

Abstract
Germline-expressed endogenous siRNAs (endo-siRNAs) transmit multigenerational epigenetic information to ensure fertility in subsequent generations. In C. elegans, nuclear RNAi ensures robust inheritance of endo-siRNAs and deposition of repressive H3K9me3 marks at target loci. How target silencing is maintained in subsequent generations is poorly understood. We discovered that morc-1 is essential for transgenerational fertility and acts as an effector of endo-siRNAs. Unexpectedly, morc-1 is dispensable for siRNA inheritance but required for target silencing and maintenance of siRNA-dependent chromatin organization. A forward genetic screen identified mutations in met-1, which encodes a H3K36 methyltransferase, as potent suppressors of morc-1(−) and nuclear RNAi mutant phenotypes. Further analysis of nuclear RNAi and morc-1(−) mutants revealed a progressive, met-1-dependent enrichment of H3K36me3, suggesting that robust fertility requires repression of MET-1 activity at nuclear RNAi targets. Without MORC-1 and nuclear RNAi, MET-1-mediated encroachment of euchromatin leads to detrimental decondensation of germline chromatin and germline mortality.
ETOC BLURB
eTOC Blurb: In C. elegans, germline immortality requires the transmission of epigenetic information via small non-coding RNAs that promote histone modifications. Weiser et al. implicate MORC-1, a highly conserved ATPase, in the transgenerational maintenance of chromatin organization downstream of small RNAs. MORC-1 prevents the encroachment of MET-1 mediated H3K26me3 into heterochromatin.
Introduction
Germline proliferation and maintenance are key processes in organismal development. The faithful transmission of genetic and epigenetic information to the next generation is essential to these processes, enabling the production of functional gametes and the subsequent generation. The ability of germ cells to undergo infinite cellular divisions makes the germline an immortal cell lineage (Smelick and Ahmed, 2005). How this immortality is achieved remains a central question in developmental biology.
The rapid development and genetic tractability of the model organism C. elegans makes it an ideal system in which to study the molecular mechanisms of germline immortality. Recent studies in C. elegans have identified epigenetic regulators that are critical for transgenerational maintenance of fertility, including regulators of DNA damage repair and telomere maintenance, as well as histone modifying enzymes (Ahmed and Hodgkin, 2000; Andersen and Horvitz, 2007; Greer et al., 2014; Katz et al., 2009; Meier et al., 2009; Zeller et al., 2016). Epigenetic regulation, such as post-translational modification of histones and DNA methylation, is a conserved mechanism for repression of repetitive elements and germline maintenance in most metazoans (Hajkova et al., 2002; Santos and Dean, 2004; Smelick and Ahmed, 2005). For example, the highly conserved family of Microrchidia (Morc) ATPases, first described in mice as a critical regulator of spermatogenesis (Inoue et al., 1999), has recently been found to repress transposons in mice and plants via regulation of chromatin superstructure (Harris et al., 2016; Moissiard et al., 2014; 2012; Pastor et al., 2014).
Although repression of deleterious genomic elements, such as transposons, is critical for germline immortality, how that repression is targeted, maintained, and transmitted to the next generation is unclear. In recent years, germline-enriched small non-coding RNAs, such as endogenous siRNAs (endo-siRNAs) and Piwi-interacting RNAs (piRNAs), have been implicated in transposon repression (Batista et al., 2008; Ketting et al., 1999; Tabara et al., 2002). In C. elegans, endo-siRNAs and piRNAs collectively regulate genome defense by targeting deleterious or foreign transcripts, such as transposons, pseudogenes, and transgenes, for repression (Ashe et al., 2012; Batista et al., 2008; Das et al., 2008; de Albuquerque et al., 2015; Han et al., 2009; Kim et al., 2005; Lee et al., 2012; Luteijn et al., 2012; Phillips et al., 2015; Shirayama et al., 2012; Tabara et al., 1999). Furthermore, these pathways are able to regulate gene expression transgenerationally via germline nuclear RNAi (Ashe et al., 2012; Buckley et al., 2012; Grishok et al., 2000; Gu et al., 2012; Luteijn et al., 2012; Shirayama et al., 2012), and, thus, are thought to transmit a memory of genomic “self” and “non-self” elements.
Recent genetic screens for nuclear RNAi defective (Nrde) and heritable RNAi defective (Hrde) mutants have identified the core factors that transmit this memory (Ashe et al., 2012; Buckley et al., 2012; Burkhart et al., 2011; Gu et al., 2012; Guang et al., 2010; 2008; Luteijn et al., 2012; Shirayama et al., 2012). These factors (HRDE-1/WAGO-9, NRDE-1, -2, and -4) comprise the germline nuclear RNA-induced silencing complex (RISC), which maintains transgenerational gene silencing through heritable, siRNA-mediated mechanisms (Ashe et al., 2012; Buckley et al., 2012; Gu et al., 2012; Ni et al., 2016; Shirayama et al., 2012). The germline nuclear RISC is guided by endo-siRNAs to nascent pre-mRNA transcripts to mediate RNA polymerase II stalling and deposition of H3K9 methylation at corresponding genomic loci (Buckley et al., 2012; Burkhart et al., 2011; Gu et al., 2012; Guang et al., 2010). Loss of the germline nuclear RNAi pathway by mutation of hrde-1 or nrde-1, -2, or -4 leads to defective inheritance of silencing by nuclear RNAi, germline mortality, and a progressive depletion of H3K9me3 marks at endogenous target sites (Buckley et al., 2012). Although previous studies have implicated the histone methyltransferases SET-25 and MET-2 in siRNA-directed H3K9 methylation (Ashe et al., 2012; Mao et al., 2015), the mechanisms by which siRNAs regulate heterochromatin deposition and maintenance remain unclear.
Here, we uncover morc-1, the single C. elegans homolog of the Morc family, as an effector of the nuclear RNAi pathway that acts downstream of the canonical NRDE factors to regulate transgenerational chromatin organization. Like hrde-1(−), nrde-1(−), -2(−), and -4(−) mutants, morc-1(−) mutants display a germline mortal phenotype. Through a forward genetic screen, we discovered that mutations in met-1, which encodes an H3K36 histone methyltransferase (Andersen and Horvitz, 2007), suppress germline mortality of morc-1(−) and nuclear RNAi mutants. Our data support a model in which loss of nuclear RNAi or morc-1 triggers met-1-dependent spread of H3K36 trimethylation and chromatin decondensation, compromising germline maintenance. We show that the integration of endo-siRNAs with germline chromatin organization via MORC-1 and the subsequent antagonism of MET-1 activity are critical for transgenerational germline maintenance.
Results
morc-1 is required for nuclear RNAi
We previously demonstrated that MORC-1 is required for transgene silencing in worms (Moissiard et al., 2012). Given that transgene silencing depends on many RNAi pathway genes (Dernburg et al., 2000; Ketting and Plasterk, 2000), we hypothesized that MORC-1 may also play a role in the germline nuclear RNAi pathway and multigenerational epigenetic inheritance. First, we investigated MORC-1 localization by introducing a 3xFlag tag at the C- terminus of MORC-1 using the CRISPR-Cas9 system (Paix et al., 2015). Immunofluorescence confirmed that MORC-1::3xFlag is expressed in the nuclei of germline and somatic cells (Figures 1A and S1A), consistent with a possible germline function for MORC-1. Indeed, we found that morc-1(−) mutants exhibit a temperature-sensitive germline mortality phenotype with a progressive decline in fertility with each generation at 25°C until they become completely sterile after 5–6 generations (Figure 1B, left). This defect is shared by mutants of the germline nuclear RISC complex, such as hrde-1(−) (Figure 1B) (Buckley et al., 2012).
Figure 1.
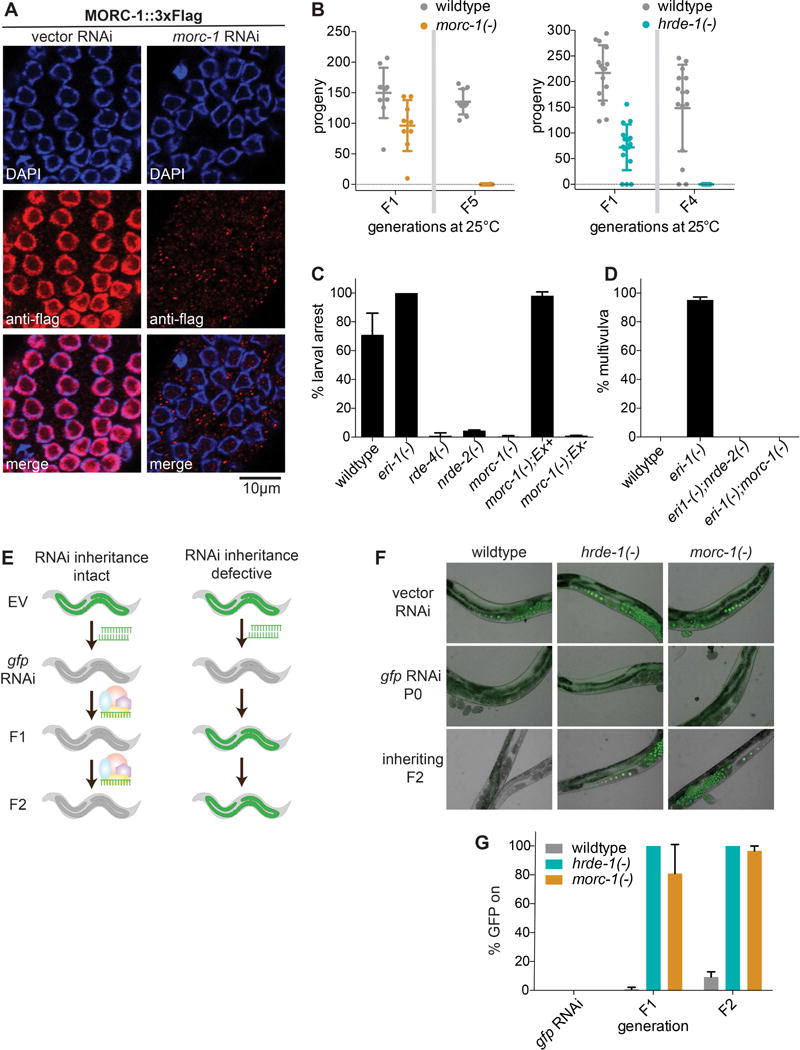
morc-1 regulates nuclear RNAi in the soma and germline. (A) Anti-Flag immunofluorescence and DAPI staining of worms expressing morc-1::3xflag at the endogenous morc-1 locus grown for two generations on empty vector or morc-1 RNAi. (B) morc-1(−) and hrde-1(−) are germline mortal at 25°C. Circles indicate the total number of viable progeny of individual, self-fertilized worms at the indicated generation. Error bars indicate mean ± SD, n≥10. (C) morc-1(−) mutants are resistant to lir-1 RNAi; this phenotype is rescued by expression of a morc-1::gfp extrachromosomal array (Ex+) driven by the ubiquitous dpy-30 promoter. Non-transgenic siblings are resistant to lir-1 RNAi. Percent larval arrest represents mean of two biological replicates ± SD; n≥85. (D) morc-1(−) suppresses the sensitivity of eri-1(−) to lin-15b RNAi after two generations on lin-15b RNAi. Percent multivulva indicates mean of three biological replicates ± SD; n≥100. (E) Model for RNAi inheritance experiments. EV denotes empty vector RNAi. (F) morc-1(−) and hrde-1(−) are defective for RNAi inheritance. On gfp RNAi, silencing of gfp is completely penetrant. Both morc-1(−) and hrde-1(−) fail to maintain gfp silencing in F1 and F2 generations. (G) Quantification of % animals with expressed GFP in (F) showing mean of two biological replicates ± SD; n≥100. See also Figure S1.
To determine if MORC-1 participates in nuclear RNAi, we tested whether morc-1(−) mutant worms recapitulate other key phenotypes of canonical nuclear RNAi mutants, including resistance to nuclear RNAi and defective RNAi inheritance (Buckley et al., 2012; Burton et al., 2011; Guang et al., 2008). We evaluated nuclear RNAi function using siRNAs that target polycistronic pre-mRNAs. For instance, the nonessential gene lir-1 is transcribed within a polycistronic pre-mRNA that includes the essential gene lin-26. siRNAs targeting lir-1 silence the lir-1/lin-26 polycistronic pre-mRNA via nuclear RNAi, triggering larval arrest and lethality due to loss of lin-26 (Guang et al., 2008). We found that morc-1(−) mutants were highly resistant to lir-1 nuclear RNAi, phenocopying nrde-2(−) mutants. This resistance was rescued with a morc-1::gfp transgene (Figure 1C). Furthermore, the genes lin-15a and lin-15b are expressed together within a polycistronic pre-mRNA, and loss of both genes is required to produce a multi-vulva (Muv) phenotype. RNAi against lin-15b is sufficient to cause Muv with almost complete penetrance in the enhanced RNAi mutant eri-1(−); this phenotype is fully suppressed in the nuclear RNAi mutant nrde-2(−) (Guang et al., 2010). Like nrde-2(−), morc-1(−) fully suppressed the enhanced sensitivity of eri-1(−) mutants to lin-15b RNAi (Figure 1D).
