46362. [DOI] [PubMed] [Google Scholar]
- Mao, Y. , Desai, S.D. , Ting, C.Y. , Hwang, J. , Liu, L.F. , 2001. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 276, 40652–40658. [DOI] [PubMed] [Google Scholar]
- Marchion, D.C. , Bicaku, E. , Daud, A.I. , Richon, V. , Sullivan, D.M. , Munster, P.N. , 2004. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J. Cell. Biochem. 92, 223–237. [DOI] [PubMed] [Google Scholar]
- McClendon, A.K. , Osheroff, N. , 2007. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 623, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco, A. , Biamonti, G. , 2007. Cellular response to etoposide treatment. Cancer Lett. 252, 9–18. [DOI] [PubMed] [Google Scholar]
- Nitiss, J.L. , Liu, Y.X. , Harbury, P. , Jannatipour, M. , Wasserman, R. , Wang, J.C. , 1992. Amsacrine and etoposide hypersensitivity of yeast cells overexpressing DNA topoisomerase II. Cancer Res. 52, 4467–4472. [PubMed] [Google Scholar]
- Relling, M.V. , Yanishevski, Y. , Nemec, J. , Evans, W.E. , Boyett, J.M. , Behm, F.G. , Pui, C.H. , 1998. Etoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemia. Leukemia. 12, 346–352. [DOI] [PubMed] [Google Scholar]
- Rogakou, E.P. , Redon, C. , Boon, C. , Johnson, K. , Bonner, W.M. , 2000. Rapid histone extraction for electrophoretic analysis. Biotechniques. 28, 38–40. 42, 46 [DOI] [PubMed] [Google Scholar]
- Roos, W.P. , Kaina, B. , 2006. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 12, 440–450. [DOI] [PubMed] [Google Scholar]
- Sedelnikova, O.A. , Rogakou, E.P. , Panyutin, I.G. , Bonner, W.M. , 2002. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res. 158, 486–492. [DOI] [PubMed] [Google Scholar]
- Shrivastav, M. , De Haro, L.P. , Nickoloff, J.A. , 2008. Regulation of DNA double-strand break repair pathway choice. Cell Res. 18, 134–147. [DOI] [PubMed] [Google Scholar]
- Soubeyrand, S. , Torrance, H. , Giffin, W. , Gong, W. , Schild-Poulter, C. , Hache, R.J. , 2001. Activation and autoregulation of DNA-PK from structured single-stranded DNA and coding end hairpins. Proc. Natl. Acad. Sci. USA. 98, 9605–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubeyrand, S. , Pope, L. , Pakuts, B. , Hache, R.J. , 2003. Threonines 2638/2647 in DNA-PK are essential for cellular resistance to ionizing radiation. Cancer Res. 63, 1198–1201. [PubMed] [Google Scholar]
- Tanaka, T. , Halicka, H.D. , Traganos, F. , Seiter, K. , Darzynkiewicz, Z. , 2007. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to cell cycle phase. Cell Cycle. 6, 371–376. [DOI] [PubMed] [Google Scholar]
- Toyoda, E. , 2008. NK314, a topoisomerase II inhibitor that specifically targets the alpha isoform. J. Biol. Chem. 283, 23711–23720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J.C. , 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell. Biol. 3, 430–440. [DOI] [PubMed] [Google Scholar]
- Willmore, E. , Frank, A.J. , Padget, K. , Tilby, M.J. , Austin, C.A. , 1998. Etoposide targets topoisomerase IIalpha and IIbeta in leukemic cells: isoform-specific cleavable complexes visualized and quantified in situ by a novel immunofluorescence technique. Mol. Pharmacol. 54, 78–85. [DOI] [PubMed] [Google Scholar]
- Wist, E. , Prydz, H. , 1979. The effect of aphidicolin on DNA synthesis in isolated HeLa cell nuclei. Nucleic Acids Res. 6, 1583–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woessner, R.D. , Mattern, M.R. , Mirabelli, C.K. , Johnson, R.K. , Drake, F.H. , 1991. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 2, 209–214. [PubMed] [Google Scholar]
- Wright, S.E. , Hines, L.H. , White, J.C. , 1990. Effects of the lipophilic anticancer drug teniposide (VM-26) on membrane transport. Chem. Biol. Interact. 75, 31–48. [DOI] [PubMed] [Google Scholar]
- Xiao, H. , Mao, Y. , Desai, S.D. , Zhou, N. , Ting, C.Y. , Hwang, J. , Liu, L.F. , 2003. The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway. Proc. Natl. Acad. Sci. USA. 100, 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, A. , Lyu, Y.L. , Lin, C.P. , Zhou, N. , Azarova, A.M. , Wood, L.M. , Liu, L.F. , 2006. A protease pathway for the repair of topoisomerase II–DNA covalent complexes. J. Biol. Chem. 281, 35997–36003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data