

Abstract
Sphingosine 1-phosphate (S1P) lyase (SPL) is an intracellular enzyme that mediates the irreversible degradation of the bioactive lipid, S1P. We have previously reported that overexpressed SPL displays anti-influenza viral activity; however the underlying mechanism is incompletely understood. In this study, we demonstrate that SPL functions as a positive regulator of IKKε to propel type I interferon (IFN)-mediated innate immune response against viral infection. Exogenous SPL expression inhibited influenza A virus (IAV) replication, which correlated with an increase in type I IFN production and interferon stimulated gene accumulation upon infection. In contrast, the lack of SPL expression led to an elevated cellular susceptibility to IAV infection. In support of this, SPL-deficient cells were defective in mounting an effective IFN response when stimulated by influenza viral RNAs. SPL augmented the activation status of IKKε, and enhanced the kinase-induced phosphorylation of IRF3 and synthesis of type I IFNs. However, S1P degradation-incompetent form of SPL also enhanced IFN responses, suggesting that SPL’s pro-IFN function is independent of S1P. Biochemical analysis revealed that SPL as well as the mutant form of SPL interact with IKKε. Importantly, when endogenous IKKε was down-regulated by an siRNA approach, SPL’s anti-influenza viral activity was markedly suppressed. This indicates that IKKε is crucial for SPL-mediated inhibition of influenza virus replication. Thus, the results illustrate the functional significance of the SPL-IKKε-IFN axis during host innate immunity against viral infection.
Introduction
The type I IFN response is a first line of innate immune system that protects the host from pathogenic viral infections (1, 2). The potent antiviral activity of type I IFNs, which include 13 isotypes of IFN-α and a single IFN-β, has been observed during infections by numerous viruses including influenza (3, 4). Influenza virus infection induces type I IFNs upon cellular sensing of viral RNA products primarily by RIG-I, which recognizes the 5′ppp moiety on influenza viral RNAs (5–7). Upon activation of RIG-I, downstream adaptor protein MAVS is engaged followed by recruitment of the kinases IKKε and TBK1 (8), both of which can phosphorylate IRF3 as well as IRF7. Phosphorylated IRF3 and IRF7 act as the major transcription factors that drive the production of IFN-β and IFN-α4 followed by the production of other IFN-α subtypes (9). Binding of IFNs to the cognate receptor (IFNAR) triggers the activation of JAK-STAT signaling pathway (10), which culminates in the transcriptional induction of an array of interferon stimulated genes (ISGs) including MX1, ISG15, OAS1, ISG56 and RIG-I. The ISG products act in concert to limit virus replication, launching the antiviral status (11). The importance of the IFN response upon viral infection is emphasized by the fact that many pathogenic viruses have evolved to antagonize the type I IFN signaling pathway to a certain extent (4, 12, 13). However, despite the presence of extensive investigation, the regulatory mechanism of type I IFN production pathway is not fully understood. Therefore, it is important to identify novel cellular factors that regulate type I IFN synthesis and unravel the detailed molecular action mode for type I IFN responses against microbial infections.
S1P lyase (SPL) is an intracellular enzyme that catalyzes the irreversible cleavage of the bioactive lipid sphingosine 1 phosphate (S1P) at its C2-C3 carbon, into by-products namely hexadecenal and phosphoethanolamine (14, 15). SPL has been reported to be localized to the endoplasmic reticulum (ER) and also found in the mitochondrial associated membrane (MAM) which is associated with both mitochondria and ER (14, 16, 17). Since S1P is a bioactive lipid (18–20), SPL has been implicated in diverse cellular processes and diseases, such as cancer, immunity, inflammation, and development (21–26). Yet, a direct role for SPL in the context of the innate immune response to virus infections has not been characterized.
We have previously published that cells constitutively overexpressing SPL were resistant to the replication of influenza A virus (IAV) (27, 28). In this study, we have characterized the contribution of endogenous SPL to type I IFN production pathway when pattern recognition receptor (PRR) such as RIG-I is stimulated following virus infection. We found that endogenous SPL is critical for eliciting an effective IFN response upon cellular recognition of influenza viral RNAs. More importantly, SPL binds to the kinase IKKε, thereby increasing its activation to promote the synthesis of type I IFN. Collectively, our results demonstrate that SPL is a host factor that augments type I IFN response during virus infection. Further, the study delineates the relationship between IKKε and SPL, which provides a mechanistic understanding behind the pro-IFN activity of SPL.
Materials and Methods
Cell lines and transfection
293T cells and human lung epithelial A549 cells, were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) as explained elsewhere (29, 30). MDCK cells were cultured in minimum essential medium Eagle (MEM; Mediatech). Cells were cultured in a CO2 incubator at 37°C, and all media were supplemented with 10% fetal bovine serum (HyClone) and penicillin (100 U/ml)/streptomycin (100 μg/ml) (Invitrogen). Lipofectamine 3000 transfection reagent (Invitrogen) was used to transfect plasmid DNA into A549 cells in 24-well plates at a concentration of 500 ng/ml of the indicated plasmid DNA by following the protocols recommended by the manufacturer. Empty vector plasmids were used to ensure equal total amounts of DNA were being transfected into each sample of transfection. Also, equal amounts of transfection reagent were used for all the samples including negative controls. For transfection of DNA into 293T cells, LipoD293 transfection reagent (Signagen) was used according to manufacturer’s instructions either in 24 well or 6 well plates Lipofectamine RNAiMax was used to perform si-RNA transfections on 293T cells in 24-well plate format. For SPL re-constitution experiments, we have used a transient transfection approach to overexpress SPL protein in the SPL-deficient cells. SPL KO cells (2 X 105) were transfected with 0.5 μg of SPL or SPL(K353L)-encoding plasmid and the expression of SPL was confirmed by Western blot analysis.
DNA vector constructs
Mammalian expression plasmids encoding WT SPL constructs including pc-hSPL and pc-hSPL-GFP and the enzymatically inactive mutant SPL constructs including pc-hSPL(K353L) and pc-hSPL(K353L)-GFP were provided by Julie D. Saba (Children’s Hospital Oakland Research Institute, California) (31). The mutation site sequences in the SPL(K353L) constructs were confirmed by DNA sequencing at the DNA core facility (University of Missouri-Columbia). Mammalian expression plasmids encoding Flag-tagged IKKε, Myc-tagged TBK1, IRF3, Flag-tagged IRF7, and luciferase reporter constructs IFN-α1/pGL3 and IFN-β/pGL3 were previously used elsewhere (8, 32). To generate HA-tagged SPL construct, SPL-coding sequence from pVB003-Flag-hSPL (23) was amplified by PCR using forward primer: 5′ CGGAATTCGCCCTAGCACAGACCTTCTGAT 3′ and reverse primer: 5′ GGGGTACCTCAGTGGGGTTTTGGAGAAC 3′. The amplified full length SPL-encoding PCR fragments were treated with EcoRI/KpnI and inserted into EcoRI/KpnI-digested pCMV-HA vector, which was provided by Dr. David Pintel (University of Missouri-Columbia). The nucleotide sequences of the subcloned full length HA-tagged SPL construct was confirmed by DNA sequencing.
Viruses
Influenza A/WSN/33 (H1N1) virus was initially provided by Yoshihiro Kawaoka (University of Wisconsin-Madison). Madin-Darby canine kidney (MDCK) epithelial cells were used to amplify the virus. Virus infection and titration studies were performed as described previously (27, 29, 30). For the infection studies, cells were infected with IAV for 1 hr and then incubated with the medium. The supernatants containing infectious viruses were harvested for the titration by plaque assay on MDCK cells. For the plaque assay, using serial dilutions of culture supernatants, viruses were adsorbed onto 3 X105 MDCK cells/ml for 1 hr, and then the cells were incubated with 2X Eagle’s minimum essential medium (EMEM; Gibco) mixed with an equal portion of 1% agarose (SeaKem ME). Cells were fixed with 25% formalin and stained with crystal violet after 2–3 days of incubation to count plaques that are generated by viral cytopathic effects.