Next, we tested whether morc-1 is required for RNAi inheritance using worms expressing a germline gfp::h2b reporter. Exposure to gfp RNAi for only one generation is sufficient to silence this reporter for many subsequent generations (Ashe et al., 2012; Buckley et al., 2012) (Figure 1E). Although gfp silencing was 100% penetrant in the parental P0 generation of morc-1(−) mutants, transgenerational maintenance of gfp silencing was defective, with most worms (67–95%) activating GFP expression in the filial F1 generation and nearly all worms (96–99%) expressing GFP by the F2 generation (Figures 1F and 1G). Taken together, these data indicate that MORC-1 functions in the germline nuclear RNAi pathway.
MORC proteins in plants, worms, and mammals are homologous to ATPases that alter chromatin superstructure (Dutta and Inouye, 2000; Iyer et al., 2008). To investigate the role of the MORC-1 ATPase domain, we generated a putative ATP hydrolysis-dead variant, MORC-1(E39A)::3xFlag, using CRISPR/Cas9 to edit the morc-1::3xflag gene at the endogenous morc-1 locus. Intriguingly, we found that the E39A variant is overexpressed relative to MORC-1(WT)::3xFlag (Figures S1B and S1C). We found that the E39A variant recapitulates the phenotypes of the morc-1(−) mutant: germline mortality (Figure S1D), resistance to lir-1 RNAi (Figure S1E), and defective RNAi inheritance (Figure S1F). The germline mortality and RNAi inheritance defects are less severe in the E39A variant compared to the deletion mutant, suggesting that it is a hypomorphic allele; therefore, we focused our subsequent studies on the morc-1(−) mutant.
morc-1 mediates endo-siRNA effector function
Based on our finding that MORC-1 is required for transgenerational silencing, we next asked where in the nuclear RNAi pathway MORC-1 functions. To determine if MORC-1 affects siRNA inheritance, we fed gfp dsRNA to adult wildtype, hrde-1(−), and morc-1(−) mutants expressing the germline gfp::h2b reporter (Figure 1E). We detected 22G siRNAs against gfp in parental (P0) as well as in the first and second inheriting generations (F1 and F2) by qRT-PCR. As expected, hrde-1(−) mutants displayed a small, but consistent, loss of siRNAs antisense to gfp at the F2 generation (Figure 2A). In contrast, morc-1(−) mutants expressed wild-type levels of siRNAs in all three generations (Figures 2A and S1G). These data suggest that unlike hrde-1, which is required for inheritance of siRNA expression in progeny, morc-1 is dispensable for siRNA biogenesis and inheritance. Thus, morc-1 functions downstream of exogenous siRNAs and the germline nuclear RISC complex.
Figure 2.
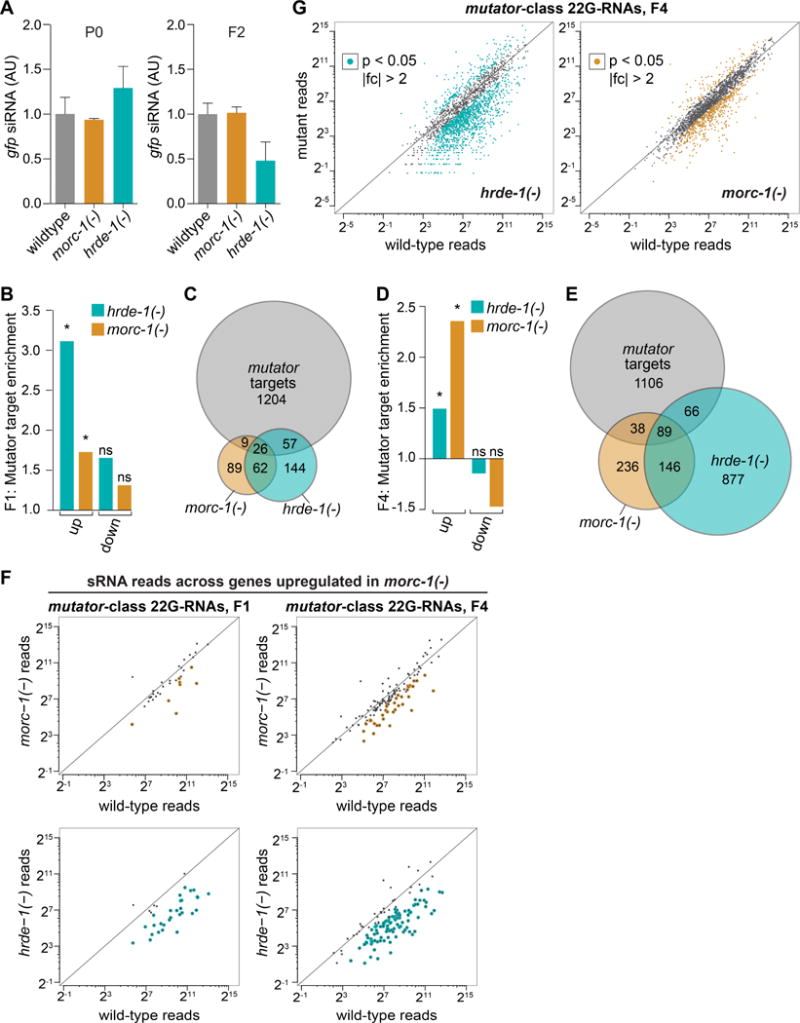
morc-1 mediates nuclear RNAi effector function. (A) morc-1 is dispensable for siRNA biogenesis and inheritance. TaqMan qPCR and quantification of a representative 22G siRNA targeting gfp normalized to U18 snoRNA at P0 and F2 generations. Levels are shown in arbitrary units with wildtype levels set to one as mean ± SD of two technical replicates. To see changes in siRNA levels between generations (i.e. wildtype not set to one), see Figure S1B. (B) Upregulated mRNAs in F1 hrde-1(−) and morc-1(−) mutants are significantly enriched for targets of mutator endo-siRNAs (hrde-1(−): p<6.6×10−16, morc-1(−): p=0.00253, Fisher’s test). Downregulated genes are not significantly enriched (ns; hrde-1(−): p=0.263, morc-1(−): p=0.785, Fisher’s test). (C) Overlap of hrde-1(−) and morc-1(−) upregulated targets with all testable mutator targets in F1 libraries. (D) Upregulated mRNAs in F4 hrde-1(−) and morc-1(−) mutants are significantly enriched for mutator targets (hrde-1(−): p=1.0758×10−8, morc-1(−): p<6.6×10−16, Fisher’s test). Downregulated genes are not enriched (hrde-1(−): p=1, morc-1(−): p=1, Fisher’s test) (E) Overlap of hrde-1(−) and morc-1(−) upregulated targets with all testable mutator targets in F4 libraries. In (C) and (E), differentially expressed mRNA lists were compared to mutator targets that had enough reads in both hrde-1(−) and morc-1(−) to test for differential expression; in a three-way comparison, not all targets are testable. (F) Quantification of 22G endo-siRNA reads across mutator targets that are upregulated in morc-1(−) mutants at F1 and F4. Upper panels: morc-1(−) vs. wildtype; yellow indicates endo-siRNAs that are >2-fold depleted, p<0.05. Lower panels: hrde-1(−) vs. wildtype; blue indicates endo-siRNAs that are >2-fold depleted, p<0.05. (G) Expression of all mutator-class 22G endo-siRNAs in late generation hrde-1(−) and morc-1(−) vs. wildtype. 22G endo-siRNAs with >2-fold change and p<0.05 in mutant vs. wildtype are highlighted (morc-1(−) vs. wildtype: yellow, hrde-1(−) vs. wildtype: blue). See also Figures S1 and S2.
To investigate whether morc-1 functions as an effector of endo-siRNAs, we performed mRNA-seq of wildtype, hrde-1(−), and morc-1(−) worms. To resolve changes in gene expression as germline function declines, we collected worms after two generations at 25°C (F1 or “early generation”) and after five generations at 25°C (F4/F5 or “late generation”) (Figure S1H). We compared mRNA-seq libraries from these samples using a significance threshold of p<0.05. Early generation morc-1(−) and hrde-1(−) worms showed upregulation of 284 and 379 mRNAs, respectively (Figure S2A), with significant enrichment for targets of the mutator pathway, which is required for amplification of 22G endo-siRNAs (Phillips et al., 2014; 2012; Zhang et al., 2011) (target enrichment in hrde-1(−): p<6.6×10−16, in morc-1(−): p=0.00253, Fisher’s test) (Figures 2B and 2C). Gene upregulation was more severe in late generation mutant worms, with 1,316 genes upregulated in morc-1(−) and 1,506 genes upregulated in hrde-1(−) (Figure S2B and Table S1). As with the early generation targets, morc-1(−) and hrde-1(−)-upregulated genes were significantly enriched for targets of the mutator endo-siRNAs (target enrichment in hrde-1(−): p=1.0758×10−8, morc-1(−): p<6.6×10−16) (Figures 2D, 2E, and Table S2). In contrast, genes that were downregulated in morc-1(−) and hrde-1(−) mutants were not enriched for mutator targets (Figures 2B, 2D, S2C, and S2D). These data indicate that morc-1 is required for silencing some endo-siRNA target genes.
In order to assess if the upregulation of the mutator targets observed in morc-1(−) was due to loss of the corresponding 22G endo-siRNAs, we performed 5′ monophosphate-independent small RNA-seq and compared 22G endo-siRNA levels in wildtype worms to morc-1(−) and hrde-1(−) mutants. We quantified 22G endo-siRNA reads across the mutator target mRNAs that were significantly upregulated in morc-1(−) mutants at early or late generation. Although some 22G endo-siRNAs were downregulated in morc-1(−) mutants compared to wildtype (Figure 2F, top), hrde-1(−) mutants exhibited a more substantial reduction of 22G endo-siRNAs corresponding to these mRNAs (Figure 2F, bottom). Next, we quantified the 22G endo-siRNAs corresponding to mutator targets that were upregulated in the hrde-1(−) mutants. Again, loss of morc-1 had much less of an effect among these 22G endo-siRNAs compared to the loss of hrde-1 (Figure S2E). Thus, we conclude that the desilencing of mutator targets in morc-1(−) mutants cannot be fully explained by a loss of endo-siRNAs, consistent with our findings regarding exogenous RNAi. Analysis of global levels of mutator-dependent 22G endo-siRNAs, irrespective of mRNA regulation, showed some endo-siRNA depletion in late generation morc-1(−) mutants compared to wildtype (Figure 2G and Tables S3 and S4). We posit that this reduction reflects the relative loss of germline tissue in morc-1(−) mutants compared to wildtype, rather than a specific role for morc-1 in regulating endo-siRNA levels. Together, our data suggest that morc-1 is required for target silencing but is dispensable for siRNA biogenesis and inheritance and, thus, must function downstream of RISC.
morc-1 is required for maintenance of H3K9me3 marks at a subset of hrde-1 targets
Given that endo-siRNA levels were largely unaffected in morc-1(−) mutants, we asked if morc-1 might regulate siRNA effector function at the level of chromatin. Based on our model that morc-1 functions downstream of hrde-1, we looked specifically at whether morc-1 is (1) required for siRNA-directed methylation of histone 3 on lysine 9 (H3K9) and (2) regulates a common set of loci with hrde-1. We performed H3K9me3 chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) on early and late generation wildtype, morc-1(−), and hrde-1(−) worms. We analyzed the H3K9me3 signal over 1 kb windows across the entire genome. Using a 1.5-fold threshold and false-discovery rate (FDR) of <0.05, we identified 57 1 kb regions that were depleted of H3K9me3 in early generation morc-1(−) mutants compared to wildtype (Figure 3A). Late generation (F4) morc-1(−) mutants exhibited a much larger defect; 206 1 kb windows were depleted of H3K9me3 in late generation morc-1(−) compared to wildtype (Figures 3A and S3A). We refer to these 206 regions as morc-1-dependent H3K9me3 loci. Importantly, these regions are strongly enriched for endo-siRNAs, confirming that morc-1 regulates H3K9me3 at endo-siRNA targets (Figure 3B).
Figure 3.
