

Summary
Dysfunction in host immune responses and pathologic alterations in the gut microbiota, referred to as dysbiosis, can both contribute to the development of inflammatory bowel disease (IBD). However, it remains unclear how specific changes in host immunity or the microbiota cause disease. We previously demonstrated that the loss of the innate immune receptor NLRP6 in mice resulted in impaired production of IL18 and increased susceptibility to epithelial-induced injury. Here, we show that NLRP6 is important for suppressing the development of spontaneous colitis in the IL10−/− mice model of IBD and that NLRP6-deficiency results in the enrichment of Akkermansia muciniphila. A. muciniphila was sufficient for promoting intestinal inflammation in both specific-pathogen free and germfree IL10−/− mice. Our results demonstrate that A. muciniphila can act as a pathobiont to promote colitis in a genetically-susceptible host and that NLRP6 is a key regulator of its abundance.
In Brief
NLRP6 is important for maintaining intestinal homeostasis. Seregin et. al. demonstrate that NLRP6 limits the colonization of mucolytic A. muciniphila, which is sufficient to induce colitis in specific pathogen free and germfree IL10−/− mice. Resistance to A. muciniphila colonization by NLRP6 is mediated by IL18.
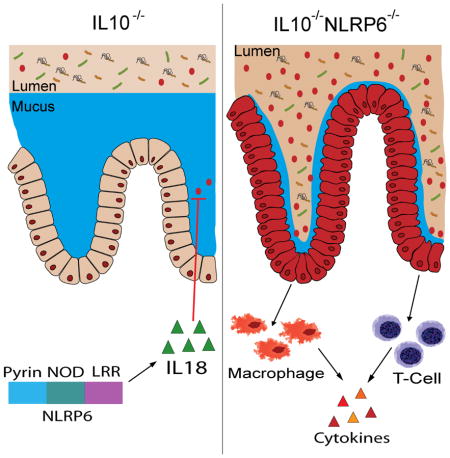
Introduction
Inflammatory bowel disease (IBD) is a significant health problem that afflicts approximately 0.5% of the general population in the Western world (Kaplan, 2015). The pathogenesis of IBD is not fully understood; however, host genetic factors, environmental exposures, and aberrant changes in the composition of the microbiota have all been implicated (Zhang and Li, 2014). In particular, the microbiome of IBD patients has been demonstrated to be of lower diversity than that of healthy controls (Joossens et al., 2011) with increased abundance of certain bacterial populations, such as Enterobacteriaceae, and increased mucosal adherence of bacteria (Johansson et al., 2008; Knights et al., 2014). In addition, defects in the immune system such as in the Nod-like receptor (NLR) family of pattern recognition receptors that are involved in the sensing of microbial and damage signals have been associated with IBD in humans and in mouse models of colitis (Rubino et al., 2012). In addition to their influence on inflammatory responses, the loss of function of these receptors has also resulted in changes in the composition of the microbiota, which in turn, may affect colitis susceptibility (Couturier-Maillard et al., 2013; Hirota et al., 2011).
NLRP6 is a member of the NLR family and has been presumed to function as part of an inflammasome based on its ability to interact with the adaptor protein ASC in overexpression assays and on the decreased levels of caspase-1 activation observed in the colon tissue of Nlrp6−/− mice (Grenier et al., 2002; Levy et al., 2015). Caspase-1 activation results in the cleavage of pro-IL1β and pro-IL18 to their mature forms (Netea et al., 2015), and consistently, NLRP6-deficient mice have impaired production of IL18 (Chen et al., 2011; Elinav et al., 2011; Levy et al., 2015). Furthermore, NLRP6 has been shown to protect mice against chemically-induced epithelial injury with dextran sulfate sodium (DSS) (Chen et al., 2011; Elinav et al., 2011; Normand et al., 2011; Seregin et al., 2016). The mechanisms involved in NLRP6-mediated protection remain to be fully elucidated, but have been related to a role in promoting the production of mature IL18, which is important for epithelial repair and in regulating the composition of the microbiota (Chen et al., 2011; Elinav et al., 2011; Levy et al., 2015). More specifically, metabolites regulated by a “healthy” microbiome have been shown to signal through NLRP6 to modulate the production of IL18 and production of antimicrobial peptides (AMPs) that are important for epithelial barrier function, resistance to DSS-induced injury, and prevention of dysbiosis (Levy et al., 2015). NLRP6 deficiency was also previously demonstrated to be associated with an increased abundance of potentially colitogenic bacteria, namely, Prevotella (Elinav et al., 2011), although it has been unclear whether the enrichment of these bacterial populations are truly NLRP6-dependent and capable of precipitating or potentiating colitis.
Studies to date that identify a role for NLRP6 function in the pathogenesis of IBD have relied on the widely-used DSS model of colitis, and is an appropriate model to examine host responses during epithelial injury and repair (Brown et al., 2007), but may not necessarily encompass all aspects of IBD pathogenesis. Here, we identify a critical role for NLRP6 in protecting mice against the development of colitis using the IL10 knockout (KO) model. IL10 is an anti-inflammatory cytokine and polymorphisms in both IL10 and IL10R have been associated with IBD (Kaser et al., 2010). IL10−/− mice develop chronic enterocolitis that is T cell dependent and characterized by upregulated pathologic Th1 responses (Davidson et al., 1996). The development of spontaneous colitis, however, is highly dependent on the composition of the gut microbiota since the severity of disease can differ between mouse facilities (Keubler et al., 2015), and germfree (GF) IL10−/− mice do not develop inflammation (Sellon et al., 1998). Thus, IL10−/− mice have been considered an alternative, and perhaps preferred model, to the DSS-induced epithelial injury and colitis model for studying IBD. We demonstrate that specific pathogen free (SPF) IL10−/−Nlrp6−/− mice develop spontaneous colitis with increased inflammation that does not occur in IL10−/− mice maintained at our facility. Moreover, IL10−/−Nlrp6−/− mice harbored an altered microbiota with increased colonization of the mucin-degrader, Akkermansia muciniphila, that is not easily transferred to co-housed IL10−/− mice, resulting in lack of transmission of colitis to co-housed IL10−/− mice. Conventionalization of GF Nlrp6−/− also resulted in increased abundance of A. muciniphila compared to that in conventionized GF wildtype (WT) mice. Enforced colonization of A. muciniphila in SPF IL10−/− mice and more importantly, monocolonization of GF IL10−/− mice with A. muciniphila were sufficient to induce spontaneous colitis. Our results demonstrate that NLRP6 specifically regulates the colonization of A. muciniphila and that A. muciniphila can act as a pathobiont to promote colitis in genetically-susceptible mice. Thus, our study highlights the ability for NLRP6 to maintain intestinal homeostasis by limiting the colonization of specific colitogenic bacteria.