CRISPR-Cas9 knockout of SPL
293T stable cell lines knocked out for SPL were generated by transduction with LentiCRISPR v2 (a gift from Feng Zhang, MIT, Massachusetts; Addgene plasmid # 52961) (33). Puromycin selection was applied followed by single cell isolation and clonal expansion. Target sequences used as guide RNA sequences were: 5′-GGTCCCATTGACGAAGATGATGG-3′ (exon 9) and 5′-GCATCACATTACTACACGCCCGG-3′ (exon 14). To verify the disruption of the SGPL1 gene open reading frame (ORF), genomic DNA from the cell lines were extracted and PCR amplified with flanking primers to amplify the target region. Primer sequences used for PCR from genomic DNA are 5′-CCCTCACTGTGGGATCACTTC-3′ and 5′-AACGGCTAGTCAACAGGAGG-3′ for Exon 9 and 5′-TGACACCCCAAGCATGAGAG-3′ and 5′-GATGTCTGTGGCAAAGGGGC-3′ for Exon 14. Among knock out (KO) cells, two cell lines were chosen and used in this study based on the loss of SPL protein expression. Exon 9 KO cells (KO-1) used in the study had a deletion of 9 and 27 nucleotides in each chromosome and exon 14 KO cells (KO-2) had an insertion of 1 and 2 nucleotides in each chromosome.
Western blot analysis and antibodies
Western blotting was performed as described elsewhere (27, 29, 30, 34). Cells were lysed using 2X sample buffer containing β-mercaptoethanol and boiled at 95°C for 10 min. Equal amounts of protein samples were loaded onto a 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel followed by transfer of resolved proteins from the gel to nitrocellulose membranes (Bio-Rad). Membrane-bound antibodies were detected using an enhanced chemiluminescence substrate (Thermo Scientific). Antibodies against influenza viral NP and M1 were purchased from Abcam; antibodies against influenza viral NS1, M2, Actin and SPL were purchased from Santa Cruz; antibodies against human GAPDH (glyceraldehyde-3-phosphate dehydrogenase), ISG56, RIG-I, pIRF3 (Ser 396), IRF3, pIKKε (Ser 172), IKKε, pTBK1 (Ser 172), TBK1, Myc tag, GFP, pSTAT1 (Tyr 701), FLAG-tag and HA tag were purchased from Cell Signaling Technology. Antibody against Influenza H1N1 NS2 was purchased from Genscript. All the data presented were repeated at least twice with independent experimental settings.
Real-time PCR
Total cellular RNA was extracted using Tri Reagent (Sigma-Aldrich) according to the manufacturer’s instructions and was treated with DNase I (Thermo Scientific) to remove contaminating DNA. Isolated RNA was reverse transcribed using random hexamers (Invitrogen) or the forward NP primer for amplifying IAV-NP negative sense NP RNA or the reverse NP primer for amplifying IAV-NP positive sense NP RNA. The resulting cDNA was then used as template for real-time quantitative PCR (qPCR) using gene-specific primers. Primers for human MX1 (5′-GTT TCC GAA GTG GAC ATC GCA-3′ and 5′-CTG CAC AGG TTG TTC TCA GC-3′), human ISG15 (5′-CGC AGA TCA CCC AGA AGA TCG-3′ and 5′-TTC GTC GCA TTT GTC CAC CA-3′), human OAS-1 (5′-GAT CTC AGA AAT ACC CCA GCC A-3′ and 5′-AGC TAC CTC GGA AGC ACC TT-3′) human IFN-β (5′-CGC CGC ATT GAC CAT CTA-3′ and 5′-GAC ATT AGC CAG GAG GTT CTC A-3′), and IAV NP (5′-TGC TTC CAA TGA AAA CAT GG-3′ and 5′-GCC CTC TGT TGA TTG GTG TT-3′) were used. qPCR was performed with SYBR green I chemistry using a Step One Plus Real-Time PCR instrument. cDNA quantities were normalized to the corresponding GAPDH RNA quantities from the same samples. All the data presented were repeated at least twice with independent experimental settings.
Luciferase reporter assay
293T cells (2 X 105) were transfected in 24-well plates with IFN-response triggering plasmids such as IKKε, IRF3 and IRF7 along with reporter plasmids 50 ng IFN-β/pGL3 reporter plasmid or IFN-α1/pGL3, as indicated along with 10 ng of pRL-CMV (Promega) encoding renilla luciferase as a transfection control in the presence of SPL or empty vector control DNA (CTR). At 24 hr post transfection, cells were lysed in the passive lysis buffer to measure the renilla and firefly luciferase activities using the dual luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Luminescence values were measured using Perkin Elmer Enspire 2300 multilabel reader.
Co-immunoprecipitation assay
293T (2 X 106) cells in 100 mm tissue culture dishes were transfected with 5 μg of GFP-SPL (gift from Dr. Julie D. Saba at Children’s Hospital Oakland Research Institute, California) and either control vector or 5 μg of Flag-IKKε. 1 day after transfection, cells were harvested in 1 ml of immunoprecipitation (IP) lysis buffer (Thermo Scientific) containing protease inhibitor cocktail and PMSF at 1 mM. Cell lysates were incubated with 20 μl of Anti DYKDDDDK G1 affinity resin overnight under rotation at 4 ° C. For the GFP or Myc-pull down experiment, plain beads were coated with α-GFP or α-MYC antibody (Cell signaling technologies), respectively and 20 μl of these beads were incubated with cell lysates overnight under rotation at 4 ° C. For the HA-pull down experiment, 20 μl of anti-HA affinity resin (Pierce) was used for incubation with cell lysates. The beads were washed three times intensively with IP lysis buffer to remove any nonspecific binding. After the final wash, sample buffer containing β-mercaptoethanol was added to the beads. The lysates were boiled for 10 mins and then used for Western blotting analysis. The experiments were independently repeated at least twice with similar results.
Confocal microscopy
293T cells were plated on 4 or 8-well chamber slides (Nunc). The following day, cells were transfected with 200 ng each of the indicated plasmids using LipoD293 transfection reagent (Signagen). At one day after transfection, cells were fixed in 4% paraformaldehyde (Fisher) and then permeabilized in 0.5% Triton X-100 (Sigma) for 10 minutes at room temperature. Samples were then blocked with 10% BSA for 30 mins followed by overnight incubation with indicated primary antibodies, anti-Flag (1:1000) antibody or anti-GFP antibody (1:100) in 3% BSA overnight at 4°C. Followed by washing, samples were incubated with Alexa Fluor 488-conjugated anti rabbit IgG (1:500, Invitrogen) or Alexa Fluor 568-conjugated anti-mouse IgG (1:500, Invitrogen) for 2 hrs, followed by staining with DRAQ5 dye (300 nM, Thermo Scientific) for 15 min at room temperature. The images were then acquired on a Leica SPE2 DM5500Q confocal microscope and analyzed with LSM Image Browser software. Representative fields are shown: each image was selected from 5–10 different fields. Results were equivalent in repeated experiments.