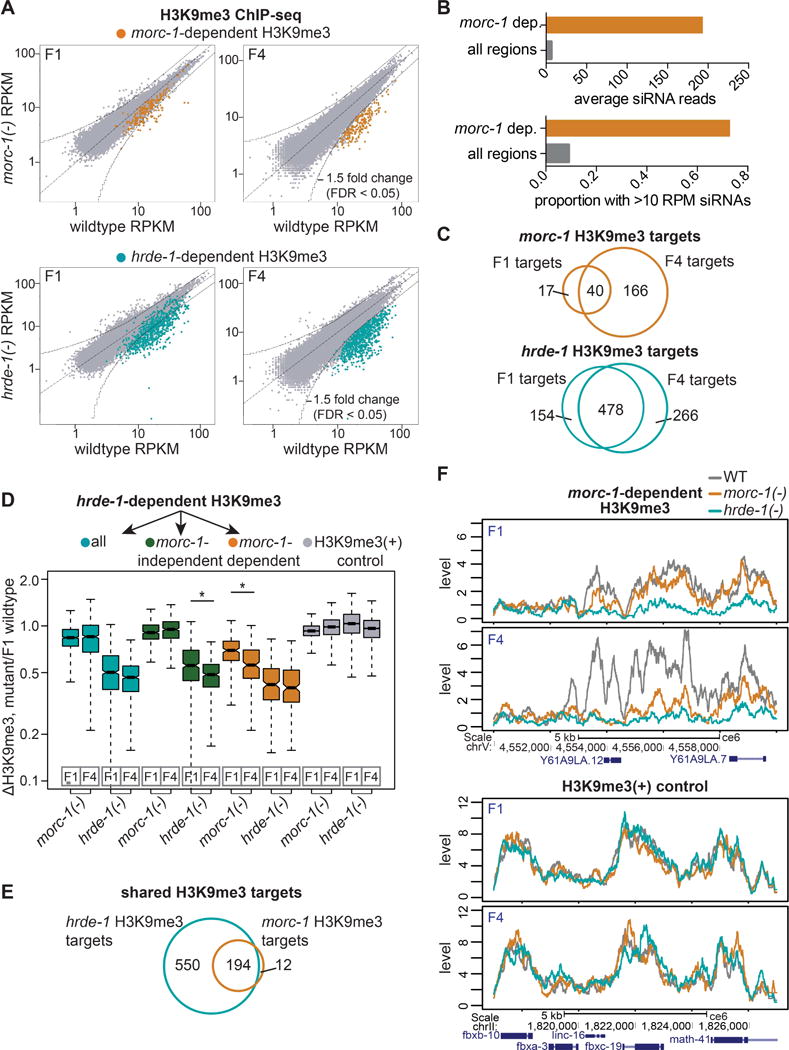
morc-1 is required for H3K9me3 maintenance at a subset of hrde-1 targets. (A) RPKM of 1 kb windows in wildtype (x-axis) vs. mutant (y-axis) H3K9me3 ChIP-seq libraries in F1 and F4 generations. Indicated in yellow and blue: regions that are >1.5-fold-depleted with FDR<0.05 in F4 morc-1(−) and hrde-1(−) for two biological replicates. (B) morc-1-dependent H3K9me3 regions are enriched for endo-siRNAs. Top: average wildtype siRNA reads across morc-1-dependent H3K9me3 1 kb regions compared to all 1 kb regions in the genome. Bottom: proportion of morc-1-dependent H3K9me3 1 kb regions with wildtype siRNA reads >10 RPM compared to 1 kb regions over the entire genome. (C) Overlap of regions that are depleted of H3K9me3 in F1 and F4 generations in morc-1(−) and hrde-1(−) vs. wildtype. (D) Box plot representing the ratio of H3K9me3 levels in the indicated mutant to F1 wildtype. Blue boxes: all hrde-1-dependent regions. Green boxes: hrde-1-dependent, morc-1-independent regions show significant, progressive H3K9me3 loss from F1 to F4 in hrde-1(−) mutants (p<2.2×10−16, Welch’s t-test). Yellow boxes: hrde-1-dependent, morc-1-dependent regions are significantly more depleted of H3K9me3 in morc-1(−) F4 compared to morc-1(−) F1 (p=8.0×10−8, Welch’s t-test). Gray boxes: H3K9me3 positive control regions. (E) Overlap of morc-1-dependent and hrde-1-dependent H3K9me3 regions at F4. (F) H3K9me3 levels in the indicated genetic background at F1 and F4 in a morc-1-dependent region (top) and an unregulated control region (bottom). See also Figures S1 and S3.
We observed a progressive increase in the number of H3K9me3-depleted sites from early to late generation morc-1(−) mutants. Furthermore, most of the H3K9me3-depleted regions in the early generation morc-1(−) mutants were also depleted in the late generation (40 of 57, Figure 3C). Relative to F1 wildtype, H3K9me3 levels at morc-1-dependent loci decreased significantly from F1 to F4 generation in morc-1(−) mutants (p=8.0×10−8, Welch’s t-test), supporting our conclusion that H3K9me3 loss in morc-1(−) mutants is progressive (Figure 3D). These findings indicate that morc-1 is required for maintenance of H3K9me3 at these regions.
Strikingly, of the 206 morc-1-dependent H3K9me3 loci, 194 were also hrde-1-dependent, indicating that morc-1 regulates a subset of hrde-1 targets (Figure 3E). Relative to F1 wildtype, these regions were more severely H3K9me3-depleted in hrde-1(−) mutants than in morc-1(−) mutants, but the effect was not progressive (Figure 3D). Thus, in morc-1(−) mutants, a subset of hrde-1 targets are progressively depleted of H3K9me3 (Figures 3D–F and S3B).
Similar to our mRNA-seq results, hrde-1 affects a larger number of sites than morc-1 in both generations (Figures 3A and 3C). We identified 744 hrde-1-dependent H3K9me3 loci from a comparison of late generation hrde-1(−) mutants and wildtype (Figure S3A). The majority of these (550) were morc-1-independent loci (Figure 3E). These regions exhibited progressive H3K9me3 depletion from F1 to F4 generation in hrde-1(−) mutants compared to wildtype (p<2.2×10−16, Welch’s t-test) but were unaffected in morc-1(−) mutants (Figures 3D and S3C). Taken together, these results indicate that morc-1 is required for maintenance of H3K9me3 at a subset of endogenous nuclear RNAi targets (Figures 3D, 3F, and S3B).
morc-1 is required for heterochromatin localization and compaction
To elucidate further the role of morc-1 and nuclear RNAi in chromatin organization, we investigated the localization of a heterochromatin reporter in embryos (Towbin et al., 2012). The high-copy gfp-lacI::lacO array is normally heterochromatinized and localized to the nuclear periphery in embryos; its proper localization is dependent on methylation at H3K9 by SET-25 and MET-2 (Towbin et al., 2012). The ubiquitously expressed GFP-LacI fusion protein binds to the lacO sites, revealing the position of the array relative to the nuclear periphery (Meister et al., 2010). We examined the localization of GFP-LacI by fluorescence microscopy in morc-1(−) (F4 generation at 25°C), nrde-2(−) (F1 generation at 25°C), and hrde-1(−) (F1 generation at 25°C) mutant embryos compared to set-25(−) and met-2(−);set-25(−) controls. Approximately 90% of GFP-LacI foci were localized to the outermost third of the nucleus in wild-type embryos, whereas only ~70% of GFP-LacI foci were correctly localized in morc-1(−), nrde-2(−), and hrde-1(−) mutant embryos (Figures 4A–4D). The degree of displacement of the array from the nuclear periphery observed in these mutants is similar to the defect that we and others have observed in set-25(−) mutants (Figure 4A) (Towbin et al., 2012). These data reveal that morc-1 and nuclear RNAi are required for proper localization of heterochromatin to the nuclear periphery.
Figure 4.
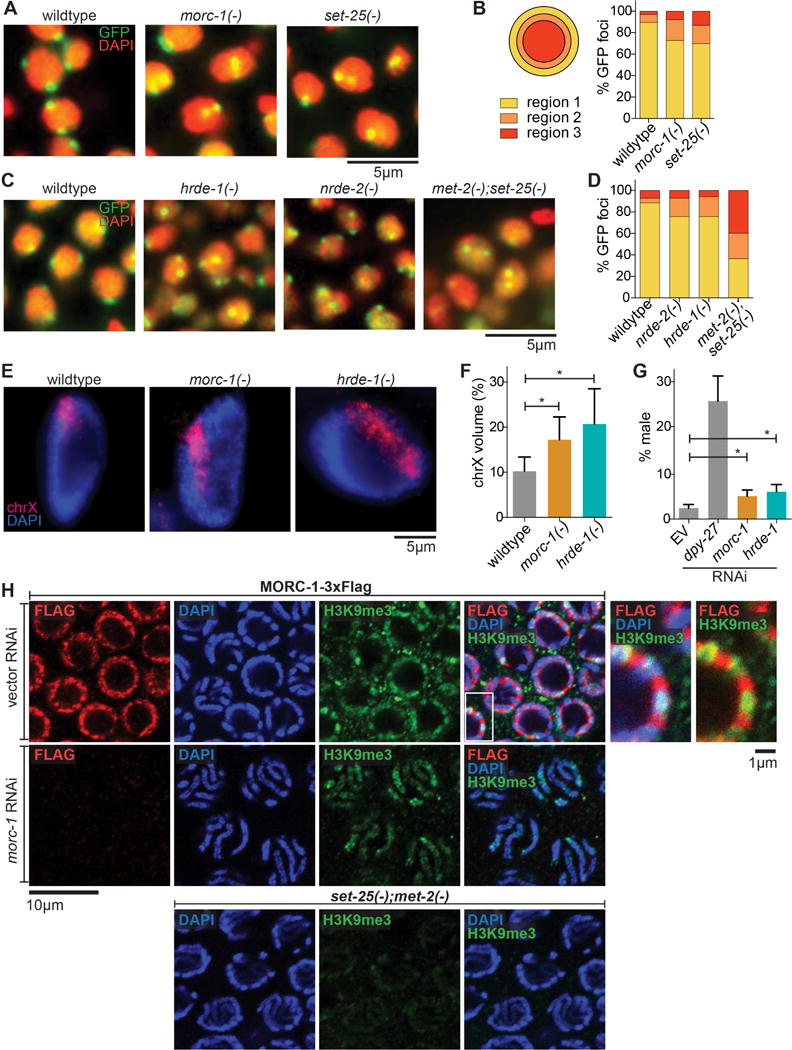
morc-1 is required for heterochromatin localization and compaction. (A) Heterochromatin localization is defective in morc-1(−) mutants. DAPI-stained embryo nuclei of the indicated genotype expressing a high-copy gwIs4[gfp-lacI::lacO] array at F4 generation at 25°C. set-25(−) is a control for mislocalization (B) Quantification is performed by determining the proportion of GFP foci in each of three concentric regions of equal volume (n≥60). (C) Localization of gwIs4 in hrde-1(−) and nrde-2(−) mutant embryos at F1 generation at 25°C using met-2(−);set-25(−) as a control for mislocalization. (D) shows quantification (n≥60) of data represented in (C). (E) Defective X chromosome compaction in intestinal nuclei of morc-1(−) and hrde-1(−) mutants. DNA FISH using a probe targeting the X chromosome and DAPI staining shows increased X chromosome volume in morc-1(−) and hrde-1(−). (F) X chromosome volume as a percentage of nuclear volume (mean±SD, n≥16) is significantly increased in morc-1(−) and hrde-1(−) intestinal nuclei (morc-1(−) vs. wildtype p=1.7×10−5, hrde-1(−) vs. wildtype p=9.6×10−6, student’s t-test). (G) RNAi against morc-1 and hrde-1 partially rescues xol-1(−);sex-1(−);him-8(−) male lethality (morc-1 vs. empty vector p=0.0053, hrde-1 vs. empty vector p=0.0018, student’s t-test). The percentage of live progeny that are male is shown as a mean of five biological replicates ± SD, n≥85. (H) Immunostaining against H3K9me3 and Flag in a strain expressing MORC-1::3xFlag on empty vector RNAi (top row). At right: high-magnification images of the region indicated by the white box with and without the DAPI channel. RNAi against morc-1 for two generations abrogates anti-Flag signal (middle row). met-2(−);set-25(−) mutants serve as a negative control for H3K9me3 staining (bottom row).
To examine the effects of morc-1 and nuclear RNAi on the chromatin structure of endogenous sequences, we performed X chromosome paint fluorescence in situ hybridization (FISH) to evaluate compaction of the X chromosome. In hermaphrodite worms, both X chromosomes are targeted by the dosage compensation complex (DCC) to decrease their transcriptional activity by half (Meyer and Casson, 1986). As a result, the X chromosomes in hermaphrodites are highly compacted, occupying about 10% of the nuclear volume despite containing 18% of the genome (Lau et al., 2014). This compaction is also dependent on methylation at H3K9 (Snyder et al., 2016). We utilized this unique feature of the hermaphrodite X chromosomes to determine whether chromatin compaction is defective in morc-1(−) and hrde-1(−) mutants. The X chromosomes in morc-1(−) and hrde-1(−) hermaphrodites each occupied 17% and 20%, respectively, of the nuclear volume compared to 10% in wildtype hermaphrodites, indicating a significant compaction defect (morc-1(−) vs. wildtype: p=1.7×10−5, hrde-1(−) vs. wildtype p=9.6×10−6, student’s t-test) (Figures 4E and 4F). Furthermore, morc-1 and hrde-1 RNAi caused a small, but significant, rescue of the male lethality triggered by aberrant targeting of the DCC to the single X chromosome in males (Carmi et al., 1998; Miller et al., 1988; Petty et al., 2009) (morc-1 vs. vector: p=0.0053, hrde-1 vs. vector: p=0.0018, student’s t-test) (Figure 4G). The magnitude of rescue achieved by knockdown of morc-1 or hrde-1 is similar to that observed with loss of H3K9 methylation (Snyder et al., 2016). RNAi against dpy-27, which encodes a DCC component, induced a more substantial rescue of male lethality, as expected (Meyer and Casson, 1986). Thus, morc-1 and hrde-1 regulate condensation of endogenous chromatin.