Results
Lack of functional NLRP6 makes IL10−/− mice prone to spontaneous colitis
We and others have previously shown that NLRP6 deficiency in mice is associated with protection against DSS-induced epithelial injury (Chen et al., 2011; Couturier-Maillard et al., 2013; Elinav et al., 2011; Seregin et al., 2016). DSS is toxic to the colonic epithelium resulting in epithelial apoptosis and mucosal ulceration that is subsequently followed by bacterial translocation and a commensal-driven inflammatory response (Kiesler et al., 2015). The DSS colitis model, therefore, has often been considered an injury model rather than a model that truly reflects IBD pathogenesis. To further delineate the role of NLRP6 in modulating intestinal inflammatory responses, we generated mice doubly deficient in NLRP6 and IL10. IL10−/− B6 mice at our facility are resistant to the development of colitis (Nagalingam et al., 2013)(Figure S1A). However, by 8 weeks of age, IL10−/−Nlrp6−/− mice developed spontaneous colitis (Figure 1A). More specifically, there was increased epithelial hyperplasia and transmural inflammation resulting in higher histological scores than WT, Nlrp6−/−, or IL10−/− mice (Figure 1A). Flow cytometric analysis confirmed increased immune cell infiltration, in particular CD3+ T cells and CD11b+Ly6chi inflammatory monocytes, into the colon lamina propria of IL10−/−Nlrp6−/− mice (Figure S1). Moreover, severe rectal prolapse reflective of underlying inflammation occurred predominantly in IL10−/−Nlrp6−/− mice, typically developing at 2–4 months of age (Figure 1B). Consistent with the increased levels of inflammation at 8 weeks of age, IL10−/−Nlrp6−/− had increases in spleen weight and size and higher levels of fecal lipocalin (Lcn-2), a surrogate marker for intestinal damage and inflammation (Sherwood, 2012), as compared to that of IL10−/− mice, which did not differ significantly from either age- and sex-matched WT and Nlrp6−/− mice, (Figures 1C and 1D). The increased Lcn-2 levels and histological scores observed with IL10−/−Nlrp6−/− mice were also associated with increased bacterial translocation to the mesenteric lymph nodes (MLNs) (Figure 1E) and increased proinflammatory cytokine production in the colonic tissue (Figure 1F). Thus, NLRP6 is critically important for suppressing colitis in IL-10−/− mice.
Figure 1. IL10−/−Nlrp6−/− mice develop significant spontaneous inflammation in the colon.

(A) Representative photographs and histological scores of colon sections from 8-week old WT, IL10−/−, Nlrp6−/− and IL10−/−Nlrp6−/− mice. Inflammation and epithelial hyperplasia are indicated by arrows. Original magnification = 200x. (B) Incidence of rectal prolapse in WT (n=116), Nlrp6−/−(n=89), IL10−/− (n=51), and IL10−/−Nlrp6−/− (n=58). (C) Spleen weights, (D) fecal lipocalin-2 levels, (E) relative levels of total bacteria/MLN in 14 week old IL10−/− or IL10−/−Nlrp6−/− mice (n=7 mice/group), and (F) mRNA expression of pro-inflammatory mediators relative to β-actin in the colons of WT (n=13), IL10−/− (n=11), Nlrp6−/− (n=12), and IL10−/−Nlrp6−/− (n=14) mice. Data are representative of three independent experiments. * - p<0.05, as compared to all other genotypes. See also Figure S1.
Cohousing of IL10−/− with IL10−/−Nlrp6−/− mice does not result in transfer of colitis susceptibility
Since the development of colitis in IL10−/− mice is, in part, facility-dependent likely reflecting differences in microbiota composition, we cohoused IL10−/− mice with IL10−/−Nlrp6−/− mice at 4 weeks of age and then monitored them for the development of colitis until 3 months of age. Interestingly, despite cohousing, IL10−/− mice did not develop significant colitis. Histological scores and fecal levels of Lcn-2 were similar to that of IL10−/− that were not cohoused, in contrast to IL10−/−Nlrp6−/− mice (Figures 2A and 2C). More importantly, IL10−/−Nlrp6−/− mice developed significant colitis with higher histological scores, increased spleen weight, and Lcn-2 levels than cohoused IL10−/− mice (Figures 2A–C). Finally, levels of IFNγ and IL1β production in the colons of IL10−/−/Nlrp6−/− also remained significantly elevated compared to cohoused IL10−/− mice (Figure 2D). We also evaluated IL10−/− and IL10−/−Nlrp6−/− littermates, and observed similar increases in spleen weight and inflammation in IL10−/−Nlrp6−/− mice compared to that in IL10−/− mice (Figure S2A–D).
Figure 2. Increased colitis susceptibility in IL10−/−Nlrp6−/− mice does not transfer to IL10−/− mice upon cohousing.

(A) Representative micrographs and histological scoring of 12 week old IL10−/− (n=6) and IL10−/−Nlrp6−/− (n=8) mice after 2 months of cohousing. Arrows point to Inflammation and hyperplasia. Original magnification = 200x. (B) Spleen weights and (C) fecal lipocalin-2 levels from cohoused IL10−/− and IL10−/−Nlrp6−/− mice. (D) Relative mRNA expression of pro-inflammatory mediators in the colon of cohoused (COH) or non-cohoused IL10−/− and IL10−/−Nlrp6−/− mice (n=8). Data are representative of two independent experiments. * - p<0.05 as compared to IL10−/− mice. See also Figure S2.