RNA interference
27mer siRNA duplexes targeting IKKε (si-IKKε) or Trilencer-27 universal scrambled negative control siRNA duplex (SCR) were purchased from Origene and used at a final concentration of 20 nM to transfect 293T cells using Lipofectamine RNAiMax transfection reagent according to the manufacturer’s instructions. 27mer siRNA duplexes targeting SPL (si-SPL) purchased from Origene was used at a final concentration of 25 nM to transfect 293T cells. Cells were harvested 2 days post si-RNA transfection and the knockdown of IKKε and SPL were confirmed by performing Western blotting analysis.
Statistical analysis
All bars represent means, error bars show standard errors of the means (SEM), and averages were compared using a bidirectional, unpaired Student’s t test. Data are representative of 2 or 3 independent experimental repetitions.
Results
SPL inhibits IAV replication, which is associated with an increased type I IFN response
We have previously observed that SPL overexpression inhibited replication of influenza A virus (IAV) (27). In that study, we had mainly used the cell line that overexpresses SPL protein constitutively. To determine whether or not the long-term constant expression of SPL is essential for the suppression of viral replication, a transient transfection experiment was performed. As shown in figure 1A, transient overexpression of SPL inhibited the expression of IAV proteins such as NS1 and M2 at both 1 MOI and 3 MOI. The result suggests that SPL displays the anti-viral activity regardless of whether SPL is temporarily or stably overexpressed. To further determine the role of endogenous SPL in viral replication, SPL-deficient cells were created by using the CRISPR/Cas9-mediated genetic approach, as described in the Methods section (33). We selected two different cell lines based on the knockout of endogenous SPL as confirmed by Western blot analysis (Fig. 1B and 1C). SPL-deficient cells of both KO-1 and KO-2 proliferated at a similar rate when compared to the SPL-sufficient wild type (WT) cells without any noticeable defects in cell growth (Fig. S1). Upon IAV infection, KO-1 and KO-2 cells displayed an enhanced susceptibility to IAV replication, as shown by a strong increase in the level of both structural and non-structural proteins of IAV such as NS2, NP, NS1, and M2 (Fig. 1B and 1C). Furthermore, we investigated the role of endogenous SPL in IAV propagation by comparing the production of infectious IAV from WT or SPL-deficient cells. As shown in Figure 1D, the lack of endogenous SPL significantly increased IAV production at both 2 dpi and 3 dpi. These data further verify the anti-viral function of endogenous SPL.
Figure 1. SPL inhibits the replication of influenza virus.

(A) A549 cells (2 X 105) were transfected with empty vector control DNA (CTR) or HA-tagged SPL (HA-SPL)-encoding DNA. One day later, cells were either mock-infected (Mock) or infected with 1 or 3 multiplicity of infection (MOI) of IAV. At 8 hr post-infection (hpi), cells were harvested for Western blot analysis to check the level of NS1, M2, HA-SPL, and GAPDH. (B and C) WT, KO-1 cells (B), and KO-2 cells (1 X 106) (C) were infected with 0.1 MOI of IAV or mock-infected. At 1 day post–infection (dpi), Western blotting was performed to detect NS2, NP, NS1, M2, SPL, and GAPDH proteins. (D) WT or KO-2 cells (2 X 105) were infected with 0.01 MOI of IAV. At 2dpi and 3dpi, plaque assay was performed to determine viral titer in the supernatant of the infected cells. Values represent mean ± SEM (n = 3/group; **, P ≤ 0.01; ***, P ≤ 0.001).
Type I IFNs are the major antiviral cytokines produced by virus-infected cells and the SPL-overexpressing cell line appeared to exhibit enhanced IFN signaling to repress IAV replication (27, 35). We further assessed the effect of SPL expression on type I IFN responses to IAV infection. Transient overexpression of SPL led to a significant increase in IFN-β production at 4 hpi when measured by qPCR (Fig. 2A). However, at 6 hpi, little increase was observed in SPL-overexpressing cells when compared to the control vector-transfected, infected cells, suggesting a temporal increase in IFN synthesis by SPL. Since ISGs are the ultimate antiviral effectors of the type I IFN response, we measured the extent of ISG upregulation. SPL enhanced the mRNA level of MX1 by approximately 2 fold (Fig. 2B). Also, SPL overexpression modestly enhanced the protein level of ISGs such as RIG-I and ISG56 (Fig. 2C). These ISGs have been reported to play important antiviral role during influenza virus infection (36, 37). Of note, in the mock-infected cells, SPL alone did not induce IFN responses, suggesting that SPL functions as a pro-IFN factor after cellular sensing of viral infection (Fig. 2A–C). Accordingly, SPL was shown to suppress the synthesis of viral NP-specific RNAs of both positive and negative sense RNAs at 6 hpi (Fig. 2D and 2E). The transient increase of IFN production by SPL at 4 hpi followed by its rapid decrease at 6hpi (Fig. 2A) could be due to the lower amounts of IFN-stimulatory viral RNAs being present in SPL-overexpressing cells compared to control cells as shown in figure 2D and 2E. Further, we have examined the type I IFN response during IAV infection in the SPL-deficient cells. Following IAV infection, endogenous SPL was shown to be critical for effective production of IFN-β (Fig. 2F) and ISGs such as ISG15 (Fig. 2G), RIG-I and ISG56 (Fig. 2H). Collectively, these data demonstrate that SPL augments type I IFN and ISG production, which correlates with a decrease in IAV replication.
Figure 2. SPL increases type I IFN response to influenza virus infection.

(A and B) A549 cells (2 X 105) were transfected with CTR (empty vector control) or SPL. At 1 day post-transfection, cells were either mock-infected or infected with 0.5 MOI of IAV. Relative RNA levels of IFN-β (A) and MX1 (B) were calculated by qPCR at 4 and 6 hpi. Values represent mean ± SEM (n = 3/group; **, P ≤ 0.01; ***, P ≤ 0.001, ****, P ≤ 0.0001, ns = not significant). (C) A549 cells (2 X 105) were transfected with empty vector control DNA (CTR) or HA-tagged SPL (HA-SPL)-encoding DNA. One day later, cells were either mock-infected (Mock) or infected with 1 or 3 MOI of IAV. At 8 hpi, cells were harvested for Western blot analysis to check the level of RIG-I, ISG56, HA-SPL, and GAPDH proteins. (D and E) A549 cells (2 X 105) were transfected with CTR (empty vector control) or SPL. At 1 day post-transfection, cells were either mock-infected or infected with 0.5 MOI of IAV. Relative levels of IAV NP negative sense RNA (D) and IAV NP positive sense RNA (E) were calculated by qPCR at 4 and 6 hpi. Values represent mean ± SEM (n = 3/group; *, P ≤ 0.05; ns = not significant). (F) WT 293T cells or KO-2 cells (2 X 105) were either mock-infected or infected with IAV at 0.01 MOI and at 2 dpi, the relative levels of IFN-β (F) and ISG15 (G) were calculated by qPCR. Values represent mean ± SEM (n = 3/group; **, P ≤ 0.01). (H) WT 293T cells or KO-2 cells (2 X 105) were either mock-infected or infected with IAV at 0.01 MOI and at 2 dpi, cells were harvested for Western blot analysis to check the level of RIG-I, ISG56, SPL, NS1, and GAPDH proteins.