Some mammalian MORC proteins directly bind H3 (Andrews et al., 2016; Li et al., 2016; Liu et al., 2016). We sought to directly identify MORC-1 genomic targets by ChIP-seq of MORC-1(WT)::3xFlag and MORC-1(E39A)::3xFlag. These experiments did not yield sufficient enrichment over background, suggesting that MORC-1 may interact with chromatin indirectly or in a highly transient manner. Intriguingly, co-immunofluorescence revealed that MORC-1::3xFlag localizes adjacent to, but not overlapping with, H3K9me3 in the distal germline (Figure 4H). Furthermore, in pachytene nuclei, which have highly condensed chromosomes that can be visualized with 4′,6-diamidino-2-phenylindole (DAPI) staining, we observed MORC-1::3xFlag at the nuclear periphery and adjacent to, but not overlapping with, the condensed chromosomes, seemingly as a barrier around condensed chromatin (Figure 4H). Together, these data indicate that MORC-1 regulates heterochromatin modifications, localization, and compaction, although it may be physically excluded from highly condensed chromatin.
Mutations in the gene encoding MET-1 suppress morc-1(−) and hrde-1(−) germline mortality
To elucidate the mechanism behind the progressive fertility defect of morc-1(−) mutants (Figure 1B), we performed a forward genetic screen to identify suppressors of morc-1(−) germline mortality. ENU-mutagenized morc-1(−) mutants were grown for 14 generations at 25°C (Figure 5A), thus selecting for suppressor mutations that enable robust fertility far beyond when unmutagenized morc-1(−) mutants become sterile (i.e. 5–6 generations at 25°C). We generated clonal lines from the most fertile worms and performed whole genome sequencing. We identified four independent alleles (xk1-4) causing missense or nonsense mutations in the gene encoding the H3K36me3 histone methyltransferase MET-1 (Andersen and Horvitz, 2007). Two of these alleles introduce substitutions in the SET domain (xk1 and xk4). The xk4 allele contains two additional substitutions outside the SET domain (Figure 5B).
Figure 5. Mutations in met-1 suppress morc-1(−) germline mortality. (A).
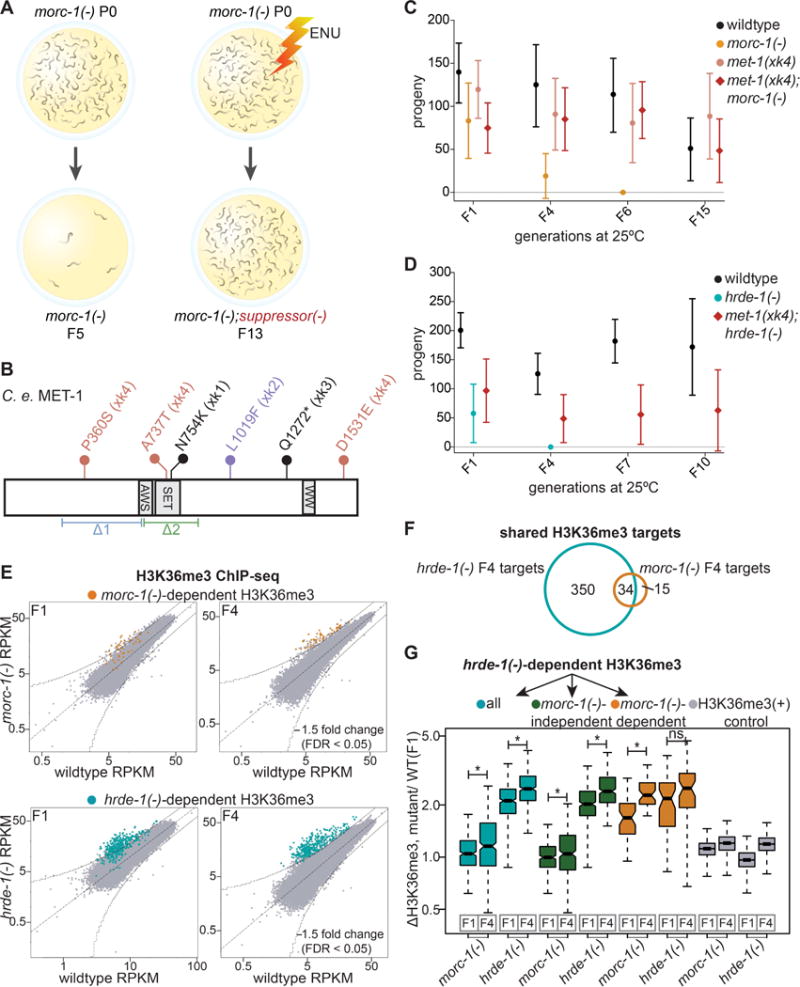
Overview of forward genetic screen to identify morc-1 suppressors. (B) Schematic of C. elegans MET-1 (isoform b). Flags indicate the alleles generated by our screen (xk1-4). The xk4 allele contains three missense mutations. Indicated below are the pre-existing deletion alleles, n4337(Δ1), and ok2172(Δ2). (C) met-1(xk4) rescues morc-1(−) and (D) hrde-1(−) germline mortality. Mean fertility ± SD is plotted for each genetic background at each generation (n=16). For brood size of individual worms in these experiments, see Figures S4B and S4C. (E) RPKM of 1 kb windows in wildtype (x-axis) vs. mutant (y-axis) H3K36me3 ChIP libraries in F1 and F4 generations. Indicated in yellow and blue are 1 kb regions >1.5-fold-enriched in F4 morc-1(−) and hrde-1(−) respectively with FDR of <0.05 in two biological replicates. (F) Overlap of morc-1(−) and hrde-1(−)-dependent H3K36me3 regions. (G) Box plot representing the ratio of H3K36me3 levels in the indicated mutant vs. F1 wildtype. hrde-1(−)-dependent regions (blue) show significant increase in H3K36me3 from F1 to F4 in both morc-1(−) (p=3.07×10−7, Welch’s t-test) and hrde-1(−) (p<2.2×10−16, Welch’s t-test) mutants. hrde-1(−)-dependent, morc-1(−)-independent regions (green) show significant increase in H3K36me3 from F1 to F4 in hrde-1(−) (p<2.2×10−16, Welch’s t-test) and morc-1(−) mutants (p=0.0012, Welch’s t-test). hrde-1(−)-dependent, morc-1(−)-dependent regions (yellow) are significantly more enriched in morc-1(−) F4 vs. morc-1(−) F1 (p=6.44×10−11, Welch’s t-test) but not significantly different in hrde-1(−) F1 vs. F4 (p=0.011, Welch’s t-test). Gray boxes show H3K36me3 levels regions with high H3K36me3 in F1 wildtype. See also Figures S4, S5, and S6.
We outcrossed the four met-1 alleles ten times and then crossed them into morc-1(−). All four met-1 alleles potently suppressed morc-1(−) germline mortality (Figures 5C, S4A, and S4B). To determine whether met-1 also suppresses the canonical nuclear RNAi mutants, we crossed met-1 alleles xk2 and xk4 into hrde-1(−) mutants. We found that both met-1 alleles also suppressed hrde-1(−) germline mortality (Figures 5D and S4C). However, the two publicly available met-1 deletions alleles, n4337 and ok2172, did not rescue morc-1(−) germline mortality (Figure S4D). Both met-1 deletion alleles, but not met-1 alleles xk2 and xk4, severely compromised fertility at 25°C (Figures 5C, S4B, and S4D). We propose that a hypomorphic allele of met-1, rather than a complete loss-of-function allele, might be required to rescue morc-1(−) germline mortality. This is consistent with our finding that depletion of met-1 by RNAi also suppressed morc-1(−) germline mortality (Figure S4E). These data suggest that at 25°C, the germline can tolerate partial, but not complete, loss of met-1 activity. Thus, depletion of MET-1 rescues germline mortality caused by loss of morc-1 or nuclear RNAi.
MET-1 mediates H3K36 trimethylation of some endo-siRNA target genes in the absence of MORC-1
We hypothesized that MET-1 might oppose nuclear RNAi by marking nuclear RNAi target genes with trimethylated H3K36, a hallmark of euchromatin. To test this hypothesis, we profiled H3K36me3 genome-wide in early and late generation wildtype, morc-1(−), and hrde-1(−) worms by ChIP-seq. We found that late generation morc-1(−) and hrde-1(−) mutants exhibited increased H3K36me3 compared to wildtype (Figure 5E). Using a 1.5-fold threshold and FDR<0.05, we identified 49 1 kb regions that are enriched for H3K36me3 in late generation morc-1(−) compared to wildtype (Figures 5E and S5A). We refer to these as morc-1(−)-dependent H3K36me3 regions. As with morc-1 regulation of H3K9me3, morc-1 regulates H3K36me3 at a subset of the loci that are also regulated by hrde-1 (Figure 5F). We observed a significant increase in H3K36me3 levels at morc-1(−)-dependent sites from early to late generations in morc-1(−) mutants (p=6.44×10−11, Welch’s t-test) (Figures 5G and S5B), similar to the progressive loss of H3K9 methylation in morc-1(−) mutants. These sites also exhibited elevated H3K36me3 levels at both generations in hrde-1(−) mutants, but there was no significant increase from early to late generation (Figure 5G).
Comparing H3K36me3 levels in hrde-1(−) to wildtype, we identified 384 hrde-1(−)-dependent H3K36me3-enriched regions (Figures 5E and S5A). Of these, the majority (350) are morc-1(−)-independent (Figure 5F). At morc-1(−)-independent loci, there was a significant gain of H3K36me3 from early to late generation in both hrde-1(−) and morc-1(−) mutants (hrde-1(−): p<2.2×10−16, morc-1(−): p=0.0012, Welch’s t-test) (Figures 5G and S5C). This suggests that morc-1 regulates a larger proportion of hrde-1(−)-dependent sites than we have identified, but not severely enough to meet our 1.5-fold threshold for defining morc-1(−)-dependent sites. As a control, we used regions that have high levels of H3K36me3 in F1 wildtype; these regions did not show regulation by hrde-1 or morc-1 at either generation (Figure 5G). Taken together, these data demonstrate that morc-1(−) and hrde-1(−) mutants progressively gain H3K36me3 at select loci.
Most regions that gained H3K36me3 in mutant backgrounds also lost H3K9me3. Of the 49 morc-1(−)-dependent H3K36me3 regions, 27 were depleted of H3K9me3 by >1.5-fold in morc1(−) mutants (Figures S6A and S6B). In hrde-1(−) mutants, 291 of the 384 H3K36me3-enriched regions were also H3K9me3-depleted (Figures S6A and S6B). For example, the endo-siRNA target bath-45 was depleted of H3K9me3 and enriched for H3K36me3 in early and late generation hrde-1(−) mutants and in late generation morc-1(−) mutants relative to wildtype (Figure 6A). Control regions showed no change in H3K36me3 enrichment (Figure S5B, bottom). Thus, most nuclear RNAi targets that gain H3K36me3 also lose H3K9me3 in morc-1(−) and hrde-1(−) mutants.
Figure 6. Expansion of met-1-dependent H3K36me3 in morc-1(−) and hrde-1(−) mutants. (A).
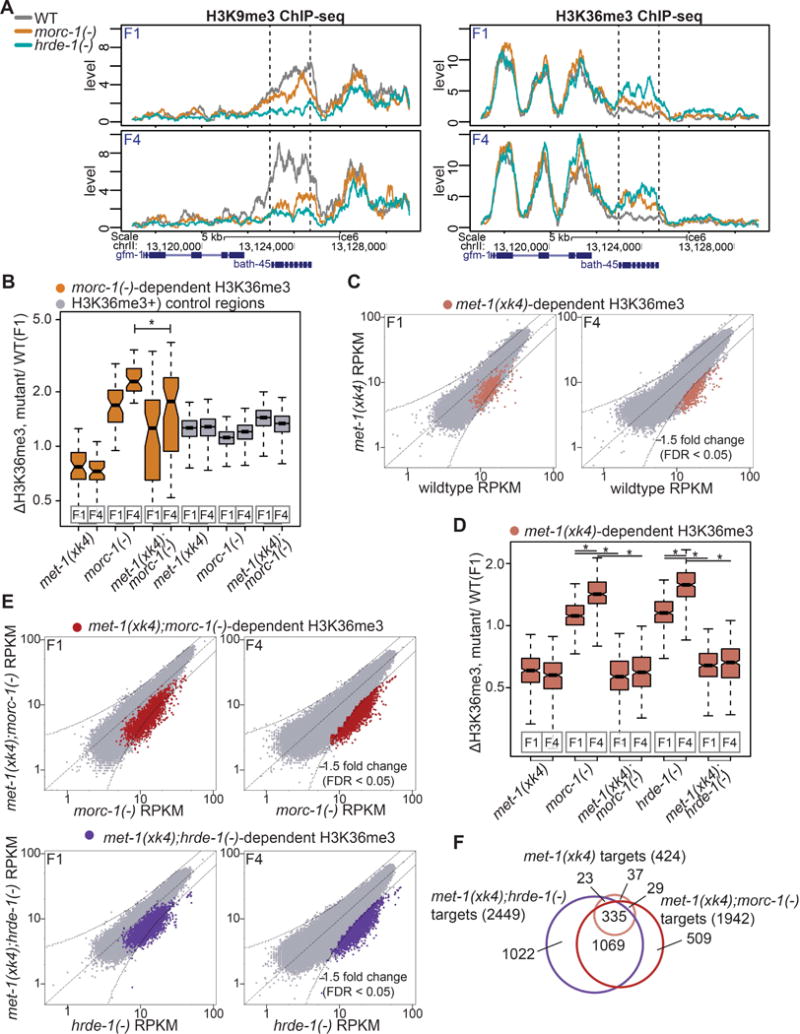
H3K9me3 (left) and H3K36me3 (right) levels in the indicated genetic background at F1 and F4 at morc-1(−)-dependent region containing bath-45, indicated by dotted lines. (B) morc-1(−)-dependent H3K36me3 regions are significantly depleted of H3K36me3 in met-1(xk4);morc-1(−) vs. morc-1(−) at F4 generation (p=2.50×10−5, Welch’s t-test). Gray boxes show H3K36me3 levels in regions with highest H3K36me3 in F1 wildtype. (C) Scatter plots showing H3K36me3 levels in met-1(xk4) vs. wildtype at early and late generation. Highlighted in pink are targets that >1.5-fold depleted in F4 met-1(xk4) compared to wildtype with FDR<0.05 in two biological replicates. (D) Box plot demonstrating that at met-1(xk4)-dependent regions, H3K36me3 levels are progressively elevated in morc-1(−) and hrde-1(−) mutants (morc-1(−) F1 vs. F4: p<2.2×10−16, hrde-1(−) F1 vs. F4: p<2.2×10−16, Welch’s t-test). H3K36me3 gain at these sites is met-1(xk4)-dependent (morc-1(−) vs. met-1(xk4);morc-1(−) at F1 and F4: p<2.2×10−16, hrde-1(−) vs. met-1(xk4);hrde-1(−) at F1 and F4: p<2.2×10−16, Welch’s t-test). (E) Scatter plots showing H3K36me3 levels in met-1(xk4);morc-1(−) vs. morc-1(−) at F1 and F4 generation and in met-1(xk4);hrde-1(−) vs. hrde-1(−) (bottom). Regions >1.5-fold depleted of H3K36me3 with FDR<0.05 in the met-1(xk4) background at F4 are highlighted in red (met-1(xk4);morc-1(−)-dependent) and purple (met-1(xk4);hrde-1(−)-dependent). (F) Overlap of 1 kb regions that are depleted in met-1(xk4) vs. wildtype, met-1(xk4);morc-1(−) vs. morc-1(−), and met-1(xk4);hrde-1(−) vs. hrde-1(−). See also Figures S5, S6, and S7.