Given the dependence on the composition of the gut microbiota for the development of colitis in IL10−/− mice, we next determined if there were differences in bacterial populations between cohoused and non-cohoused IL10−/− and IL10−/−Nlrp6−/− mice by performing 16S rRNA sequencing of bacterial DNA extracted from the stool of these mice. Despite cohousing, there remained significant differences in overall community structure between IL10−/− and IL10−/−Nlrp6−/− mice (Figure S2E–F). As observed in IBD patients, the microbiome of IL10−/−Nlrp6−/− mice was also reduced in richness and α-diversity (Figure 3A) (Willing et al., 2010). We used the linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011) method to identify operational taxonomic units (OTUs) that were the most differentially abundant between IL10−/− and IL10−/−Nlrp6−/− mice. These OTUs, which represent bacterial sequences that are at least 97% identical to each other, also reflected bacterial populations that did not effectively transfer between mice genotypes. Relative to either non-cohoused or cohoused mice, Bacteroides (OTU63357), Prevotella (OTU63571), Mucispirillum (OTU66), Akkermansia (OTU2), and Helicobacter (OTU7) were more highly abundant in IL10−/−Nlrp6−/− mice in contrast to IL10−/− mice (Figures 3B and 3C). There were also several bacterial populations that were significantly more abundant in IL10−/− mice compared to IL10−/−Nlrp6−/− mice, although these were not differentially abundant in cohoused mice (Figures 3B and 3C). As the gut microbiome is typically maternally transmitted (Stappenbeck and Virgin, 2016) to further rule out the possibility that microbiome differences between IL10−/− and IL10−/−Nlrp6−/− were due to independent breeding of these colonies, we performed similar analyses using IL10−/− and IL10−/−Nlrp6−/− littermates. Based on average θyc distances, the community structures of IL10−/− and IL10−/−Nlrp6−/− littermates were significantly different (Figure S2G–H). LEfSe analysis between IL10−/− and IL10−/−Nlrp6−/− littermates revealed that Prevotella (OTU63571) and Akkermansia (OTU2), but not Helicobacter (OTU7) were more abundant in IL10−/−Nlrp6−/− mice (Figure S2I). Altogether, these results strongly suggest that IL10−/−Nlrp6−/− mice harbor multiple bacterial populations that are differentially abundant compared to IL10−/− mice despite cohousing or use of littermates, providing a possible explanation for colitis development in IL10−/−Nlrp6−/− mice.
Figure 3. Inflammatory phenotype of IL10−/−Nlrp6−/− mice correlates with altered microbiome not corrected by cohousing.

(A) Inverse Simpson’s α-diversity index and observed community richness as measured by number of operational taxonomic units (OTUs), are shown for non-cohoused, cohoused (at 35 and 60 days), and littermate IL10−/− and IL10−/−Nlrp6−/− mice. LEfSe analysis shows bacteria that were most differentially abundant between (B) non-cohoused or (C) cohoused IL10−/− and IL10−/−Nlrp6−/− mice and indicate the effect size of differentially abundant OTUs in the colon. Data are representative of two independent experiments, n=8 for non-cohoused groups, n=6 and n=8 for cohoused IL10−/− and IL10−/−Nlrp6−/− mice respectively. * - p<0.05 as compared to IL10−/− group. See also Figure S2.
NLRP6 regulates the colonization of Akkermansia muciniphila
The above results suggest that NLRP6 deficiency in IL10−/− mice is associated with an altered microbiome that cannot be completely transferred to co-housed mice. However, as only IL10−/−Nlrp6−/− mice develop significant colitis, it is possible that the observed differences in microbial composition are secondary to inflammation. To determine more directly whether NLRP6 regulates the composition of the gut microbiome, we generated germfree (GF) Nlrp6−/− mice and compared the colonization of these mice with GF WT mice conventionalized with the microbiome of WT SPF mice. Stool samples were collected from recolonized GF WT (gWT) and Nlrp6−/− (gNlrp6−/−) mice over a two-week period and microbial communities that established in these mice were analyzed based on 16S rRNA sequences. Both groups of mice received the same donor bedding and fecal material resulting in similar community structures shortly after conventionalization (day 3) (Figure S3A,B). However, after two weeks, the microbiome that established in gNlrp6−/− mice exhibited less α-diversity and richness compared to that of gWT mice (Figures 4A and 4B). β-Diversity analysis using θyc also demonstrated that the microbial community structure of gNlrp6−/− mice was distinct from that of gWT mice (Figure 4C). Consistently, significant differences in the relative abundances of several OTUs were found between gWT and gNlrp6−/− mice. Based on LEfSe analysis, Akkermansia was the most differentially abundant OTU with significantly increased levels in gNlrp6−/− mice (Figure 4D). The majority of these differentially abundant bacterial populations did not overlap with the OTUs found to be significantly different between cohoused or non-cohoused IL10−/− and IL10−/−Nlrp6−/− mice (Figure 3B and 3C). We then examined the number of OTUs that were differentially abundant between gWT versus gNlrp6−/− mice, non-cohoused IL10−/− versus IL10−/−Nlrp6−/− mice, and cohoused IL10−/− versus IL10−/−Nlrp6−/− mice that were associated with an LDA score of 2 or higher by LEfSe analysis (Figure 4E). Only two OTUs were significantly different in relative abundance between all three pairwise comparisons, that being OTU97, which belongs to an unclassified bacterial phylum, and OTU2, Akkermansia. Based on the 16S rRNA sequence, OTU2 was identified to be the mucin-degrader Akkermansia muciniphila (Figure 4E). Quantitative PCR with primers specific to A. muciniphila confirmed the increased relative abundance of this bacterium in gNlrp6−/− compared to gWT as well as in cohoused and non-cohoused IL10−/−Nlrp6−/− mice compared to IL10−/− mice (Figures 4F, G and S3F). The increased relative abundance of A. muciniphila associated with NLRP6 deficiency was unlikely to be secondary to inflammation since fecal lipocalin, calprotectin, and cytokine levels were not significantly elevated or different between gWT and gNlrp6−/− mice and neither mice developed frank colitis (Figure S3C–E and data not shown). Furthermore, increased levels of Akkermansia were also observed in younger 5 week old IL10−/−Nlrp6−/− mice which had low levels of lipocalin compared to that of older mice (Figure S4A,B). Altogether, these results strongly suggest that NLRP6 regulates the composition of the gut microbiota and, in particular, the relative abundance of A. muciniphila.
Figure 4. Conventionalized GF Nlrp6−/− mice have altered microbiota associated with reduced α-diversity and richness and dramatically increased A. muciniphila colonization.