Endogenous SPL is important for efficient induction of IFN response upon cellular sensing of influenza viral RNAs
Although SPL enhanced IFN responses during IAV infection, the effect appeared to be modest (Fig. 2B and 2C). This could be due to the presence of dynamic antagonistic interaction between viral components and host cellular factors of the IFN system during the IAV replication process (30, 38–44). To overcome this, we opted for an alternative method to trigger IFN response other than IAV infection. Hence, viral RNAs (vRNAs) that were purified from IAV-infected cells were used to stimulate IFN production. Transfection of cells with influenza vRNAs was reported to activate RIG-I signaling pathway by recognition of triphosphate moieties on the viral RNAs, leading to an induction of type I IFNs (5, 7, 45, 46). Cellular RNAs were isolated from uninfected cells (designated as cRNA) and used as a negative control. Following transfection with vRNAs, cells deficient in endogenous SPL (KO-1 and KO-2 cells) were much less efficient in synthesizing IFN-β mRNA than the SPL-sufficient WT cells (Fig. 3A). The decreased production of IFNs caused by SPL-deficiency resulted in the decreased level of ISG-specific mRNAs being upregulated such as ISG15 (Fig. 3B) and OAS1 (Fig. 3C). Accordingly, SPL deficiency markedly suppressed the protein expression of ISGs such as RIG-I and ISG56 (Fig. 3D and Fig. 3E). Consistent with these findings, when SPL was downregulated by an siRNA, viral RNA-induced expression of RIG-I and ISG56 was impaired (Fig. 3F). These data clearly demonstrate that endogenous SPL is important for efficient production of type I IFNs and ISG products, which may in turn establish optimal antiviral condition upon cellular recognition of IAV RNAs.
Figure 3. SPL is important for mounting an effective IFN response upon cellular recognition of influenza viral RNAs.

(A, B, and C) WT 293T cells, KO-1, or KO-2 cells (1 X 106) were either transfected with RNAs isolated from IAV-infected cells (viral RNAs, vRNA) or RNAs isolated from non-infected cells (cellular RNAs, cRNA) at a concentration of 2.5 7mu;g/ml. At 1 day after transfection, relative mRNA levels of IFN-β (A), ISG15 (B), and OAS1 (C) were determined by performing qPCR and plotted as fold induction over cRNA-treated samples. Values represent mean ± SEM (n = 3/group; ***, P ≤ 0.001; ****, P ≤ 0.0001). (D and E) WT 293T cells, KO-1 cells (D), and KO-2 cells (1 X 106) (E) were either transfected with vRNA or cRNA. After 1 day, Western blotting analysis was performed to detect RIG-I, ISG56, SPL, and GAPDH proteins. (F) 293T cells were transfected with siRNA specific to SPL (si-SPL) or non-specific scrambled siRNA (SCR). At 24 hours post-transfection, cells were either transfected with vRNA or cRNA at a concentration of 1.25 μg/ml. One day later, the levels of RIG-I, ISG56, SPL, and GAPDH were analyzed by Western blotting.
SPL interacts with IKKε and augments IKKε activation that leads to enhanced type I IFN production
IKKε and TBK1 phosphorylate and activate IRF3, which functions as a transcription factor to direct type I IFN synthesis (8, 9). To define the molecular mechanism behind SPL’s pro-IFN activity, we investigated whether SPL regulates IKKε or TBK1 activation, which leads to the activation of IRF3. While TBK1 was auto-phosphorylated and induced IRF3 phosphorylation, SPL had no effect on the level of TBK1 auto-phosphorylation or TBK1-mediated IRF3 phosphorylation (Fig. S2A), suggesting that SPL does not affect TBK1-mediated IFN responses. In contrast, SPL increased the level of activated form of IKKε (pIKKε at Ser172) and IKKε-mediated phosphorylation of IRF3 (Fig. 4A). Further, SPL increased the level of pIKKε in a dose-dependent manner (Fig. 4B). These data indicate that SPL promotes the activation of IKKε, but not that of TBK1. To further investigate if SPL regulates IKKε activation, we employed the luciferase reporter assay to measure the promoter activities of IFN-α/β. This allows us to determine if SPL has the ability to influence IKKε-induced IFN synthesis. As shown in Figure 4C, SPL significantly increased IKKε-mediated activation of IFN-β promoter. Also, SPL promoted IKKε-mediated activation of IFN-α promoter compared to the control when stimulated along with IRF3 (Fig. 4D) or IRF7 (Fig. 4E). However, SPL overexpression in the absence of IKKε was not capable of inducing transcriptional activation of type I IFN (Fig. 4C–E). We have further investigated SPL’s function in promoting IKKε activation by using SPL-deficient cells. As shown in Figure 4F, SPL deficiency resulted in the strong inhibition of IKKε auto-phosphorylation. Consistently, we have also observed a decrease in IRF3 phosphorylation (Fig. 4F) and IFN-β production (Fig. 4G) in the absence of endogenous SPL. Taken together, these findings indicate that SPL enhances the stimulation of IKKε, leading to the increased activation of transcription factors IRF3 and IRF7 that ultimately enhances the synthesis of IFN-α/β.
Figure 4. SPL increases IKKε activation to enhance IKKε-mediated IFN induction.

(A) 293T cells (2 X 105) were transfected with increasing doses of IKKε expression plasmid (5, 25, or 125 ng) in the presence of 200 ng of IRF3 plasmid or empty vector control plasmid (-) in the presence of 200 ng of SPL plasmid or the empty vector control plasmid (-), as indicated. After 10 hr, cells were harvested and Western blotting was performed to detect pIKKε, IKKε, pIRF3, IRF3, and GAPDH. (B) 293T cells (1 X 106) were transfected with 25 ng of IKKε with increasing doses of SPL plasmid (0.2, 0.4, 0.6, 0.8, or 1 μg). Ten hr later, Western blot analysis was performed to detect pIKKε, IKKε, SPL, or GAPDH. Both the short exposure (SE) and long exposure (LE) blots of SPL are provided as indicated. (C) 293T cells were transfected with 50 ng of IFN-β reporter plasmid along with 10 ng of IKKε plasmid in the presence of SPL or empty vector (CTR). At 1 day post-transfection, luciferase assay was performed. (D and E) 293T cells were transfected with 100 ng of IFN-α1 reporter plasmid along with 10 ng of IKKε and IRF3 (D) in the presence of 250 ng of SPL or empty vector control (CTR). In (E), IRF7 was used instead of IRF3. At 1 day post-transfection, cells were harvested and luciferase assay was performed. Relative luciferase values are shown. Values represent mean ± SEM (n = 3/group; **, P ≤ 0.01; ****, P ≤ 0.0001; ns = not significant). (F) WT 293T cells or SPL KO-2 cells (2 X 105) were transfected with 50 ng of IKKε expression plasmid or empty vector control plasmid (-). Thirty two hours later, Western blot analysis was performed to detect pIKKε, IKKε, pIRF3, IRF3, and GAPDH. (G) WT 293T cells or SPL KO-2 cells (2 X 105) were transfected with 50 ng of IKKε expression plasmid or empty vector control plasmid (0 hr). Twenty four hours later, relative mRNA levels of IFN-β were determined by performing qPCR. Results are plotted as fold induction of IFN-β in WT and KO-2 samples over the value from each samples transfected with empty vector control plasmid (0 hr). Values represent mean ± SEM (n = 3/group; **, P ≤ 0.01; ***, P ≤ 0.001).