To assess how morc-1 and hrde-1 affect H3K9me3 and H3K36me3 marks over larger genomic intervals, we plotted relative H3K9me3 and H3K36me3 coverage 20 kb up and downstream from the 5′ end of the 744 hrde-1-dependent H3K9me3 loci (Figure S6C). For visual clarity, we assigned the 5′ end of the 1 kb regions to position zero and plotted average coverage at 10 bp resolution. We performed the same analysis with the 206 morc-1-dependent H3K9me3 loci (Figure S6D). We observed that in the wildtype background, both hrde-1-dependent and morc-1-dependent loci occupy local maxima of H3K9me3 enrichment and local minima of H3K36me3 enrichment. In morc-1(−) and hrde-1(−) mutants, we observed strong depletion of H3K9me3 over a span of several kb and concurrent enrichment of H3K36me3 to a level similar to the flanking regions or to a local maximum (Figures S6C and S6D). Taken together, these data show that loci that lose H3K9me3 in hrde-1(−) and morc-1(−) mutants also gain H3K36me3, suggesting that HRDE-1 and MORC-1 may repress encroachment of H3K36me3 from neighboring loci. Intriguingly, we observed that in the distal germline, H3K36me3 localizes to the nuclear periphery adjacent to H3K9me3, very similar to MORC-1::3xFlag (Figure S6E), suggesting that MORC-1 may associate with H3K36-methylated chromatin.
To determine whether the enrichment of H3K36me3 in morc-1(−) and hrde-1(−) backgrounds is met-1-dependent, we performed H3K36me3 ChIP-seq in early (F1) and late generation (F4) met-1(xk4), met-1(xk4);morc-1(−), and met-1(xk4);hrde-1(−) mutants (Figure S6F). First, we compared H3K36me3 levels at the 49 sites with morc-1(−)-dependent H3K36me3 enrichment. Indeed, the H3K36me3 enrichment observed at these loci in late generation morc-1(−) mutants was partially suppressed in met-1(xk4);morc-1(−) mutants (p=2.50×10−5, Welch’s t-test) (Figure 6B). Second, we compared H3K36me3 levels at the 384 loci with hrde-1(−)-dependent H3K36me3 enrichment. At these loci, the H3K36me3 enrichment observed in late generation hrde-1(−) mutants was also partially suppressed in met-1(xk4);hrde-1(−) mutants (F1: p=3.28×10−14, F4: p=1.15×10−10, Welch’s t-test) (Figure S7A). We then asked whether met-1 contributes to loss of H3K9me3 or upregulation of endo-siRNA targets. By H3K9me3 ChIP-seq, we found that H3K9me3 levels, including at morc-1-dependent loci, were not affected by met-1(xk4) (Figure S7B). By qRT-PCR, we observed that met-1(xk4) slightly suppressed upregulation of some endo-siRNA targets in late generation morc-1(−), but the targets were still overexpressed relative to wildtype (Figure S7C). met-1(xk4) does not affect endo-siRNA target upregulation in hrde-1(−) mutants or the expression levels of germline-expressed genes that are not targeted by endo-siRNAs (Figures S7C and S7D).
Taken together, these data indicate that upon loss of morc-1 or hrde-1, the primary role of MET-1 is the deposition of H3K36me3 marks. The finding that H3K9me3 marks are not restored in met-1(xk4);morc-1(−) mutants suggests that it is not loss of H3K9 methylation per se that drives germline mortality, but rather an imbalance of heterochromatin and euchromatin that is incompatible with transgenerational germline maintenance.
morc-1 restricts met-1-dependent H3K36 trimethylation genome-wide
Having shown that the ectopic H3K36me3 observed in morc-1(−) and hrde-1(−) mutants is partially met-1-dependent, we next wanted to identify met-1-dependent H3K36 methylated regions in wildtype worms with an intact nuclear RNAi pathway. Therefore, we compared H3K36me3 in met-1(xk4) and wildtype worms by ChIP-seq. We observed a progressive loss of H3K36me3 from early to late generation in met-1(xk4) mutants compared to wildtype (Figure 6C), confirming that the xk4 allele is in fact hypomorphic and suggesting that met-1 participates in transgenerational maintenance of H3K36me3 marks. We identified 424 loci that were depleted of H3K36me3 in met-1(xk4) compared to wildtype (Figure S6F). We observed a progressive increase in H3K36me3 in morc-1(−) and hrde-1(−) mutants compared to F1 wildtype at these regions (morc-1(−) F1 vs. F4: p<2.2×10−16, hrde-1(−) F1 vs. F4: p<2.2×10−16, Welch’s t-test). The increased H3K36me3 at these loci is met-1-dependent (Figure 6D). Thus, MET-1 activity at its normal targets in wildtype worms is increased upon loss of morc-1 or hrde-1.
Next, we wanted to determine if the distribution of met-1-dependent H3K36 methylated regions is altered in morc-1(−) or hrde-1(−) backgrounds by comparing H3K36me3 levels in met-1(xk4);morc-1(−) vs. morc-1(−) and met-1(xk4);hrde-1(−) vs. hrde-1(−) mutants. We identified 1,942 regions that were depleted of H3K36me3 in met-1(xk4);morc-1(−) compared to morc-1(−) and 2,449 loci that were depleted of H3K36me3 in met-1(xk4);hrde-1(−) compared to hrde-1(−) (Figures 6E and S6F). These data indicate that loss of morc-1 or hrde-1 dramatically increases the number of met-1-dependent loci (Figure 6F). Taken together, these data suggest that the chromatin decompaction induced by loss of morc-1 or hrde-1 is associated with an increase in met-1-dependent H3K36 trimethylation throughout the genome.
met-1 depletion restores chromatin organization in morc-1(−) mutants
We next asked how the increased H3K36 methylation in morc-1(−) affects germline chromatin by whole-mount DAPI staining of adult wildtype, morc-1(−), and met-1(xk4);morc-1(−) worms in the F5 generation at 25°C. All of the morc-1(−) mutants were sterile and exhibited pleiotropic germline defects. Consistent with the decondensation of the X chromosome in intestinal nuclei of morc-1(−) hermaphrodites (Figure 4E), distal germline nuclei were enlarged and lacked the uniformity or regular spacing of wildtype germline nuclei (Figure 7A). In the loop region and the proximal germline, we did not observe the highly-condensed chromosomes or the single-file line of nuclei characteristic of diplotene and diakinetic nuclei, suggesting failure to properly complete meiotic prophase (Figure 7A). In contrast, these defects were largely restored in met-1(xk4);morc-1(−) mutants (Figure 7A). Thus, in the absence of MORC-1, a partial loss of the histone H3K36 methyltransferase MET-1 and reduction of H3K36me3 are sufficient to compensate for reduced transgenerational maintenance of H9K9me3 and restore germline immortality and chromatin organization.
Figure 7. Germline chromatin decompaction in morc-1(−) mutants is met-1-dependent. (A).
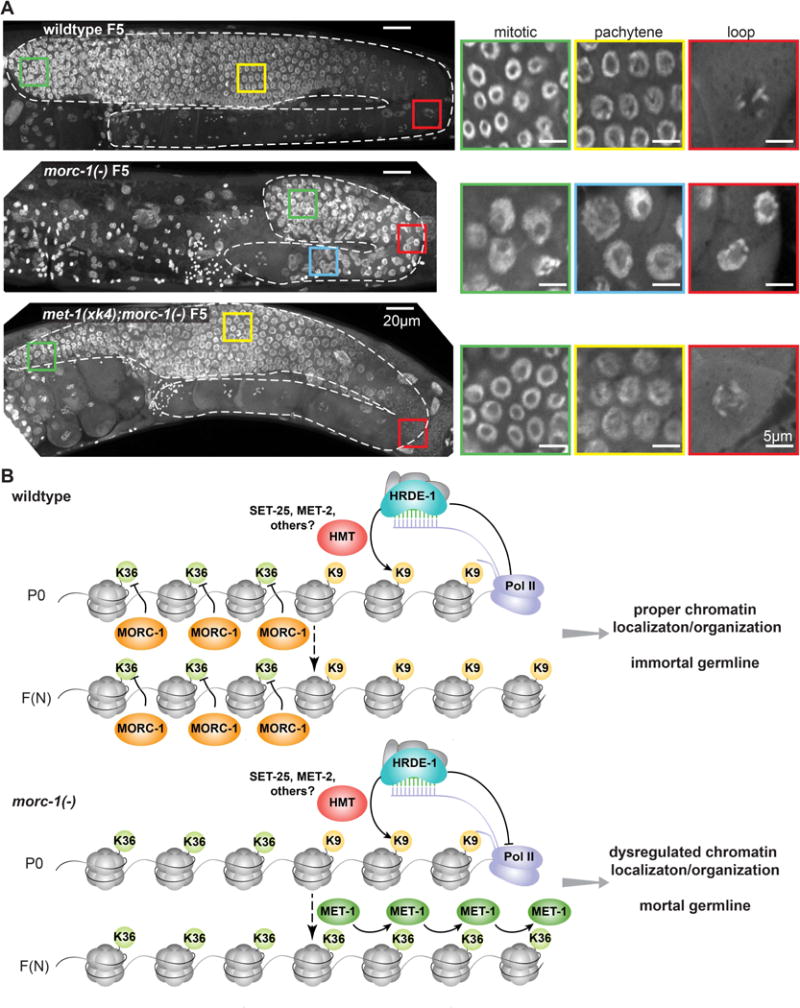
Whole mount DAPI staining of wildtype, morc-1(−), and met-1(xk4);morc-1(−) worms at F5 at 25°C. morc-1(−) worms are sterile. Left: maximum projection images from top to bottom of the germline. The germline is outlined in dashed white lines; colored boxes indicate location of insets shown at right. Right: single plane from the indicated region. Due to chromatin disorganization in morc-1(−) mutants, we cannot distinguish mitotic and pachytene regions based on nuclear morphology. (B) Model for MORC-1-mediated regulation of transgenerational chromatin marks at HRDE-1-target loci. In wildtype worms, MORC-1 restrains the spread of H3K36me3 into H3K9me3-marked endo-siRNA targets, allowing H3K9me3 to be maintained in subsequent generations. In morc-1(−) mutants, MET-1 mediates the spread of H3K36me3 to these targets, causing progressive loss of H3K9me3 and germline mortality.
Discussion
Here, we demonstrate that morc-1 functions as a critical link between endo-siRNAs and heritable chromatin modifications to ensure germline integrity. We found that morc-1 is essential for H3K9me3 maintenance at a subset of endogenous nuclear RNAi targets (Figure 3). Why do some nuclear RNAi targets require MORC-1 for H3K9me3 maintenance while others do not? Our data indicate that hrde-1(−) mutants are more severely depleted of H3K9me3 at morc-1-dependent loci than at morc-1-independent loci (Figure 3D). Based on our anchored analysis showing that morc-1-dependent H3K9me3 sites reside in local maxima of H3K9me3 and local minima of H3K36me3 (Figure S6D), we speculate that proximity to euchromatin may contribute to susceptibility to loss of heterochromatin marks, thus necessitating a protective factor, such as MORC-1, to prevent the spread of H3K36me3.