(A) Time course for inverse Simpson’s α-diversity index and (B) observed community richness are shown for gWT and gNlrp6−/− mice. (C) Average θyc distance within or between groups of gWT and gNlrp6−/− mice. (D) LEfSe results show most differentially abundant OTUs. (E) Venn diagram illustrating the number of bacterial OTUs that were differentially abundant (LDA score > 2) between the indicated mice. (F) Relative abundance of A. muciniphila in the colons of 12-week old non-cohoused or cohoused IL10−/− and IL10−/−Nlrp6−/− mice normalized to total bacteria. (G) Relative abundance of A. muciniphila in colons of gWT and gNlrp6−/− mice (day 15 after conventionalization); n=8 for non-cohoused groups, n=6 and n=8 for cohoused IL10−/− and IL10−/−Nlrp6−/− mice respectively; n=5, n=8 for gWT and gNlrp6−/− mice respectively. * - p<0.05 as compared to corresponding gWT group (A–C) or to IL10−/− group (F). See also Figure S3.
IL18 modulates abundance of A. muciniphila
We and others have previously demonstrated that NLRP6 deficiency is associated with impairment in IL18 production (Chen et al., 2011; Elinav et al., 2011; Levy et al., 2015; Seregin et al., 2016). Consistently, we observed reduced IL18 levels in plasma and colons of IL10−/−Nlrp6−/− mice as compared to IL10−/− mice, although this was not associated with reduced levels of caspase-1, caspase-8, or caspase-11 activation (Figure S4C–F), suggesting a different mechanism for the cleavage of pro-IL-18 in this context, which has been previously reported (Netea et al., 2015). In fact, in adult colitic IL10−/−Nlrp6−/− mice, there are increased levels of caspase-1 activation compared to that in IL-10−/− mice consistent with increased inflammation and IL-1β production (Figure S4F). To determine if IL18 limits the relative abundance of A. muciniphila, we measured levels of A. muciniphila colonization in IL18−/− and IL-18R−/− mice. We observed significantly increased levels of A. muciniphila in IL18−/−, but not IL1β−/− or IL1R−/− mice (Figure 5A). We next determined whether restoration of IL18 could reduce A. muciniphila colonization in SPF Nlrp6−/− mice by administering recombinant IL18 (rIL18) or rIL1β over three consecutive days and monitoring the relative abundance of A. muciniphila by qPCR. Within a few days after the last rIL18 injection, there was a dramatic reduction in A. muciniphila colonization to levels similar to that in SPF WT mice, which did not occur with rIL1β (Figures 5B and S4G). These results indicate that one mechanism by which NLRP6 regulates the colonization of A. muciniphila is through the production of IL18.
Figure 5. rIL18 is sufficient to reduce A. muciniphila colonization in Nlrp6−/− mice.

(A) Relative abundance of A. muciniphila from fecal samples collected from adult WT, IL18−/−, IL18R−/− (n=8 mice/genotype), IL1β−/−, IL1R−/−, IL10−/−, Nlrp6−/− and IL10−/−Nlrp6−/− mice (n=5 mice/genotype) and (B) Nlrp6−/− mice before and after treatment with rIL-18 (treatment days indicated by arrows). Data are representative of two independent experiments. * - p<0.05 compared to WT. See also Figure S4.
Increased colonization of A muciniphila is sufficient to trigger significant colitis in IL10−/− mice
Previous studies suggested that Nlrp6−/− mice have increased levels of Prevotella and TM7 compared to WT mice (Elinav et al., 2011), although it was not clear whether these observations were made in littermates and NLRP6-dependent. In our gWT and gNlrp6−/− mice recolonized with the microbiota of SPF WT donors, which contained both Prevotella and TM7, the relative abundance of Prevotella and TM7, as determined by qPCR, was not significantly different (Figure S3G).
We next determined whether the enrichment of A. muciniphila that occurred in the absence of NLRP6 signaling contributed to the development of colitis in the context of IL10 deficiency in mice. To address this, we isolated a murine A. muciniphila strain and orally gavaged A. muciniphila into SPF IL10−/− mice weekly over 7 weeks to maintain relatively stable colonization levels (~3%) during this period (Figure S5A). As a control for specificity, we gavaged with the same frequency and dose a separate cohort of SPF IL10−/− mice with Bacteroides acidifaciens (Figure S5B) since its relative abundance was similar to that of A. muciniphila (~1–10%), but not significantly different between conventionalized GF WT and Nlrp6−/− mice (Figure S3H). SPF IL10−/− mice gavaged with A. muciniphila lost weight compared to mice gavaged with B. acidifaciens (Figure 6A). In addition, A. muciniphila-gavaged IL10−/− mice developed significant colitis based on histological scoring compared to either control IL10−/− mice or IL10−/− mice gavaged with B. acidifaciens (Figure 6B). Consistently, A. muciniphila-gavaged IL10−/− mice exhibited splenomegaly, increased colon weight/length ratios, dramatically elevated fecal Lcn-2 levels and increased bacterial translocation to the MLNs (Figures 6C–F). Importantly, there was significantly increased production of multiple pro-inflammatory mediators compared to either IL10−/− mice gavaged with B. acidifaciens or IL10−/− control mice (Figure 6G).
Figure 6. A. muciniphila gavaged into SPF IL10−/− mice triggers colitis.

SPF IL10−/− mice were left untreated or gavaged weekly with 2×108 CFU/ml of A. muciniphila or B. acidifaciens. (A) Percent weight change of age- and sex-matched groups of mice. (B) Representative micrographs and histological scores of colon sections from IL10−/− + no gavage, IL10−/− + A. muciniphila or IL10−/− + B. acidifaciens mice (200x). (C) Spleen weights (left) and size (right), (D) colon inflammation index (weight/length), (E) fecal lipocalin-2 levels as measured by ELISA, (F) levels of total bacteria/MLN and (G) relative mRNA expression of various pro-inflammatory mediators in the colons from IL10−/− + no gavage, IL10−/− + A. muciniphila or IL10−/− + B. acidifaciens mice as determined by qPCR with β-actin used as the housekeeping gene control. Data are representative of three independent experiments, n=11, n=13, n=12 for IL10−/− control, IL10−/− + A. muciniphila or IL10−/− + B. acidifaciens groups of mice respectively. * - p<0.05 as compared to IL10−/− + B. acidifaciens group. See also Figure S5.