To further explore the mode of action behind SPL-mediated IKKε activation, we tested the possible interaction between SPL and IKKε. Co-immunoprecipitation (co-IP) was performed wherein IKKε was pulled down and the ability of SPL to co-precipitate in the IP fraction was assessed. The data shown in Figure 5A demonstrated that GFP-tagged SPL interacted with IKKε, whereas GFP itself did not bind to IKKε. The interaction between SPL and IKKε was confirmed by performing the reverse co-IP, i.e., pulling down of SPL followed by detection of IKKε in the immunoprecipitated fraction (Fig. 5B). However, SPL did not bind to the closely related kinase TBK1 (Fig. S2B and S2C). Furthermore, we performed confocal microscopy analysis to visualize the localization patterns of SPL and IKKε. SPL has been reported to be localized to cytoplasmic organelles such as ER (17, 31). IKKε displayed a diffuse localization pattern throughout the cytoplasm (Fig. 5C, Flag-IKKε). Interestingly, when SPL and IKKε were co-expressed, a substantial fraction of IKKε was re-distributed into ‘punctate’ structures in the cytosol where SPL was co-localized (Fig 5C, GFP-SPL + Flag-IKKε). However, IKKε did not undergo this type of re-distribution when co-expressed with GFP (Fig. 5C, GFP + Flag-IKKε), which was used as a negative control. These results led us to conclude that SPL interacts with IKKε, which could facilitate IKKε activation by increasing the phosphorylation status of IKKε. Since we have used the GFP-SPL fusion protein in the aforementioned biochemical assays of co-IP and confocal microscopy, we have also tested the function of GFP-SPL to ensure that GFP-SPL remains functional and displays the same phenotype as the untagged SPL. GFP-SPL was proven to enhance the auto-phosphorylation of IKKε similar to the untagged SPL, whereas GFP protein did not affect IKKε phosphorylation (Fig. 6A). In support of this observation, GFP-SPL, but not GFP, increased IFN-β production upon IAV infection (Fig. 6B).
Figure 5. SPL interacts with IKKε.

(A) 293T cells were transfected with Flag-tagged IKKε (Flag-IKKε) in the presence of GFP-tagged SPL (GFP-SPL) or GFP expressing plasmid (GFP) or an empty control Flag vector (-). 24 hr after transfection, co-immunoprecipitation (IP) was carried out using anti-FLAG affinity resin and Western blotting analysis was performed to detect Flag-IKKε, GFP-SPL and GFP in the pull-down fractions (IP: FLAG) and in the whole cell lysates (Input). (B) 293T cells were transfected with Flag-IKKε in the presence of either an empty control vector (-) or HA-SPL. After 24 hr, co-IP was carried out using anti-HA coated affinity resin. Western blotting analysis was conducted to detect Flag-IKKε and HA-SPL in the pull-down fraction (IP: HA) as well as in the whole cell lysates (Input). (C) Flag-IKKε and GFP-SPL were either transfected alone or co-transfected together into 293T cells. Flag-IKKε and GFP expressing plasmid (GFP) were co-transfected into cells (GFP + Flag-IKKε) as a negative control. At 24 hr post-transfection, cells were stained with DRAQ5 to detect nuclei, which are shown in the merged images, and with antibodies against Flag (α-Flag) to detect IKKε and GFP (α-GFP) to detect SPL by confocal microscopy. Cells in the square boxes in the far left panels are shown in detail. White arrows indicate the punctate-like structure. Representative confocal images are shown. Scale bar represents 10 μm.
Figure 6. GFP fusion does not impair the function of SPL in enhancing IKKε activation and IFN production.

(A) 293T cells (1 X 106) were transfected with 25 ng of IKKε in the presence of 0.2 or 0.8 μg of SPL plasmid (SPL) or GFP-tagged SPL plasmid (GFP-SPL) or GFP expressing plasmid (GFP). Ten hr later, Western blot analysis was performed to detect pIKKε, Flag-IKKε, SPL, GFP, and GAPDH. (B) A549 cells (2 X 105) were transfected with CTR (empty vector control) or GFP expressing plasmid (GFP) or GFP tagged SPL (GFP-SPL). At 1 day post-transfection, cells were either mock-infected or infected with 0.5 MOI of IAV. Relative RNA levels of IFN-β were calculated by qPCR at 6 hpi. Values represent mean ± SEM (n = 3/group; **, P ≤ 0.01; ns = not significant).
IKKε is important for the antiviral effect exhibited by SPL upon IAV infection
Next, we attempted to ascertain the functional relevance of the SPL-IKKε axis during IAV infection. For this purpose, we downregulated endogenous IKKε using an si-RNA approach in the presence or absence of SPL upon IAV infection. As expected, SPL displayed an antiviral effect in the scrambled control si-RNA-treated cells (Fig. 7A) and inhibited the synthesis of viral proteins M1 and NP. However, this antiviral effect of SPL was almost abrogated when endogenous IKKε was knocked-down (Fig. 7A). This observation signifies the importance of IKKε in mediating the antiviral effect exhibited by SPL. In order to further confirm if a similar effect is also seen in the production of infectious viruses, we performed a plaque assay from the supernatants of the infected cells. In support of the results from the viral protein expression, downregulation of IKKε nullified SPL’s ability to repress infectious IAV production (Fig. 7B). Furthermore, we have repeated the IKKε-knockdown experiment on A549 cells to confirm our results. SPL’s anti-viral effect was also abolished when endogenous IKKε was knocked-down in A549 cells (Fig. 7C). Additionally, SPL was found to induce the phosphorylation of IKKε at 6 hpi, which must have contributed to SPL’s antiviral function. When IKKε was downregulated by using si-RNA, this phosphorylated fraction of IKKε did not appear (Fig. 7C), which supports the result that SPL acts on IKKε activation to inhibit IAV replication. Taken together, our data further demonstrate that SPL’s anti-viral activity is dependent on IKKε.
Figure 7. IKKε is critical for the anti-viral function of SPL during IAV infection.
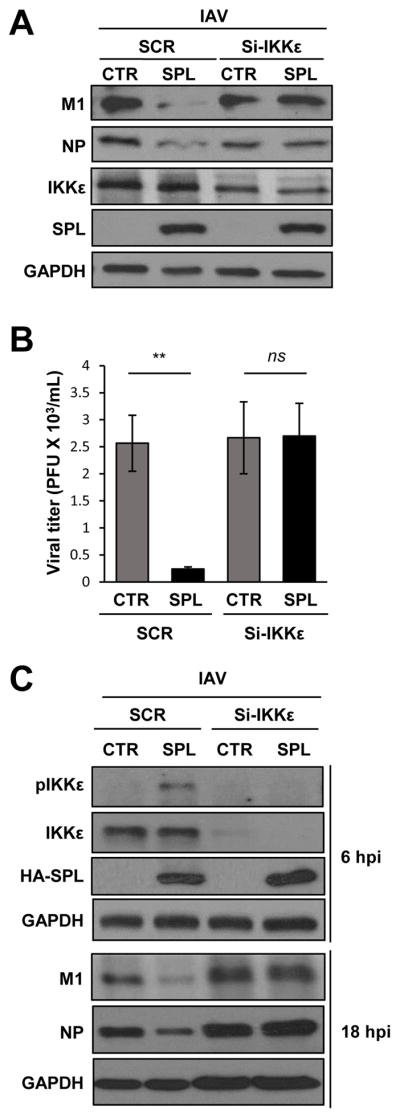
(A) SPL KO-1 cells (2 X 105) were transfected with si-RNA specific to IKKε (si-IKKε) or non-specific scrambled (SCR) si-RNA in the presence of SPL or empty vector control (CTR). Transfected cells were infected with IAV at an MOI of 0.1. At 1 dpi, Western blotting was performed to detect M1, NP, IKKε, HA-SPL, and GAPDH. (B) KO-2 cells (2 X 105) were transfected with si-IKKε or non-specific scrambled (SCR) si-RNA in the presence of SPL or empty vector control (CTR). Transfected cells were infected with IAV at an MOI of 0.1 and then at 24 hpi, the supernatants were collected to quantify viral titers by plaque assay on MDCK cells. Three separate samples of virus-infected cells per group were used for each condition. Values are means ± SEM (n = 3/group; **, P ≤ 0.01). (C) A549 cells (2 X 105) were transfected with si-RNA specific to IKKε (si-IKKε) or non-specific scrambled (SCR) si-RNA in the presence of SPL or empty vector control (CTR). Transfected cells were infected with IAV at an MOI of 0.5. At 6 hpi, Western blotting was performed to detect pIKKε, IKKε, HA-SPL, and GAPDH and at 18 hpi, Western blotting was performed to detect viral M1, NP, and GAPDH.