We propose a model in which MORC-1 restrains the spread of H3K36me3 into a subset of regions that are targeted by HRDE-1 and endo-siRNA for heterochromatinization (Figure 7B), thereby protecting heterochromatin compaction and localization. A role in restricting the spread of H3K36me3 would be consistent with the striking MORC-1 localization pattern observed in pachytene nuclei, where MORC-1, like H3K36me3, is adjacent to H3K9me3-marked regions at the nuclear periphery (Figures 4H and S6E). Because the anti-H3K36me3 and anti-Flag antibodies were both raised in mouse, we could not directly test colocalization of MORC-1::3xFlag and H3K36me3, but these data are consistent with an association of MORC-1 with H3K36 methylated chromatin.
We posit that the imbalance arising from loss of heterochromatin and expansion of euchromatin is likely a major driver of the germline mortality phenotype of morc-1(−) mutants. In support of this model (Figure 7B), we identified mutations in the gene encoding the H3K36me3 histone methyltransferase MET-1 as potent suppressors of morc-1(−) and hrde-1(−) germline mortality (Figures 5A–D and S4). Thus, in a morc-1(−) mutant, H3K36me3 expansion drives loss of H3K9me3, germline chromatin disorganization, and germline mortality (Figure 7B). The expansion of H3K36me3 can be mitigated by mutating met-1, thus restoring the balance of heterochromatin and euchromatin without restoring H3K9 methylation at endo-siRNA targets (Figure S7E). These studies highlight an essential role for nuclear RNAi and MORC-1 in transgenerational control of chromatin structure and emphasize the critical role of chromatin architecture in multigenerational germline maintenance.
In support of a role for MORC-1 in restraining the expansion of H3K36me3, we observed a progressive increase in H3K36me3 in morc-1(−) mutants compared to that in wildtype (Figures 5E and 5G). While the number of sites that meet our 1.5-fold threshold for enrichment of H3K36me3 in morc-1(−) mutants is small (49), we posit that this is reflective of a larger trend for several reasons. First, we observe a dramatic increase in the number of met-1(xk4)-dependent loci in the morc-1(−) background compared to in the wildtype background (Figure 6F). This would be consistent with these loci exhibiting a small amount of met-1-dependent H3K36me3 in wildtype, gain of H3K36me3 in morc-1(−) mutants, and depletion to below wildtype levels in met-1(xk4);morc-1(−) mutants. In such cases, it is possible that only the depletion in met-1(xk4);morc-1(−) mutants would be severe enough to meet our 1.5-fold threshold. Second, our sample collection method for ChIP-seq studies selected against mutants with the strongest enrichment of H3K36me3. Due to the large populations of worms required to perform robust ChIP, it was necessary to isolate embryos at each generation through hypochlorite treatment of gravid worms. Because the penetrance of germline mortality is quite variable in intermediate generations, this strategy enriched our samples for the most fertile worms. The potent suppression of morc-1(−) germline mortality by four met-1 alleles highlights the substantial impact of H3K36me3 on the fertility of morc-1(−) mutants. For experiments that did not require such large late generation samples, such as germline mortality assays and DAPI staining for germline morphology, we propagated worms by picking progeny to new plates before they reached adulthood. At these larval stages, we could not distinguish fertile and sterile worms and therefore did not introduce any selective pressure to enrich mutant populations for the most fertile worms.
Not only have we identified a downstream effector of the nuclear RNAi pathway, but also the first suppressor of a fertility defect of nuclear RNAi mutants, met-1. Our findings indicate that germline mortality may be actively regulated and not merely a stochastic decline in germline function. Furthermore, our study suggests that met-1 is an important regulator of the chromatin landscape in C. elegans even when the nuclear RNAi pathway is intact, as we observed a dramatic increase in the number of loci that are H3K36me3-depleted in met-1(xk4) compared to wildtype from F1 to F4 generation (Figure 6C). This suggests that MET-1 contributes to transgenerational maintenance of H3K36me3: a role that has been previously ascribed solely to the other H3K36 histone methyltransferase, MES-4 (Furuhashi et al., 2010; Rechtsteiner et al., 2010). Although mes-4 did not emerge from our screen as a candidate morc-1(−) suppressor, it may also contribute to the spread of H3K36me3 in morc-1(−) and hrde-1(−) mutants. Because loss of mes-4 causes maternal-effect sterility, it is unlikely that our screen, which was based on rescue of germline mortality, would have identified mes-4. Due to the progressive nature of the morc-1(−) mutant phenotypes and the complete penetrance of the maternal-effect-sterility resulting from mutation or RNAi knockdown of mes-4, we are currently unable to address whether mes-4 antagonizes morc-1. MES-4 and two H3K27 methyltransferases, MES-3 and MES-6, were recently implicated in nuclear RNAi-dependent H3K27 trimethylation (Mao et al., 2015).
Perhaps the most striking defect we observed in morc-1(−) mutants is the extensive disorganization and decondensation of germline chromatin in the late generation (Figure 7A). Our data suggest that the severely decondensed nuclei in the distal germline are not able to complete meiotic prophase. In fact, the entire germline is so abnormal that we cannot accurately distinguish the mitotic and meiotic zones in the distal germline. These defects are likely to be major contributors to germline mortality observed in morc-1(−) mutants. Although we cannot distinguish whether chromatin decompaction is the cause or consequence of euchromatic encroachment in morc-1(−) mutants, the restoration of germline architecture in late generation met-1(xk4);morc-1(−) mutants suggests that some degree of decompaction is driven by met-1-mediated H3K36me3 deposition. The dramatic increase in the number of loci that are regulated by met-1 in morc-1(−) and hrde-1(−) mutants (Figure 6F) as well as the increased H3K36me3 at sites that are normally regulated by met-1 (Figure 6D) suggest that nuclear RNAi and morc-1 repress met-1 activity at a large number of sites. In fact, our data suggest that morc-1 represses met-1 at a much larger number of sites than it regulates via H3K9me3 maintenance, highlighting a new role for nuclear RNAi in regulation of euchromatic marks.
Finally, our work suggests that endo-siRNAs are required not only for silencing discrete genetic loci, but also for maintaining chromatin organization. We have identified MORC-1 as a major effector and MET-1 as a major antagonist of this function. We expect that future studies will uncover additional factors connecting endo-siRNAs to global regulation of chromatin architecture.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John K. Kim (jnkim@jhu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strains were maintained using standard procedures at 20°C unless otherwise noted. Bristol N2 strain was used for wildtype control. All strains used in this study are listed in Key Resource Table. Worms were fed OP50 E. coli for all experiments that did not involve RNAi. RNAi experiments were performed with HT115 E. coli and the empty vector L4440 as a control.
METHOD DETAILS
Construction of transgenic strains
The ubiquitous morc-1::gfp extrachromosomal array was made using a 4.1 kb dpy-30 promoter, 2.9 kb of morc-1 genomic coding sequence with no termination codon, 0.9 kb gfp coding sequence, and 0.6 kb of tbb-2 3′UTR. This was injected at a concentration of 1.0 ng/μL with 2.0 ng/μL myo2::rfp co-injection marker. The morc-1::3xflag strain was made by standard Crispr/Cas9 methods (Paix et al., 2015). crRNA and repair template sequences are listed in Key Resource Table.
RNAi sensitivity assays
Bacterial RNAi clones were grown from the Ahringer library (Kamath et al., 2003). Synchronized L1 worms were plated on the indicated RNAi clone. Sensitivity to lir-1 RNAi was determined by counting arrested or dead worms 72 and 96 h after plating (to account for any developmental delay). Sensitivity to lin-15b RNAi was determined by counting adult worms showing multiple vulvas after two generations on lin-15b RNAi.
Germline mortality assays
Worms were maintained at 20°C, embryos were isolated through hypochlorite preparation, and hatched overnight in M9. Synchronized L1 worms were plated on OP50 at 25°C and designated as the P0 generation. Twenty of the progeny (F1 generation) of these worms were transferred to a new plate at the L2 or L3 stage. This was repeated for each generation. To test fertility in a given generation, L2 or L3 hermaphrodites were singled to new OP50 plates, and their live progeny were counted. The met-1 RNAi clone was generated by inserting 713 bp of met-1 coding sequence into the L4440 vector using SacII and KpnI restriction enzymes. The met-1 fragment was made with primers TGAATGGTACCTCAAATCCACTCTGC and ATCATCCGCGGCGCTTCAACTGCTAA.
RNAi inheritance assays
Synchronized L1 worms (P0 generation) were plated on empty vector L4440 or gfp RNAi. P0 gravid adults were subjected to hypochlorite treatment to generate F1 embryos. Synchronized L1 worms from F1 and subsequent generations were plated on OP50. At each generation, gravid adults were imaged with an Olympus BX61 microscope or collected in TriReagent (ThermoFisher) for RNA extraction. Germline nuclear GFP brightness was categorically scored as on or off.
Western blotting
Worms were grown on empty vector L4440 or morc-1 RNAi. 40 gravid worms were collected in Tris-glycine SDS sample buffer (ThermoFisher), run on 8% Novex WedgeWell Tris-Glycine precast gels (ThermoFisher), and transferred to PVDF membrane (Millipore). Antibodies used include Sigma F1804 (anti-Flag) and Abcam ab12079 (anti-H3) at 1:1,000 and 1:5,000, respectively. Western blot development was performed using a LI-COR Odyssey Fc according to the manufacturer’s instructions using Odyssey Blocking Buffer and IRDye secondary antibodies at 1:15,000 dilution (LI-COR).
RNA extraction, library preparation, and sequencing
Worms were collected in TriReagent (ThermoFisher) and subjected to three freeze-thaw cycles. After phase-separation using 1-bromo-3-chloropropane (BCP), RNA was precipitated in isopropanol at −30°C for 2 h. RNA was pelleted by centrifugation at 21,000 × g for 30 min at 4°C. After two washes in 70% ethanol and one wash in 75% ethanol, the pellet was resuspended in water. mRNA libraries were made using Illumina TruSeq Stranded mRNA Library Prep Kit for NeoPrep and an Illumina NeoPrep Library Prep machine according to manufacturer’s instructions. Phosphate independent small RNA libraries were made as described in (Phillips et al., 2014) using RNA 5′ polyphosphatase (Illumina) and the Illumina TruSeq Small RNA Library Prep Kit. Libraries were sequenced on the Illumina HiSeq 2000 platform.
Quantitative RT-PCR
TaqMan small RNA probes were made by Applied Biosystems. cDNA was made using 50 ng of total RNA using Multiscribe Reverse Transcriptase (Applied Biosystems). Analysis was performed using a Realplex Thermocycler (Eppendorf) with TaqMan Universal PCR Master Mix and No AmpErase® UNG (Applied Biosystems). A TaqMan small RNA probe detecting U18 was used for normalization of gfp 22G levels. Assays for mRNA levels were performed with Absolute Blue SYBR Green (ThermoFisher) and normalized to eft-2 using CFX63 Real Time System Thermocyclers (Biorad).
Heterochromatin localization assays
Worms expressing the gwIs4 reporter in the indicated backgrounds were maintained at 20°C. L4 hermaphrodites were picked to plates at 25°C to lay the P0 generation embryos. P0 L4s were picked to new plates to produce the F1 generation. At the specified generation (F4 for morc-1(−), F1 for nrde-2(−) and hrde-1(−)), embryos were dissected from day one adults, freeze-cracked, and DAPI stained as described in (Seydoux and Dunn, 1997). Embryos were imaged in GFP and DAPI channels with an Olympus BX61 microscope at 60× magnification across 10 μm of Z-stacks at 250 nm intervals. Deconvolution was performed in Huygens Essential software.
X chromosome staining
DNA FISH probes were prepared and used as described in (Csankovszki et al., 2004). In brief, adult worms were dissected in sperm salts (50 mM PIPES pH 7.0, 25 mM KCl, 1 mM MgSO4, 45 mM NaCl, 2 mM CaCl2) and fixed with 2% paraformaldehyde (PFA) for 5 min in a humid chamber. After freeze-cracking, slides were washed 3 times in PBST (1× PBS, 1 mM EDTA, 0.5% Triton X-100) for 10 min. Slides were then incubated in increasing concentrations of ethanol; that is, 2 min each in 70%, 80%, 95%, and 100% ethanol. FISH probes (10μL) were added to each slide, and then it was denatured on a 95°C heat block for 3 min. Slides were incubated in a humid chamber at 37°C overnight. The following washes were performed at 39°C: 5 min in 2xSSC/50% formamide (three times), 5 min in 2xSSC (three times), 10 min in 1xSSC (once). The final wash was performed in 4xSSC with 0.01 μg/mL DAPI for 15 min at room temperature. Slides were mounted with Vectashield (Vector Labs) and imaged on an Olympus BX61 microscope at 60× magnification.
Male rescue
him-8(−);xol-1(−);sex-1(−) embryos were isolated with hypochlorite and grown on the indicated RNAi clone or empty vector RNAi (L4440) and grown at 20°C. Worms were transferred as young adults to a new plate of the same RNAi clone (three worms per plate, four plates per RNAi clone) and allowed to lay embryos overnight. The next morning, adult worms were removed and embryos were counted. One day later, unhatched (dead) embryos were counted. Over the next two days, male worms were counted. Rescue of male lethality was calculated as the proportion of live embryos that developed into male worms.