In murine colitis models and in IBD patients, there is penetration of the mucus layer that normally separates bacteria from the colonic mucosa, which may contribute to the development of colitis (Johansson et al., 2014). A. muciniphila is a mucin-degrader and, therefore, one possible mechanism by which A. muciniphila contributes to colitis development in IL10−/− mice is by reducing the thickness of the mucus, thereby predisposing mice to bacterial penetration into the intestinal mucosa to induce inflammation. Consistently, greater bacterial translocation to the MLNs was observed in IL10−/−Nlrp6−/− and in A. muciniphila-gavaged IL10−/− mice compared to that in IL-10−/− mice and B. acidifaciens-gavaged IL-10−/− mice, respectively (Figures 1E and 6F). In addition, IL10−/−Nlrp6−/− and IL10−/− mice gavaged with A. muciniphila had a reduced mucus layer compared to IL10−/− and IL10−/− mice gavaged with B. acidifaciens based on Alcian blue staining of Methacarn-fixed colon sections (Figures S5C–E). We also observed a thinner mucus layer in Nlrp6−/− and IL18−/− mice that have elevated levels A. muciniphila (Figure S5E). These differences, however, were not due to a reduction in the number of goblet cells (Figure S5F) or in reduced expression of MUC2, a major component of mucus (Figure S5G). Furthermore, fluorescent in-situ hybridization (FISH) revealed the presence of A. muciniphila in the inner mucus layer in close proximity to the colon epithelium of IL10−/− mice gavaged with A. muciniphila (Figure S5H). Altogether, these results support the colitogenic properties of A. muciniphila, which can promote colitis in SPF IL10−/− mice when a threshold of colonization has been crossed.
It remained unclear if A. muciniphila promotes colitis directly or induces inflammatory responses indirectly by affecting the colonization of other potential colitogenic bacterial populations. We, therefore, monocolonized GF IL10−/− mice with either A. muciniphila or B. acidifaciens. Both bacterial strains efficiently colonized GF IL10−/− mice after a single oral gavage and maintained high levels of colonization for the duration of the experiment (Figure 7A). Similar to what was observed with SPF IL10−/− mice gavaged with A. muciniphila, GF IL10−/− mice colonized with A. muciniphila were unable to gain weight unlike GF IL10−/− mice colonized with B. acidifaciens (Figure 7B). A. muciniphila colonization also resulted in significantly increased histological scores and fecal Lcn-2 levels, which was associated with increased bacterial translocation consistent with its mucin-degrading properties (Figure 7D–E). Although GF IL10−/− colonized with B. acidifaciens resulted in the upregulation of some proinflammatory cytokines as compared to GF IL10−/− mice controls, A. muciniphila-monocolonized GF IL10−/− mice induced them to a significantly greater extent and also upregulated additional cytokines including IL-6 and IL12p40 (Figure 7F).
Figure 7. A. muciniphila is sufficient to trigger inflammation in GF IL10−/− mice.

(A) GF IL10−/− mice were monocolonized with A. muciniphila or B. acidifaciens and specific colonization confirmed by qPCR. (B) Percent weight change, (C) fecal lipocalin-2 levels, (D) histological inflammatory scores, and (E) normalized levels of total bacteria/MLN in monoassociated GF IL10−/− mice. (F) mRNA expression of various pro-inflammatory mediators relative to β-actin in the colons of the indicated mice. n=11 for GF IL10−/− + A. muciniphila and GF IL10−/− + B. acidifaciens groups, and n=6 for GF IL10−/− mice. * - p<0.001 compared to GF IL10−/− + B. acidifaciens (B, C) or to GF IL10−/− (F).
Consistent with the colitogenic properties of A. muciniphila, stimulation of bone marrow-derived macrophages (BMDMs) isolated from IL10−/− mice with heat-killed A. muciniphila extracts resulted in significantly increased production of IL-6 and IL-12p40 compared to that with B. acidifaciens extracts (Figures S6–7). A. muciniphila culture supernatants also upregulated cytokines to a greater extent than B. acidifaciens, but to a lesser degree than bacterial extracts (Figure S6A), The immunostimulatory activity of A. muciniphila extracts was not affected by either proteinase K or DNase/RNase treatment suggesting that the major immunostimulatory molecule from A. muciniphila is not a protein or nucleic acid (Figure S6A). Furthermore, the major immunostimulatory activity contained in A. muciniphila extracts required MyD88 and TLR4 signaling (Figure S6B), and therefore, likely to be lipopolysaccharide (LPS). To confirm, we purified the outer membrane (OM) as well as LPS from A. muciniphila and B. acidifaciens and demonstrated that both the OM and LPS derived from A. muciniphila upregulated cytokine production in IL10−/− BMDMs to a greater extent than that from B. acidifaciens (Figure S6C–D). Moreover, although unstimulated CD11b+Ly6G− colonic myeloid cells isolated from the LP of IL10−/− mice have generally high basal levels of inflammatory cytokines, stimulation with LPS purified from A. muciniphila resulted in significantly higher cytokine responses than that from B. acidifaciens (Figure S7B). Altogether, these results strongly suggest that A. muciniphila that normally colonizes the mouse gut can act as a pathobiont to promote colitis in a genetically-susceptible host.
Discussion
IBD is a devastating chronic disease that remains without a cure. Studies using mouse models of colitis have demonstrated that the pathogenesis of IBD is multifactorial and involves a complex interplay between host genetics, environment factors, including the gut microbiome, and the immune system. The current study further highlights the contributions of all three. We have demonstrated that NLRP6 protects mice deficient in IL10 from the development of colitis. NLRP6 was previously shown to be important for resistance against epithelial damage and subsequent inflammation in a chemically-induced epithelial injury model using DSS. In the DSS model, the onset of inflammation in NLRP6-deficient mice was associated with reduced production of IL18. IL18 deficiency, in turn, has been suggested to impair epithelial restitution after DSS-induced injury, which is consistent with IL18 being upstream of MyD88 signaling that is important for commensal-induced reparative immune responses (Rakoff-Nahoum et al., 2004; Salcedo et al., 2010). In the current study, using an alternative model of IBD that is not dependent on epithelial injury and repair, we demonstrate that NLRP6 protects against the development of colitis in IL10−/− mice by limiting the colonization of colitogenic A. muciniphila.