SPL enhances type I IFN response independent of its enzymatic activity
The enzymatic function of SPL is to degrade intracellular S1P. Hence, we investigated if the S1P-metabolizing activity of SPL is needed for SPL to exert its pro-IFN function. In order to examine that, we used the mutant form of SPL, SPL(K353L), which cannot degrade S1P due to a point mutation in its cofactor binding site (31), in our experiments. Interestingly, SPL(K353L) displayed similar anti-viral activity as the WT SPL to inhibit IAV replication in the SPL-deficient cells (Fig. 8A). Thus, SPL’s function to impair IAV replication seems to be independent of its ability to degrade intracellular S1P. Next, we investigated if the mutant SPL is capable of increasing IKKε stimulation. As shown in Figure 8B, SPL(K353L) can increase IKKε activation in a dose-dependent manner similar to that of the WT SPL (Fig. 4B). In line with these data, co-IP experiments revealed that mutant SPL can also bind to IKKε (Fig. 8C and 8D). These data collectively indicate that SPL acts as a pro-IFN factor independent of its enzymatic activity of degrading cellular S1P.
Figure 8. SPL enhances type I IFN response independent of its enzymatic activity.

(A) WT 293T cells or KO-1 cells (2 X 10 5) were transfected with empty vector control DNA (CTR), WT SPL, or mutant SPL [SPL(K353L)], as indicated. One day later, transfected cells were infected with IAV at 0.1 MOI. Western blotting analysis was performed at 1 dpi to detect NS2, NS1, SPL or GAPDH proteins. (B) 293T cells (1 X 106) were transfected with 25 ng of Flag-tagged IKKε with increasing doses of SPL(K353L) plasmid (0.4, 0.6, 0.8, or 1 μg). 10 hr post-transfection, Western blotting analysis was performed to detect pIKKε, IKKε, SPL or GAPDH proteins. (C) 293T cells were transfected with GFP-tagged SPL(K353L) [GFP-SPL(K353L)] and either an empty control Flag vector (-) or Flag-tagged IKKε (Flag-IKKε). 24 hr after transfection, IP was carried out using anti-FLAG affinity resin and Western blotting analysis was performed to detect Flag-IKKε and GFP-SPL(K353L) in the pull-down fractions (IP: FLAG) and in the whole cell lysates (Input). (D) 293T cells were transfected with Flag-IKKε in the presence of either an empty control vector (-) or GFP-SPL(K535L). After 24 hr, co-IP was carried out using anti-GFP coated affinity resin. Western blotting analysis was conducted to detect Flag-IKKε and GFP-SPL(K353L) in the pull-down fraction (IP: GFP) as well as in the whole cell lysates (Input).
Discussion
In this study, we demonstrate that SPL is critical for the host innate immunity by promoting efficient type I IFN response and establishment of an antiviral state. Mechanistically, SPL augments IFN production by binding and activating the kinase IKKε that phosphorylates IRF3. We delineate the relationship between SPL and IKKε and their interplay to promote IFN-α/β production.
Both IKKε and TBK1 are known to activate the transcription factors IRF3/IRF7 to induce type I IFNs (8). However, the commonality and difference of these two kinases as to their activity in the IFN production pathway are incompletely understood. While TBK1 is constitutively expressed in most cell types, IKKε is more predominantly detected in immune cells and its expression is often upregulated upon stimulation with LPS or inflammatory cytokines (47). Also, studies performed with TBK1-deficient and IKKε-deficient mouse embryonic fibroblasts (MEFs) have established a redundant role for both IKKε and TBK1 in IFN response upon poly (I:C) treatment, while IKKε had a dispensable role in IFN production upon LPS treatment (48). Interestingly, TBK1-deficient MEFs could still mount an IFN response to poly (I:C) treatment, which was abolished in IKKε−/− TBK1−/− double deficient MEFs, indicating that IKKε does play a definitive role in IFN production (48). Thus, the function of IKKε may be more reliant on the type of stimulus or cellular condition. Our findings show that SPL regulates IKKε, but not TBK1. The differential effect that SPL has on TBK1 and IKKε activation is further substantiated by our co-IP data, which provides mechanistic evidence that SPL does bind to IKKε, but not TBK1. Determining the in-depth mechanism for SPL-mediated specific regulation of IKKε may extend our understanding of the differential functions of IKKε and TBK1 in directing the synthesis of type I IFNs.
In the confocal microscopy analysis, we have observed that IKKε re-localizes into punctate cytoplasmic bodies (Fig. 5C). The nature of these cytoplasmic bodies remains unknown at this time. The punctate-like structure of IKKε was also observed when IKKε was co-expressed with another IKKε-binding protein, TRIM6 (49). It was proposed that IKKε re-localizes into TRIM6-ubiquitin-rich bodies for enhanced IKKε activation. Therefore, it is likely that IKKε gathers to form the punctate-like structure when it binds to other cellular proteins for activation of IFN signaling. Whether TRIM6 is involved in SPL-mediated regulation of IKKε activity and/or localization will be interesting to investigate. Also, it remains unknown as to what triggers SPL to interact with IKKε. We have observed that SPL expression level does not change upon stimulation by IFN. However, there is a possibility that upon cellular sensing of viral RNAs, SPL undergoes post-translational modification. Once modified, SPL may then exert its function as a pro-IFN factor by interacting with IKKε. Alternatively, upon cellular recognition of viral RNAs, activated IKKε may recruit SPL into the IFN pathway. These possibilities are currently under investigation.
Influenza virus was reported to strive to counterattack the potent antiviral IFN response (50, 51). For instance, influenza viral NS1 was shown to antagonize the type I IFN system in multiple ways (6, 50). However, it remains unknown if IAV protein such as NS1 regulates SPL’s pro-IFN function or if SPL modulates the viral protein’s anti-IFN effect. Delineating the molecular mechanism behind this dynamic regulation will help us understand IAV-host interaction pathways.
IKKε−/− mice were hyper-susceptible to influenza infection with increased virus titer in the lungs compared to wild type control mice (52). Also, respiratory syncytial virus and vesicular stomatitis virus were shown to induce IKKε expression and/or activation, which contributed to IRF3 activation in response to these infections (53, 54). Furthermore, Ebola virus VP35 protein and nucleoprotein of lymphocytic choriomeningitis virus were reported to interact with IKKε to prevent IKKε from binding to IRF3, thereby antagonizing the cellular IFN response (55, 56). Thus, IKKε is a key player for eliciting the innate immune response in the context of multiple virus infections. By using the IKKε-knockdown approach, we conclusively demonstrated the importance of IKKε in SPL-mediated anti-influenza viral action (Fig. 7). Our study linking SPL to IKKε and the type I IFN response can be extrapolated to the context of other pathogenic virus infections where the significance of IKKε in the virus lifecycle has already been established.