Chromatin immunoprecipitation and sequencing
Frozen worm pellets (500 μL packed volume) were ground into powder and crosslinked with 2% formaldehyde in RIPA buffer (1xPBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) for 10 min at 4°C. Crosslinking was quenched using 100 μL 1M Tris-Cl pH 7.0 per 1 mL 2% formaldehyde in RIPA buffer. Crosslinked chromatin was sonicated using a Diagenode Bioruptor for three 8-min cycles on high amplitude, 30 sec on/off or a Diagenode BioruptorPico for three 3-min cycles, 30 sec on/off. Chromatin was nutated overnight at 4°C with 2 μg of the designated antibody and then for 2 h with 50μL Protein A (for H3K9me3 ChIP) or Protein G (for H3K36me3 ChIP) Dynabeads (Invitrogen). After three washes with 800 μL LiCl buffer (100 mM Tris-Cl pH 7.5, 500 mM LiCl, 1% NP-40, 1% sodium deoxycholate), chromatin was decrosslinked in worm lysis buffer (0.1 M Tris-Cl pH 7.5, 0.1 M NaCl, 50 mM EDTA, 1% SDS) for at least 4 h at 65°C. DNA was extracted by phenol-chloroform and dissolved in TE buffer. RNAse A (Invitrogen) treatment was performed for at least 1 h at 37°C. Antibodies used were Abcam ab8898 (H3K9me3) and WAKO 300-95289 (H3K36me3). For each ChIP sample, an input library was generated from 10% of the amount of chromatin in the IP. These samples were decrosslinked, and DNA extraction of input samples was performed as described above.
Fifty-one DNA ChIP-seq libraries were sequenced and used in this study. ChIP-seq libraries were made using NuGen Ovation Ultralow Library Systems v2 according to the manufacturer’s instructions. DNA ChIP-seq libraries for different samples were labeled with 6 nt index, pooled together, and sequenced. All libraries were sequenced using Illumina HiSeq 2000 platform: 50 nt single-end run with index sequencing. Demultiplexed raw data was provided by the sequencing provider in fastq format.
Immunofluorescence microscopy
Dissected adult worms were fixed in 2% formaldehyde and 1× sperm salts (50 mM PIPES pH 7.0, 25 mM KCl, 1 mM MgSO4, 45 mM NaCl, 2 mM CaCl2) for 5 min in a humid chamber. Samples were then freeze-cracked, fixed in methanol for 10 min at −20°C, and washed three times in PBST (PBS+0.2% Twee n-20). Slides were incubated with primary antibody in a humid chamber overnight at 4°C. Slides were washed three times in PBST and then incubated with secondary antibody in a humid chamber for 1 h at room temperature. Slides were washed three times in PBST, stained with 0.01 μg/mL DAPI in PBST for 15 min, and mounted with Vectashield (Vectorlabs H-1000). All washes were performed at room temperature. Slides were imaged at 63× magnification on a Zeiss LSM700 confocal microscope. Antibodies used were Abcam ab8898 (rabbit anti-H3K9me3), Sigma F1804 (mouse anti-Flag), and WAKO 300-95289 (mouse anti-H3K36me3); secondary antibodies were purchased from Invitrogen.
Genetic screen
Starting with a morc-1(−);pie1P-gfp-h2b strain, worms in the fourth larval stage were mutagenized in 0.6 mM N-ethyl-N-nitrosourea (ENU) for 4 h. After three washes in M9, 100 worms (P0 generation) were transferred to each of 32 plates. Worms were grown for 5 days at 20°C. Adult worms (F1 generation) were bleached to isolate embryos (F2 generation), which were plated, grown at 25°C, and then bleached to isolate embryos (F3 generation). Worms were maintained at 25°C and bleached at adulthood for every generation until F14. Ten larval worms were singled from each of the remaining fertile pools (29 of 32), and their progeny were counted. The most fertile worms of each pool were then propagated as clonalized lines. Genomic DNA was extracted from 20 clonalized lines, representing 17 pools (three pools were represented by two lines each) using the Gentra Puregene Tissue kit (Qiagen). Genomic paired-end DNA libraries were made using the Illumina Truseq Nano DNA LT Library Prep Kit according to the manufacturer’s instructions and sequenced on the Illumina Hi Seq 2000 platform.
Whole-mount DAPI staining
Worms were nutated in M9 in 1.5 mL microtubes for 15 min at room temperature, then centrifuged at 2,300 × g for 30 s. The supernatant was discarded, and the pellet was resuspended in 1 mL ice cold methanol and nutated at room temperature for 10 min. Worms were washed twice with 1 mL PBST (1xPBS with 0.1% Tween-20). Worms were centrifuged at 2,300 × g for 30 seconds, resuspended in 1 mL PBST with 100 μg/mL DAPI, and nutated at room temperature for 30 min. Worms were then centrifuged at 2,300 × g for 30 sec, pipetted onto a microscope slide, and mounted with Vectashield. Slides were imaged with a Zeiss LSM700 confocal microscope at 63× magnification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless specified below, quantitative data are displayed as mean with standard deviation shown as error bars; the n values are shown in the figure legends. Statistical tests and results are presented in Results and corresponding figure legends. For qRT-PCR, germline mortality assays, and western blot, at least two independent experiments were carried out; one representative biological replicate is presented. RNAi sensitivity, RNAi inheritance, and male rescue data are presented as means of biological replicates, with the number of replicates specified in the figure legend. Western blot quantification was performed using a LICOR Odyssey Fc according to the manufacturer’s instructions.
Small RNA sequencing data analysis
Raw qseq files were converted to fastq format using custom Awk scripts. Small RNAs were parsed from adapter sequences, and reads 18–32 nt long passing a Q30 threshold were aligned to the C. elegans genome (WS230) using CASHX v. 2.3 (Fahlgren et al., 2009). Total read counts for Mutator-class siRNAs (Phillips et al., 2014; Zhang et al., 2011) were analyzed for differential expression using DESeq2 (Love et al., 2014) in F1 wildtype (n=3), F4 wildtype (n=3), F1 morc-1(tm6048) (n=3), F4 morc-1(tm6048) (n=3), F1 hrde-1(tm1200) (n=2), and F4 hrde-1(tm1200) (n=2). Scatter plots were generated using R.
mRNA sequencing data analysis
Raw qseq files were converted to fastq format using custom Awk scripts. Reads were adapter-trimmed, quality filtered (Q30) using Trimmomatic v. 0.35 (Bolger et al., 2014), and aligned to the C. elegans genome and transcriptome (WS230) using TopHat2 (Kim et al., 2013). Differential gene expression analysis was done using CuffDiff2 (Trapnell et al., 2013). Two biological replicates per condition were processed. Plots were generated using R and CummeRBund v. 2.12.1. Gene enrichment analysis was done in R using Fisher’s exact test, and p-values were corrected for multiple comparisons using the Bonferroni adjustment. Venn diagrams were generated using BioVenn (Hulsen et al., 2008).
ChIP-seq analysis
H3K9me3 or H3K36me3 ChIP-seq sequencing results in fastq format were aligned to the WS190(ce6) genome using Bowtie 0.12.7 (Langmead et al., 2009). All analysis was performed using Python and R. RPKM value for each 1 kb was calculated by the number of reads that perfectly aligned to both strands of 1 kb-non-overlapping regions normalized by the sequencing depth of the library in millions. All analysis excluded misannotated regions and morc-1(−)-duplication region (chromosome V position 2350000-2570000bp). All types of targets were identified as 1.5-fold change with FDR<0.05 (Maniar and Fire, 2011) and as an overlap of two replicates. A table with RPKM values and lists of targets was deposited in NCBI (GEO accession number: GSE89887). Two-region Venn diagrams were generated using VennDiagram package in R; three-region Venn diagram was generated using http://www.benfrederickson.com/venn-diagrams-with-d3.js/. Exemplary regions were plotted at 1 bp resolution, normalized by the sequencing depth of the corresponding library. Anchor plots were calculated +/− 20 kb around start of each 1 kb region of corresponding target list and plotted with smoothing window of 10 bp.
Genome sequence data processing
Raw paired-end fastq files were demultiplexed based on 6 nt barcodes and aligned to the C. elegans genome (version WS220) using Bowtie2 (Langmead and Salzberg, 2012) and the following parameters: -q –phred64 -N 1 –end-to-end. Alignment SAM files were filtered using samtools (Li et al., 2009) to only include alignments with a bit FLAG containing 2, which indicates that each segment aligned properly, and then converted to BAM format. Alignment BAM files were sorted (SortSam), named (AddOrReplaceReadGroups), and indexed (BuildBamIndex) using functions from the PICARD suite of tools for manipulating high-throughput sequencing data (https://github.com/broadinstitute/picard). Alignment BAM files were realigned to improve indel calling using RealignerTargetCreator and IndeRealigner from the Genome Analysis ToolKit (GATK) suite of tools (McKenna et al., 2010). Duplicate reads were marked using PICARD MarkDuplicates, and the resulting BAM files were used with GATK’s UnifiedGenotyper command to call variants.
Identification of suppressor mutations
Variants (SNPs) were combined from replicate unmutagenized morc-1(−) strains using GATK’s CombineVariants. To identify variants in each mutagenized strain that do not appear in the unmutagenized morc-1(−) strain, GATK’s SelectVariants function was used to subtract combined unmutagenized morc-1(−) variants from each mutagenized strain. Unique variants from all mutagenized strains were filtered using SnpSift (Cingolani et al., 2012) to retain only variants with a high quality score (QUAL≥30) and covered by at least five reads (DP≥5). To determine the effect of each variant in each strain on encoded proteins, SnpEff (Cingolani et al., 2012) was used to annotate each variant and predict its impact on any encoded protein. Genes of interest in the genetic screen were defined as genes that contained different variants with “high” impact on the protein-coding sequence (e.g. non-synonymous replacement, start codon loss/gain) across two or more of the mutagenized strains. met-1, for example, contained four different variants in six different mutagenized strains that originated from four of the original 32 pools.
Heterochromatin localization assays
DAPI-labeled nuclear diameters were measured in ImageJ by averaging three diameter measurements, and nuclei were divided into three zones of equal volume. The distance from nuclear boundary to the center of GFP foci was measured in ImageJ, and array localization was assigned to Zone 1 (peripheral third), Zone 2 (intermediate third), or Zone 3 (interior third). At least two independent experiments were performed, except for hrde-1(−) and nrde-2(−) mutants, which were performed once. Where multiple replicates were performed, quantification is shown for one representative experiment.
X chromosome staining
The volume of intestinal nuclei and the X chromosome were quantified as described in (Lau et al., 2014) using Slidebook 5.0. For each channel, a user-defined intensity threshold was used to generate a mask that distinguished signal from background noise. The volume of each nucleus was determined by the number of voxels (three-dimensional pixels) with DAPI signal above the threshold. A secondary mask was generated to define X chromosome paint signal, and the volume of the X chromosome was determined by the number of voxels present in both masks. Two independent experiments were performed; one representative experiment is shown. In the figure legend, n indicates the number of nuclei quantified: these represent at least 8 worms per genotype.
DATA AND SOFTWARE AVAILABILITY
The mRNA-seq, sRNA-seq, ChIP-seq, and genomic DNA data have been deposited in NCBI under GEO accession number: GSE89887.
Supplementary Material
Table S1: all regulated mRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Table S2: all regulated mutator target mRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Table S3: all regulated mutator-class 22G endo-siRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Table S4: all regulated 22G endo-siRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Highlights.
MORC-1 mediates nuclear RNAi effector function downstream of RISC
MORC-1 enforces transgenerational maintenance of H3K9me3 at endo-siRNA targets
MORC-1 prevents the MET-1-mediated encroachment of H3K36me3 into heterochromatin
Antagonism between MORC-1 and MET-1 preserves germline immortality
Acknowledgments
We thank Susan Gasser and Shohei Matani for providing strains and Khursheed Wani for generating the MORC-1::3xFlag and E39A strains. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Mahnaz Akhavan and the UCLA Broad Stem Cell Research Center High Throughput Biosequencing Core for Illumina sequencing. We thank Donald Moerman and Stephane Flibotte for help analyzing genomic DNA libraries and Gloria Ha for illustrations. This work was supported with grants from the American Cancer Society RSG RMC-125264, NIH HG004276-01, Pew Charitable Trusts (to JKK); NIH T32-HD007505 and NIH T32-GM007315 (to NEW); NIH T32-GM007544-34 (to DXY); NIH GM60398 (to SEJ); NIGMS R01GM111752 (to SGG); NIH 1R25GM119775-01, Boettcher Foundation 003614-00002 (to TAM); NIH R01 GM093173 (to RCC); NIH R01 GM079533 (to GC). SEJ is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: Conceptualization of the study: NEW, JKK, and SEJ; experimental design: NEW, DXY, and JKK; heterochromatin localization studies: JK and RCC; X chromosome staining and male rescue: NEW, MJS, and GC; all other experiments: NEW and DXY; library preparation and sequencing: SF and SEJ; ChIP-seq computational analysis: NK and SGG; RNA-seq computational analysis: KCB and TAM; computational analysis of genome sequencing: MAF; NEW and JKK wrote the manuscript.