Previous studies have shown that NLRP6 deficiency is associated with expanded colonization of Prevotella and TM7 and antibiotic treatment of Nlrp6−/− mice reduced its relative abundance and ameliorated colitis (Elinav et al., 2011). Cohousing Nlrp6−/− with WT mice resulted in transfer of bacteria and transmission of susceptibility to DSS-induced colitis (Elinav et al., 2011). These studies were the first indication that NLRP6 can regulate the composition of the gut microbiota to resist DSS-induced colitis. However, these studies only established an association between Prevotella colonization and NLRP6 deficiency, and it remained unclear whether any differences in microbiome composition were due to colony-dependent, but NLRP6-independent differences. We, therefore, generated GF Nlrp6−/− mice and compared the resulting microbiome composition to that of GF WT mice after conventionalization. Our results demonstrated that despite colonization with the same donor microbiota, the microbiomes that established in gWT and gNlrp6−/− were distinct, suggesting that NLRP6 does indeed regulate the composition of the gut microbiota. Interestingly, there were no differences in the relative abundance of Prevotella or TM7 as previously reported even though the input donor microbiota harbored both bacterial populations, suggesting that NLRP6 does not regulate the abundances of these bacteria (Elinav et al., 2011). A significant increase in members of the family Porphyromonadaceae was also reported to be in enriched in Asc−/− mice (Henao-Mejia et al., 2012), and this increase was less clearly related to NLRP6 and colitis susceptibility. In the current study, we see different OTUs belonging to the family of Porphyromonodaceae that are either increased or decreased in abundance in our conventionalized germfree Nlrp6−/− mice compared to WT, and none of them were among the most differentially abundant bacterial populations in IL-10 versus IL-10/NLRP6 DKO mice. Thus, it is not clear the relative importance of different Porphyromonodaceae strains on colitis. Importantly, we found an expansion of A. muciniphila in gNlrp6−/− mice. A. muciniphila was also significantly more abundant in IL10−/−Nlrp6−/− mice compared to IL10−/− mice. Cohousing IL10−/−Nlrp6−/− with IL10−/− mice over several weeks did not result in transfer of A. muciniphila or colitis-susceptibility to IL10−/− mice, indicating that lack of disease transmissibility with cohoused mice does not necessarily rule out microbiome contributions.
A. muciniphila is a Gram-negative, strictly anaerobic bacterium belonging to the Verrucomicrobia phylum. It is capable of degrading mucin and is also very abundant in the human gut (Derrien et al., 2004). Our study shows that repeated oral gavage of A. muciniphila resulted in increased colitis in SPF IL10−/− mice and, furthermore, monocolonization of GF IL10−/− mice resulted in increased inflammation as evidenced by histological evaluation, elevated lipocalin levels and induction of proinflammatory mediators, effectively fulfilling Koch’s postulates. A potentially negative role for A. muciniphila within the intestine is also supported by studies demonstrating its increased abundance in mice that develop higher tumor burdens in a model of colitis-associated tumorigenesis (Baxter et al., 2014). The presence of A. muciniphila also exacerbated Salmonella-induced colitis and is positively correlated with ulcerative colitis patients with active pouchitis and IBD patients that are treatment-resistant (Ganesh et al., 2013; White et al., 2009; Zella et al., 2011). Other small clinical studies of IBD patients, however, have shown contradictory results (Png et al., 2010; Rajilic-Stojanovic et al., 2013), and therefore studies of larger scale will clearly be needed to confirm the clinical relevance of A. muciniphila in IBD.
The mechanism by which A. muciniphila promotes colitis remains to be fully elucidated. Given its mucin-degrading properties, we hypothesized that A. muciniphila degrades the mucus layer, thereby allowing greater microbial access to the intestinal mucosa and facilitating commensal-driven inflammation in the setting of IL-10 deficiency. Indeed, we observed a thinner mucus layer in SPF IL10−/− mice orally administered A. muciniphila associated with increased total bacterial translocation and the induction of inflammation likely driven by bacteria not limited to A. muciniphila in the setting of IL10 deficiency. How other bacterial populations can contribute to colitis in SPF IL10−/− mice and whether the increased abundance of A. muciniphila contributes to the pathogenicity of other bacteria will need to be further investigated. However, our data suggests that A. muciniphila can itself be colitogenic since it is sufficient to cause inflammation in monocolonized GF IL10−/− mice. As compared to B. acidifaciens, which was incapable of inducing colitis in GF or SPF IL10−/− mice, A. muciniphila induced significantly higher levels of pro-inflammatory cytokines in the colon, indicating its colitogenic potential. Consistently, A. muciniphila extracts can induce robust cytokine production by IL10−/− BMDMs in contrast to lower levels of cytokine production induced by B. acidifaciens. It was previously shown that colitis in IL-10−/− mice is largely driven by colonic macrophages via MyD88 signaling likely triggered by intestinal bacteria (Hoshi et al., 2012). Our data suggests that A. muciniphila, and in particular, its LPS, can induce significantly higher levels of cytokine production in IL-10−/− BMDMs and in IL-10−/− CD11b+Ly6G− colonic myeloid cells within the lamina propria than B. acidifaciens. The combination of A. muciniphila’s mucin-degrading ability and its highly immunstimulatory LPS activity likely contributes to the development of colitis in GF IL-10−/− mice that does not occur with B. acidifaciens. It is important to note that the colitis in GF IL-10−/− mice monoassociated with A. muciniphila is not as severe as that observed in SPF IL-10−/− mice gavaged with A. muciniphila and suggests that the level of colitogenicity of A. muciniphila may be context-dependent. Identifying other bacterial populations that may interact and synergize with A. muciniphila to promote colitis in IL-10−/− mice would be an important future endeavor. Indeed, it has been previously demonstrated that monoassociation of GF IL-10−/− mice with different bacterial populations resulted in varying degrees of colitis (Kim et al., 2005).
Our data indicate that NLRP6 limits the colonization of A. muciniphila in an IL18- dependent manner. Both IL18−/− and IL18R−/− mice have increased A. muciniphila and furthermore, administration of rIL18 reduced the relative abundance of A. muciniphila in Nlrp6−/− mice. Both IL18−/− and IL18R−/− mice also experience increased severity of DSS-induced colitis (Takagi et al., 2003). In addition, our results are consistent with previously published reports of microbiome changes observed in mice deficient in the AIM2 inflammasome. Similar to Nlrp6−/− mice, AIM2−/− mice are susceptible to DSS-induced colitis, have impaired IL18 production, and also have increased colonization of A. muciniphila (Hu et al., 2015; Man et al., 2015). In addition, it was recently demonstrated that conventionalized GF Nlrp6−/− mice had a microbiome that was different from that of recolonized GF WT mice although it was not determined if recolonized GF Nlrp6−/− mice were enriched for any colitogenic bacteria (Levy et al., 2015). Administration of rIL18 also caused additional changes in the microbiome of Nlpr6−/− mice that did not occur in WT mice (Levy et al., 2015). Whether NLRP6 regulates the composition of other disease-inducing or protective bacteria through IL18 warrants further investigation. Nlrp6−/− mice also had reduced levels of AMP production that were IL18-dependent (Levy et al., 2015). Although AMPs may have selective bactericidal activity (Hooper et al., 2003), it is unclear whether this is the primary mechanism by while NLRP6 regulates A. muciniphila abundance. Our study also does not rule out the possibility that NLRP6 affects A. muciniphila colonization indirectly such as by influencing the abundance of other bacterial populations.