Our data suggests that the mutant SPL can display antiviral potential to the same extent as the wild type SPL during IAV infection. This data indicates that the S1P-degrading activity of SPL is not necessary for it to exert its pro-IFN function. This is consistent with the prior observation that sphingosine analogs or S1P receptor 1 (S1P1R) agonist did not affect the replication of influenza virus in vitro and in vivo (27, 28, 57–59). The protein-protein interaction between SPL and IKKε appears to be important and sufficient for SPL to increase IKKε activation to promote type I IFN response. However, the sphingosine analogs or S1P1R agonist could regulate cytokine responses during influenza virus infection (57–60). Also, it was recently reported that the agonist of S1P1R or exogenous S1P can directly induces degradation of type I IFN receptor (IFNAR) to inhibit IFN amplification on plasmacytoid dendritic cells (61). This could be an important mechanism for the regulation of inflammatory response, given that S1P has been shown to affect cytokine/chemokine responses in diverse conditions (62–64). Conceivably, SPL may cause degradation of S1P to prevent S1P1R-mediated IFNAR1 degradation, thereby leading to preservation of intact IFN pathway. However, the regulation of intracellular SPL may result in altered physiological conditions different from exogenous stimulation of S1P1R. Further, our data indicate that the phenotypic and functional interaction between SPL and IKKε does not require SPL’s enzymatic activity. Perhaps, cellular detection of viral RNAs guides the action of SPL into IKKε-mediated type I IFN pathway in the virus-infected cells. Thus, it is conceivable that SPL has a dual role in regulating host immune responses via S1P-dependent and S1P-independent mechanisms. The intricate mechanisms for sphingolipid system-host immunity interaction need to be further investigated by using diverse systems, including the regulation of S1P-metabolizing enzymes as well as S1PR activation.
It was reported that SPL KO mice do not survive for more than 2–3 weeks after birth (65). More recently, conditional SPL KO mice have been developed that can be utilized to study the cell-type specific function of SPL in vivo (66). Investigating the function of SPL in the mice model will allow us to determine the importance of SPL during IAV infection in vivo. Further, the role of SPL in primary human respiratory epithelial cells or immune cells remains to be defined.
Type I IFNs are cellular factors critical for the host innate immune responses against most viral infections. Thereby, understanding the precise mechanism of the signal transduction events contributing to the establishment of this defense system has great impact on the field of viral immunology and infectious diseases. More importantly, the research on SPL-IKKε-IFN axis could extend our knowledge of virus-host interaction and immuno-regulatory pathways, which also has translational potential for designing new therapeutic interventions for the treatment of viral infections.
Supplementary Material
Acknowledgments
This work was supported by NIH/NIAID Grant R01AI091797 (BH), NIH R01 AI108861 (JH), and NIH GM110776 (MCJ).
We thank Julie Saba (Children’s Hospital Oakland Research Institute) for kind provision of SPL plasmids. We thank David Pintel (University of Missouri-Columbia) for providing the pCMV-HA plasmid. We also thank Greg Lambert and Michael Baldwin (University of Missouri-Columbia) for help with confocal microscopy.
Footnotes
Disclosures
The authors declare that they have no competing interests.
References
- 1.Seo YJ, Hahm B. Type I Interferon Modulates the Battle of Host Immune System Against Viruses. Advances in applied microbiology. 2010;73C:83–101. doi: 10.1016/S0065-2164(10)73004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann HH, Schneider WM, Rice CM. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol. 2015;36:124–138. doi: 10.1016/j.it.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isaacs A, Lindenmann J. Virus interference. I. The interferon. By A. Isaacs and J. Lindenmann, 1957. J Interferon Res. 1987;7:429–438. doi: 10.1089/jir.1987.7.429. [DOI] [PubMed] [Google Scholar]
- 4.Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nature reviews Immunology. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 5.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic Sensing of Viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Killip MJ, Fodor E, Randall RE. Influenza virus activation of the interferon system. Virus research. 2015;209:11–22. doi: 10.1016/j.virusres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. Journal of virology. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 9.Sato Mitsuharu. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Responseto Viruses for IFN-a/b Gene Induction. Immunity. 2000 doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 10.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 11.Schoggins JW. Interferon-stimulated genes: roles in viral pathogenesis. Curr Opin Virol. 2014;6:40–46. doi: 10.1016/j.coviro.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beachboard DC, Horner SM. Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol. 2016;32:113–119. doi: 10.1016/j.mib.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gale M, Jr, Sen GC. Viral evasion of the interferon system. J Interferon Cytokine Res. 2009;29:475–476. doi: 10.1089/jir.2009.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilar A, Saba JD. Truth and consequences of sphingosine-1-phosphate lyase. Advances in biological regulation. 2012;52:17–30. doi: 10.1016/j.advenzreg.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourquin F, Riezman H, Capitani G, Grütter MG. Structure and Function of Sphingosine-1-Phosphate Lyase, a Key Enzyme of Sphingolipid Metabolism. Structure. 2010;18:1054–1065. doi: 10.1016/j.str.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Rolando M, Escoll P, Buchrieser C. Legionella pneumophila restrains autophagy by modulating the host’s sphingolipid metabolism. Autophagy. 2016;12:1053–1054. doi: 10.1080/15548627.2016.1166325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda M, Kihara A, Igarashi Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5’-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun. 2004;325:338–343. doi: 10.1016/j.bbrc.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 18.Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nature Reviews Drug Discovery. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MB. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–662. doi: 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- 20.Newton J, Lima S, Maceyka M, Spiegel S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Experimental Cell Research. 2015;333:195–200. doi: 10.1016/j.yexcr.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fyrst H, Saba JD. Sphingosine-1-phosphate lyase in development and disease: sphingolipid metabolism takes flight. Biochim Biophys Acta. 2008;1781:448–458. doi: 10.1016/j.bbalip.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends in Molecular Medicine. 2007;13:210–217. doi: 10.1016/j.molmed.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Min J, Van Veldhoven PP, Zhang L, Hanigan MH, Alexander H, Alexander S. Sphingosine-1-phosphate lyase regulates sensitivity of human cells to select chemotherapy drugs in a p38-dependent manner. Molecular cancer research : MCR. 2005;3:287–296. doi: 10.1158/1541-7786.MCR-04-0197. [DOI] [PubMed] [Google Scholar]
- 24.Li G, Foote C, Alexander S, Alexander H. Sphingosine-1-phosphate lyase has a central role in the development of Dictyostelium discoideum. Development. 2001;128:3473–3483. doi: 10.1242/dev.128.18.3473. [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Wessels D, Daniels KJ, Alexander H, Alexander S, Soll DR. Sphingosine-1-phosphate plays a role in the suppression of lateral pseudopod formation during Dictyostelium discoideum cell migration and chemotaxis. Cell Motil Cytoskeleton. 2004;59:227–241. doi: 10.1002/cm.20035. [DOI] [PubMed] [Google Scholar]
- 26.Herr DR, Fyrst H, Phan V, Heinecke K, Georges R, Harris GL, Saba JD. Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development. 2003;130:2443–2453. doi: 10.1242/dev.00456. [DOI] [PubMed] [Google Scholar]