References
- Ahmed S, Hodgkin J. MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature. 2000;403:159–164. doi: 10.1038/35003120. [DOI] [PubMed] [Google Scholar]
- Andersen EC, Horvitz HR. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development. 2007;134:2991–2999. doi: 10.1242/dev.009373. [DOI] [PubMed] [Google Scholar]
- Andrews FH, Tong Q, Sullivan KD, Cornett EM, Zhang Y, Ali M, Ahn J, Pandey A, Guo AH, Strahl BD, et al. Multivalent Chromatin Engagement and Inter-domain Crosstalk Regulate MORC3 ATPase. Cell Rep. 2016;16:3195–3207. doi: 10.1016/j.celrep.2016.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe A, Sapetschnig A, Weick EM, Mitchell J, Bagijn MP, Cording AC, Doebley AL, Goldstein LD, Lehrbach NJ, Le Pen J, et al. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell. 2012;150:88–99. doi: 10.1016/j.cell.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley BA, Burkhart KB, Gu SG, Spracklin G, Kershner A, Fritz H, Kimble J, Fire A, Kennedy S. A nuclear Argonaute promotes multigenerational epigenetic inheritance and germline immortality. Nature. 2012;489:447–451. doi: 10.1038/nature11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart KB, Guang S, Buckley BA, Wong L, Bochner AF, Kennedy S. A pre-mRNA-associating factor links endogenous siRNAs to chromatin regulation. PLoS Genet. 2011;7:e1002249. doi: 10.1371/journal.pgen.1002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NO, Burkhart KB, Kennedy S. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc Natl Acad Sci USa. 2011;108:19683–19688. doi: 10.1073/pnas.1113310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi I, Kopczynski JB, Meyer BJ. The nuclear hormone receptor SEX-1 is an X-chromosome signal that determines nematode sex. Nature. 1998;396:168–173. doi: 10.1038/24164. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G, McDonel P, Meyer BJ. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science. 2004;303:1182–1185. doi: 10.1126/science.1092938. [DOI] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Albuquerque BFM, Placentino M, Ketting RF. Maternal piRNAs Are Essential for Germline Development following De Novo Establishment of Endo-siRNAs in Caenorhabditis elegans. Dev Cell. 2015;34:448–456. doi: 10.1016/j.devcel.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Zalevsky J, Colaiácovo MP, Villeneuve AM. Transgene-mediated cosuppression in the C. elegans germ line. Genes Dev. 2000;14:1578–1583. [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Fahlgren N, Sullivan CM, Kasschau KD, Chapman EJ, Cumbie JS, Montgomery TA, Gilbert SD, Dasenko M, Backman TWH, Givan SA, et al. Computational and analytical framework for small RNA profiling by high-throughput sequencing. Rna. 2009;15:992–1002. doi: 10.1261/rna.1473809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi H, Takasaki T, Rechtsteiner A, Li T, Kimura H, Checchi PM, Strome S, Kelly WG. Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin. 2010;3:15. doi: 10.1186/1756-8935-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Beese-Sims SE, Brookes E, Spadafora R, Zhu Y, Rothbart SB, Aristizábal-Corrales D, Chen S, Badeaux AI, Jin Q, et al. A histone methylation network regulates transgenerational epigenetic memory in C. elegans. Cell Rep. 2014;7:113–126. doi: 10.1016/j.celrep.2014.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Tabara H, Mello CC. Genetic requirements for inheritance of RNAi in C. elegans. Science. 2000;287:2494–2497. doi: 10.1126/science.287.5462.2494. [DOI] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, Fire A. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat Genet. 2012;44:157–164. doi: 10.1038/ng.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465:1097–1101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Pavelec DM, Burkhart KB, Harding S, Lachowiec J, Kennedy S. An Argonaute transports siRNAs from the cytoplasm to the nucleus. Science. 2008;321:537–541. doi: 10.1126/science.1157647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- Han T, Manoharan AP, Harkins TT, Bouffard P, Fitzpatrick C, Chu DS, Thierry-Mieg D, Thierry-Mieg J, Kim JK. 26G endo-siRNAs regulate spermatogenic and zygotic gene expression in Caenorhabditis elegans. Proc Natl Acad Sci USa. 2009;106:18674–18679. doi: 10.1073/pnas.0906378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CJ, Husmann D, Liu W, Kasmi FE, Wang H, Papikian A, Pastor WA, Moissiard G, Vashisht AA, Dangl JL, et al. Arabidopsis AtMORC4 and AtMORC7 Form Nuclear Bodies and Repress a Large Number of Protein-Coding Genes. PLoS Genet. 2016;12:e1005998. doi: 10.1371/journal.pgen.1005998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue N, Hess KD, Moreadith RW, Richardson LL, Handel MA, Watson ML, Zinn AR. New gene family defined by MORC, a nuclear protein required for mouse spermatogenesis. Hum Mol Genet. 1999;8:1201–1207. doi: 10.1093/hmg/8.7.1201. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Abhiman S, Aravind L. MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biol Direct. 2008;3:8. doi: 10.1186/1745-6150-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Katz DJ, Edwards TM, Reinke V, Kelly WG. A C. elegans LSD1 demethylase contributes to germline immortality by reprogramming epigenetic memory. Cell. 2009;137:308–320. doi: 10.1016/j.cell.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Plasterk RH. A genetic link between co-suppression and RNA interference in C. elegans. Nature. 2000;404:296–298. doi: 10.1038/35005113. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp TH, van Luenen HG, Plasterk RH. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell. 1999;99:133–141. doi: 10.1016/s0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Gabel HW, Kamath RS, Tewari M, Pasquinelli A, Rual JF, Kennedy S, Dybbs M, Bertin N, Kaplan JM, et al. Functional genomic analysis of RNA interference in C. elegans. Science. 2005;308:1164–1167. doi: 10.1126/science.1109267. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AC, Nabeshima K, Csankovszki G. The C. elegans dosage compensation complex mediates interphase X chromosome compaction. Epigenetics Chromatin. 2014;7:31. doi: 10.1186/1756-8935-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Gu W, Shirayama M, Youngman E, Conte D, Mello CC. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell. 2012;150:78–87. doi: 10.1016/j.cell.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yen L, Pastor WA, Johnston JB, Du J, Shew CJ, Liu W, Ho J, Stender B, Clark AT, et al. Mouse MORC3 is a GHKL ATPase that localizes to H3K4me3 marked chromatin. Proc Natl Acad Sci USa. 2016:201609709. doi: 10.1073/pnas.1609709113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tempel W, Zhang Q, Liang X, Loppnau P, Qin S, Min J. Family-wide Characterization of Histone Binding Abilities of Human CW Domain-containing Proteins. J Biol Chem. 2016;291:9000–9013. doi: 10.1074/jbc.M116.718973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn MJ, van Bergeijk P, Kaaij LJT, Almeida MV, Roovers EF, Berezikov E, Ketting RF. Extremely stable Piwi-induced gene silencing in Caenorhabditis elegans. Embo J. 2012;31:3422–3430. doi: 10.1038/emboj.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniar JM, Fire AZ. EGO-1, a C. elegans RdRP, modulates gene expression via production of mRNA-templated short antisense RNAs. Curr Biol. 2011;21:449–459. doi: 10.1016/j.cub.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Zhu C, Zong D, Weng C, Yang X, Huang H, Liu D, Feng X, Guang S. The Nrde Pathway Mediates Small-RNA-Directed Histone H3 Lysine 27 Trimethylation in Caenorhabditis elegans. Curr Biol. 2015;25:2398–2403. doi: 10.1016/j.cub.2015.07.051. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B, Barber LJ, Liu Y, Shtessel L, Boulton SJ, Gartner A, Ahmed S. The MRT-1 nuclease is required for DNA crosslink repair and telomerase activity in vivo in Caenorhabditis elegans. Embo J. 2009;28:3549–3563. doi: 10.1038/emboj.2009.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P, Towbin BD, Pike BL, Ponti A, Gasser SM. The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev. 2010;24:766–782. doi: 10.1101/gad.559610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BJ, Casson LP. Caenorhabditis elegans compensates for the difference in X chromosome dosage between the sexes by regulating transcript levels. Cell. 1986;47:871–881. doi: 10.1016/0092-8674(86)90802-0. [DOI] [PubMed] [Google Scholar]
- Miller LM, Plenefisch JD, Casson LP, Meyer BJ. xol-1: a gene that controls the male modes of both sex determination and X chromosome dosage compensation in C. elegans. Cell. 1988;55:167–183. doi: 10.1016/0092-8674(88)90019-0. [DOI] [PubMed] [Google Scholar]
- Moissiard G, Bischof S, Husmann D, Pastor WA, Hale CJ, Yen L, Stroud H, Papikian A, Vashisht AA, Wohlschlegel JA, et al. Transcriptional gene silencing by Arabidopsis microrchidia homologues involves the formation of heteromers. Proc Natl Acad Sci USa. 2014;111:7474–7479. doi: 10.1073/pnas.1406611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, et al. MORC family ATPases required for heterochromatin condensation and gene silencing. Science. 2012;336:1448–1451. doi: 10.1126/science.1221472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Kalinava N, Chen E, Huang A, Trinh T, Gu SG. A transgenerational role of the germline nuclear RNAi pathway in repressing heat stress-induced transcriptional activation in C. elegans. Epigenetics Chromatin. 2016;9:3. doi: 10.1186/s13072-016-0052-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. High Efficiency, Homology-Directed Genome Editing in Caenorhabditis elegans Using CRISPR-Cas9 Ribonucleoprotein Complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Stroud H, Nee K, Liu W, Pezic D, Manakov S, Lee SA, Moissiard G, Zamudio N, Bourc’his D, et al. MORC1 represses transposable elements in the mouse male germline. Nat Commun. 2014;5:5795. doi: 10.1038/ncomms6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty EL, Collette KS, Cohen AJ, Snyder MJ, Csankovszki G. Restricting dosage compensation complex binding to the X chromosomes by H2A.Z/HTZ-1. PLoS Genet. 2009;5:e1000699. doi: 10.1371/journal.pgen.1000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Brown KC, Montgomery BE, Ruvkun G, Montgomery TA. piRNAs and piRNA-Dependent siRNAs Protect Conserved and Essential C. elegans Genes from Misrouting into the RNAi Pathway. Dev Cell. 2015;34:457–465. doi: 10.1016/j.devcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Montgomery BE, Breen PC, Roovers EF, Rim YS, Ohsumi TK, Newman MA, van Wolfswinkel JC, Ketting RF, Ruvkun G, et al. MUT-14 and SMUT-1 DEAD box RNA helicases have overlapping roles in germline RNAi and endogenous siRNA formation. Curr Biol. 2014;24:839–844. doi: 10.1016/j.cub.2014.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Montgomery TA, Breen PC, Ruvkun G. MUT-16 promotes formation of perinuclear mutator foci required for RNA silencing in the C. elegans germline. Genes Dev. 2012;26:1433–1444. doi: 10.1101/gad.193904.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtsteiner A, Ercan S, Takasaki T, Phippen TM, Egelhofer TA, Wang W, Kimura H, Lieb JD, Strome S. The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet. 2010;6:e1001091. doi: 10.1371/journal.pgen.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F, Dean W. Epigenetic reprogramming during early development in mammals. Reproduction. 2004;127:643–651. doi: 10.1530/rep.1.00221. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Dunn MA. Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development. 1997;124:2191–2201. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D, Mello CC. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelick C, Ahmed S. Achieving immortality in the C. elegans germline. Ageing Res Rev. 2005;4:67–82. doi: 10.1016/j.arr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Snyder MJ, Lau AC, Brouhard EA, Davis MB, Jiang J, Sifuentes MH, Csankovszki G. Anchoring of Heterochromatin to the Nuclear Lamina Reinforces Dosage Compensation-Mediated Gene Repression. PLoS Genet. 2016;12:e1006341–33. doi: 10.1371/journal.pgen.1006341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Tabara H, Yigit E, Siomi H, Mello CC. The dsRNA binding protein RDE-4 interacts with RDE-1, DCR-1, and a DExH-box helicase to direct RNAi in C. elegans. Cell. 2002;109:861–871. doi: 10.1016/s0092-8674(02)00793-6. [DOI] [PubMed] [Google Scholar]
- Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150:934–947. doi: 10.1016/j.cell.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller P, Padeken J, van Schendel R, Kalck V, Tijsterman M, Gasser SM. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat Genet. 2016:1–13. doi: 10.1038/ng.3672. [DOI] [PubMed] [Google Scholar]
- Zhang C, Montgomery TA, Gabel HW, Fischer SEJ, Phillips CM, Fahlgren N, Sullivan CM, Carrington JC, Ruvkun G. mut-16 and other mutator class genes modulate 22G and 26G siRNA pathways in Caenorhabditis elegans. Proc Natl Acad Sci USa. 2011;108:1201–1208. doi: 10.1073/pnas.1018695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: all regulated mRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Table S2: all regulated mutator target mRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Table S3: all regulated mutator-class 22G endo-siRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.
Table S4: all regulated 22G endo-siRNAs in hrde-1(−) and morc-1(−) mutants. Related to Figure 2.