In summary, our data provide critical insights into how NLRP6 maintains intestinal homeostasis and establish a role for NLRP6 in limiting the colonization of IBD-inducing bacteria, in particular, the mucolytic A. muciniphila, which can promote colitis in both SPF and GF IL10−/− mice. Resistance to A. muciniphila accumulation by NLRP6 signaling is mediated at least in part by IL18. Our findings have significant clinical implications as they illustrate the ability of innate immune and cytokine signaling to affect colonization levels of bacteria that in the right context can have direct impact on colitis susceptibility. As the ligand for NLRP6 remains unknown, future studies to identify upstream regulators and the mechanism by which downstream effectors modulate the gut microbiome and IL18 production would be important for the development of strategies to maintain intestinal health and prevent IBD. The current work provides the necessary framework for these studies.
Experimental Procedures
Animals
SPF WT, Nlrp6−/−, IL10−/−, IL18−/−, IL18r1−/−, IL1β−/−, and IL1R−/− mice (all B6 background) were bred in-house at the University of Michigan animal facility. Nlrp6−/−IL10−/− mice were generated by crossing Nlrp6−/− females with IL10−/− males and then intercrossing F1 heterozygotes. Adult male or female 8–14 week-old mice were used except in cohousing experiments in which 4-week-old SPF IL10−/− and IL10−/− Nlrp6−/− mice were cohoused at 1:1 ratio for up to 8 weeks. GF WT, IL10−/− and Nlrp6−/− mice were housed and bred at the GF mouse facility, and sterility was regularly verified by aerobic and anaerobic cultures, Gram stains and qPCR. GF mice were conventionalized using bedding/feces pooled from several cages of SPF WT mice. gWT or gNlrp6−/− mice were housed in 2–3 separate cages each to control for potential cage effects. Animal studies were conducted under protocols approved by the University of Michigan Committee on the Use and Care of Animals.
Treatment of mice with rIL18
Nlrp6−/− mice were administered 1 μg of sterilely filtered rIL18 (MBL) i.p. in 200 μl PBS for 3 consecutive days.
Assessment of colon inflammation
Colons were flushed free of feces, opened longitudinally, and jelly-rolled for formalin fixation and paraffin embedding. Histological assessment of H&E sections was performed in a blinded fashion by a pathologist (KAE) using a previously described scoring system (Seregin et al., 2016).
Reverse transcription and qPCR
Total RNA was isolated from colon tissue using the Nucleospin RNA kit (Machery-Nagel). cDNA synthesis was performed using iScript (Bio-Rad), and cDNA was used for qPCR using the SYBR Green Master Mix (Applied Biosystems) on the ABI 7900HT (Applied Biosystems). Bacterial DNA was isolated from fecal samples using the MoBio PowerSoil DNA Isolation Kit. Relative abundance of bacterial populations in stool samples was quantified by qPCR and normalized to the universal 16S rRNA gene EUB primers. Primer sequences are shown in Supplementary table I.
ELISA
Lcn-2 levels were measured from the supernatants of feces homogenized in PBS at 100 mg/ml and diluted to a range of 1:500 to 1:5000 using the Lcn-2/NGAL ELISA kit (R&D Systems).
Oral gavage of bacteria
A. muciniphila and B. acidifaciens were isolated from WT C57BL/6 mice. Specifically, cecal contents from mice were collected under strictly anaerobic conditions immediately after euthanasia, homogenized in anaerobic modified chopped-meat carbohydrate broth (CMCB) (Hehemann et al., 2012), and plated as serial dilutions onto either brain heart infusion (BHI) agar with 10% horse serum and gentamicin (200 μg ml−1) or agar-solidified CMCB with gentamicin. Plates were cultured at 37° C anaerobically (85% N2, 10% H2, 5% CO2) in a Coy anaerobic chamber and several hundred colonies representing various morphologies were picked into 96-well plates containing liquid CMCB with gentamicin. Growth kinetics at 600nm were monitored for three days using a plate handling device connected to an absorbance reader (Biotek, Winooski, VT) as previously described (Martens et al., 2011) and the resulting growth profiles used to distinguish culture wells that were likely to contain different organisms based on unique profiles. Sequencing of V4 of 16S rRNA was used to confirm identity of bacterial species. Prior to gavage, bacterial strains were grown anaerobically at 37° C overnight in CMCB. IL-10−/− mice were gavaged with A. muciniphila or B. acidifaciens at a dose 2×108 CFU/mouse in 200 μl media.
Isolation of Bacterial DNA and 16S rRNA Sequences Analyses
DNA was isolated from fecal samples and processed as previously described and further detailed in supplemental methods (Kozich et al., 2013; Schloss et al., 2009). FASTQ sequence data can be obtained from the Sequence Read Archive at NCBI (Accession number SRP076233).
Bacterial translocation
Total DNA was extracted from MLNs using the MoBio PowerSoil DNA Isolation Kit and bacterial load quantified by qPCR using the universal 16S rRNA gene primers (EUB) in 20 ng DNA.
Statistical analysis
Statistically significant differences were determined using two-way ANOVA with a Bonferroni post-hoc test (time x genotype, p<0.05) (e.g., weight changes or fecal Lcn-2 time courses), by one-way ANOVA with a Student-Newman-Keuls post-hoc test (p value < 0.05) (e.g., qPCR analysis), or by 2-tailed Student’s unpaired t test when only 2 groups are compared (e.g., fecal Lcn-2 levels). Kruskal–Wallis one-way analysis of variance non-parametric test was used for non-continuous variables (histology scoring). Differences in bacterial community structure were analyzed using AMOVA in mothur (Excoffier et al., 1992). Data are shown as mean ± SEM. Statistical analyses were performed using GraphPad Prism6 and mothur software.
Supplementary Material
Highlights.
NLRP6 deficiency promotes colitis in IL10−/− mice.