- 27.Seo YJ, Blake C, Alexander S, Hahm B. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J Virol. 2010;84:8124–8131. doi: 10.1128/JVI.00510-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seo YJ, Alexander S, Hahm B. Does cytokine signaling link sphingolipid metabolism to host defense and immunity against virus infections? Cytokine Growth Factor Rev. 2011;22:55–61. doi: 10.1016/j.cytogfr.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo YJ, Pritzl CJ, Vijayan M, Bomb K, McClain ME, Alexander S, Hahm B. Sphingosine kinase 1 serves as a pro-viral factor by regulating viral RNA synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection. PloS one. 2013;8:e75005. doi: 10.1371/journal.pone.0075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia C, Vijayan M, Pritzl CJ, Fuchs SY, McDermott AB, Hahm B. Hemagglutinin of Influenza A Virus Antagonizes Type I Interferon (IFN) Responses by Inducing Degradation of Type I IFN Receptor 1. J Virol. 2016;90:2403–2417. doi: 10.1128/JVI.02749-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reiss U, Oskouian B, Zhou J, Gupta V, Sooriyakumaran P, Kelly S, Wang E, Merrill AH, Jr, Saba JD. Sphingosine-phosphate lyase enhances stress-induced ceramide generation and apoptosis. J Biol Chem. 2004;279:1281–1290. doi: 10.1074/jbc.M309646200. [DOI] [PubMed] [Google Scholar]
- 32.Gale M, Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. The E3 Ubiquitin Ligase Triad3A Negatively Regulates the RIG-I/MAVS Signaling Pathway by Targeting TRAF3 for Degradation. PLoS Pathog. 2009;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vijayan M, Seo YJ, Pritzl CJ, Squires SA, Alexander S, Hahm B. Sphingosine kinase 1 regulates measles virus replication. Virology. 2014;450–451:55–63. doi: 10.1016/j.virol.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vijayan M, Hahm B. Influenza viral manipulation of sphingolipid metabolism and signaling to modulate host defense system. Scientifica (Cairo) 2014;2014:793815. doi: 10.1155/2014/793815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell Type-Specific Involvement of RIG-I in Antiviral Response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Pichlmair A, Lassnig C, Eberle CA, Górna MW, Baumann CL, Burkard TR, Bürckstümmer T, Stefanovic A, Krieger S, Bennett KL, Rülicke T, Weber F, Colinge J, Müller M, Superti-Furga G. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nature Immunology. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 38.Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, Fernandez-Sesma A, Palese P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011;7:e1002067. doi: 10.1371/journal.ppat.1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, Garcia-Sastre A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol. 2000;74:11566–11573. doi: 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riegger D, Hai R, Dornfeld D, Manz B, Leyva-Grado V, Sanchez-Aparicio MT, Albrecht RA, Palese P, Haller O, Schwemmle M, Garcia-Sastre A, Kochs G, Schmolke M. The nucleoprotein of newly emerged H7N9 influenza A virus harbors a unique motif conferring resistance to antiviral human MxA. J Virol. 2015;89:2241–2252. doi: 10.1128/JVI.02406-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liedmann S, Hrincius ER, Guy C, Anhlan D, Dierkes R, Carter R, Wu G, Staeheli P, Green DR, Wolff T, McCullers JA, Ludwig S, Ehrhardt C. Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun. 2014;5:5645. doi: 10.1038/ncomms6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graef KM, Vreede FT, Lau YF, McCall AW, Carr SM, Subbarao K, Fodor E. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol. 2010;84:8433–8445. doi: 10.1128/JVI.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Chen H, Sutton T, Obadan A, Perez DR. Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J Virol. 2014;88:10432–10447. doi: 10.1128/JVI.01383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis e Sousa C. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Killip MJ, Fodor E, Randall RE. Influenza virus activation of the interferon system. Virus research. 2015 doi: 10.1016/j.virusres.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimada T. IKK-i, a novel LPS-inducible kinase that is related to IkB kinases. 1999 doi: 10.1093/intimm/11.8.1357. [DOI] [PubMed] [Google Scholar]
- 48.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. The Roles of Two IκB Kinase-related Kinases in Lipopolysaccharide and Double Stranded RNA Signaling and Viral Infection. The Journal of Experimental Medicine. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajsbaum R, Versteeg Gijs A, Schmid S, Maestre Ana M, Belicha-Villanueva A, Martínez-Romero C, Patel Jenish R, Morrison J, Pisanelli G, Miorin L, Laurent-Rolle M, Moulton Hong M, Stein David A, Fernandez-Sesma A, tenOever Benjamin R, García-Sastre A. Unanchored K48-Linked Polyubiquitin Synthesized by the E3-Ubiquitin Ligase TRIM6 Stimulates the Interferon-IKKε Kinase-Mediated Antiviral Response. Immunity. 2014;40:880–895. doi: 10.1016/j.immuni.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Sastre A. Induction and evasion of type I interferon responses by influenza viruses. Virus research. 2011;162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van de Sandt CE, Kreijtz JH, Rimmelzwaan GF. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses. 2012;4:1438–1476. doi: 10.3390/v4091438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 53.Indukuri H, Castro SM, Liao SM, Feeney LA, Dorsch M, Coyle AJ, Garofalo RP, Brasier AR, Casola A. Ikkepsilon regulates viral-induced interferon regulatory factor-3 activation via a redox-sensitive pathway. Virology. 2006;353:155–165. doi: 10.1016/j.virol.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 54.tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC, Lin R, Hiscott J. Activation of TBK1 and IKK Kinases by Vesicular Stomatitis Virus Infection and the Role of Viral Ribonucleoprotein in the Development of Interferon Antiviral Immunity. J Virol. 2004;78:10636–10649. doi: 10.1128/JVI.78.19.10636-10649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prins KC, Cardenas WB, Basler CF. Ebola Virus Protein VP35 Impairs the Function of Interferon Regulatory Factor-Activating Kinases IKK and TBK-1. J Virol. 2009;83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martinez-Sobrido L, de la Torre JC, Kunz S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKepsilon. J Virol. 2012;86:7728–7738. doi: 10.1128/JVI.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsolais D, Hahm B, Walsh KB, Edelmann KH, McGavern D, Hatta Y, Kawaoka Y, Rosen H, Oldstone MB. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci U S A. 2009;106:1560–1565. doi: 10.1073/pnas.0812689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146:980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M, Kawaoka Y, Rosen H, Oldstone MB. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci U S A. 2011;108:12018–12023. doi: 10.1073/pnas.1107024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marsolais D, Hahm B, Edelmann KH, Walsh KB, Guerrero M, Hatta Y, Kawaoka Y, Roberts E, Oldstone MB, Rosen H. Local not systemic modulation of dendritic cell S1P receptors in lung blunts virus-specific immune responses to influenza. Molecular pharmacology. 2008;74:896–903. doi: 10.1124/mol.108.048769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teijaro JR, Studer S, Leaf N, Kiosses WB, Nguyen N, Matsuki K, Negishi H, Taniguchi T, Oldstone MB, Rosen H. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-alpha autoamplification. Proc Natl Acad Sci U S A. 2016;113:1351–1356. doi: 10.1073/pnas.1525356113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Degagne E, Saba JD. S1pping fire: Sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clin Exp Gastroenterol. 2014;7:205–214. doi: 10.2147/CEG.S43453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagahashi M, Hait NC, Maceyka M, Avni D, Takabe K, Milstien S, Spiegel S. Sphingosine-1-phosphate in chronic intestinal inflammation and cancer. Advances in biological regulation. 2014;54:112–120. doi: 10.1016/j.jbior.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalez-Cabrera PJ, Brown S, Studer SM, Rosen H. S1P signaling: new therapies and opportunities. F1000Prime Rep. 2014;6:109. doi: 10.12703/P6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allende ML, Bektas M, Lee BG, Bonifacino E, Kang J, Tuymetova G, Chen W, Saba JD, Proia RL. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J Biol Chem. 2011;286:7348–7358. doi: 10.1074/jbc.M110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zamora-Pineda J, Kumar A, Suh JH, Zhang M, Saba JD. Dendritic cell sphingosine-1-phosphate lyase regulates thymic egress. J Exp Med. 2016;213:2773–2791. doi: 10.1084/jem.20160287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.