NLRP6 controls colonization of A. muciniphila in an IL18-dependent manner.
Akkermansia muciniphila is sufficient to induce colitis in germfree IL10−/− mice.
Acknowledgments
We appreciate assistance from Joel Whitfield from the University of Michigan ELISA Core, the Flow Cytometry Core and the Germfree Animal Facility. This work was supported by the National Institutes of Health (NIH) grants R01CA166879 and R21CA191744 and American Cancer Society Research Scholar Grant awarded to CGY, and NIH grants F32CA200144 and UL1TR000433 from the National Center for Advancing Translational Sciences awarded to SSS.
Footnotes
Disclosure
The authors have declared no conflicts of interest.
Author contributions
Conceptualization, G.Y.C. and S.S.S.; Methodology, G.Y.C. and S.S.S.; Investigation, S.S.S., N.G., B.S., J.C., J.M., N.A.P., N.T.B., L.Z, P.D.S., E.C.M, K.A.E.; Writing – Original Draft, G.Y.C. and S.S.S.; Writing – Review & Editing, G.Y.C., S.S.S., E.C.M., P.D.S., K.A.E.; Funding Acquisition, G.Y.C. and S.S.S.; Resources, K.A.E., P.D.S., E.C.M., L.Z.; Supervision, G.Y.C. and S.S.S.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014;2:20. doi: 10.1186/2049-2618-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, Stappenbeck TS. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. The Journal of clinical investigation. 2007;117:258–269. doi: 10.1172/JCI29159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011;186:7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. The Journal of clinical investigation. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson NJ, Leach MW, Fort MM, Thompson-Snipes L, Kuhn R, Muller W, Berg DJ, Rennick DM. T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med. 1996;184:241–251. doi: 10.1084/jem.184.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh BP, Klopfleisch R, Loh G, Blaut M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PLoS One. 2013;8:e74963. doi: 10.1371/journal.pone.0074963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, Carlson A, Merriam S, Lora JM, Briskin M, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002;530:73–78. doi: 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- Hehemann JH, Kelly AG, Pudlo NA, Martens EC, Boraston AB. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc Natl Acad Sci U S A. 2012;109:19786–19791. doi: 10.1073/pnas.1211002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359–1372. doi: 10.1002/ibd.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- Hoshi N, Schenten D, Nish SA, Walther Z, Gagliani N, Flavell RA, Reizis B, Shen Z, Fox JG, Iwasaki A, et al. MyD88 signalling in colonic mononuclear phagocytes drives colitis in IL-10-deficient mice. Nat Commun. 2012;3:1120. doi: 10.1038/ncomms2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Peng L, Kwak YT, Tekippe EM, Pasare C, Malter JS, Hooper LV, Zaki MH. The DNA Sensor AIM2 Maintains Intestinal Homeostasis via Regulation of Epithelial Antimicrobial Host Defense. Cell reports. 2015;13:1922–1936. doi: 10.1016/j.celrep.2015.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjovall H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63:281–291. doi: 10.1136/gutjnl-2012-303207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- Kaplan GG. The global burden of IBD: from 2015 to 2025. Nature reviews Gastroenterology & hepatology. 2015;12:720–727. doi: 10.1038/nrgastro.2015.150. [DOI] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keubler LM, Buettner M, Hager C, Bleich A. A Multihit Model: Colitis Lessons from the Interleukin-10-deficient Mouse. Inflamm Bowel Dis. 2015;21:1967–1975. doi: 10.1097/MIB.0000000000000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesler P, Fuss IJ, Strober W. Experimental Models of Inflammatory Bowel Diseases. Cellular and molecular gastroenterology and hepatology. 2015;1:154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, Tyler AD, van Sommeren S, Imhann F, Stempak JM, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome medicine. 2014;6:107. doi: 10.1186/s13073-014-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell. 2015;163:1428–1443. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Zhu Q, Zhu L, Liu Z, Karki R, Malik A, Sharma D, Li L, Malireddi RK, Gurung P, et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell. 2015;162:45–58. doi: 10.1016/j.cell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9:e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalingam NA, Robinson CJ, Bergin IL, Eaton KA, Huffnagle GB, Young VB. The effects of intestinal microbial community structure on disease manifestation in IL-10−/− mice infected with Helicobacter hepaticus. Microbiome. 2013;1:15. doi: 10.1186/2049-2618-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol. 2015;33:49–77. doi: 10.1146/annurev-immunol-032414-112306. [DOI] [PubMed] [Google Scholar]
- Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A. 2011;108:9601–9606. doi: 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105:2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- Rajilic-Stojanovic M, Shanahan F, Guarner F, de Vos WM. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis. 2013;19:481–488. doi: 10.1097/MIB.0b013e31827fec6d. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rubino SJ, Selvanantham T, Girardin SE, Philpott DJ. Nod-like receptors in the control of intestinal inflammation. Curr Opin Immunol. 2012;24:398–404. doi: 10.1016/j.coi.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O’HUigin C, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seregin SS, Golovchenko N, Schaf B, Chen J, Eaton KA, Chen GY. NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury. Mucosal Immunol. 2016 doi: 10.1038/mi.2016.55. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RA. Faecal markers of gastrointestinal inflammation. J Clin Pathol. 2012;65:981–985. doi: 10.1136/jclinpath-2012-200901. [DOI] [PubMed] [Google Scholar]
- Stappenbeck TS, Virgin HW. Accounting for reciprocal host-microbiome interactions in experimental science. Nature. 2016;534:191–199. doi: 10.1038/nature18285. [DOI] [PubMed] [Google Scholar]
- Takagi H, Kanai T, Okazawa A, Kishi Y, Sato T, Takaishi H, Inoue N, Ogata H, Iwao Y, Hoshino K, et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scandinavian journal of gastroenterology. 2003;38:837–844. doi: 10.1080/00365520310004047. [DOI] [PubMed] [Google Scholar]
- White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS computational biology. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854. e1841. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185:4912–4920. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zella GC, Hait EJ, Glavan T, Gevers D, Ward DV, Kitts CL, Korzenik JR. Distinct microbiome in pouchitis compared to healthy pouches in ulcerative colitis and familial adenomatous polyposis. Inflamm Bowel Dis. 2011;17:1092–1100. doi: 10.1002/ibd.21460. [DOI] [PubMed] [Google Scholar]
- Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World journal of gastroenterology. 2014;20:91–99. doi: 10.3748/wjg.v20.i1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.