

Abstract
There are several in silico programs that endeavor to predict the functional impact of an individual’s sequence variation at splice donor/acceptor sites, but experimental confirmation is problematic without a source of RNA from the individual that carries the variant. With the aid of an exon trapping vector, such as pSPL3, an investigator can test whether a splice site sequence change leads to altered RNA splicing, through expression of reference and variant mini-genes in mammalian cells and analysis of the resultant RNA products.
Keywords: Splicing, Mutation, Exon, Trapping, Expression, Transcript, Variant, Splice
Background
We wished to experimentally test the functional impact of two splice donor site variants, c.760+2T>C and c.3300+2delT, identified in the TEK gene ( Souma et al., 2016 ). As is often the case, samples of cells or mRNA were not available from the individuals carrying these sequence variants, so we utilized the exon trapping method to serve as a functional test. DNA samples were available from patients for PCR amplification of the genomic regions of interest. If patient gDNA samples are unavailable, sequence variants can also be incorporated into wild-type sequence by methods such as PCR-based site-directed mutagenesis.
The exon trapping approach was originally developed to identify unknown exons within long stretches of genomic DNA ( Duyk et al., 1990 ). The pSPL3 exon trapping vector was created to increase the efficiency and reliability of exon identification, and also allowed larger genomic fragments to be screened ( Church et al., 1994 ; Nisson et al., 1994 ). The pSPL3 vector contains a small artificial gene composed of an SV40 promoter, an exon-intron-exon sequence with functional splice donor and acceptor sites, and a late polyadenylation signal. Within the single intron a multiple cloning site is located, into which a genomic fragment of interest is inserted to create a mini-gene expression construct.
In our example, patient and control genomic DNA fragments from the TEK gene were PCR amplified and cloned between pSPL3 vector exons V1 and V2 using XhoI and BamHI restriction sites. COS-7 cells were then transfected with the mini-gene constructs and the resulting RNA content purified. mRNA transcripts were then reverse transcribed into cDNA. Using vector exon-specific primers, cDNAs produced from the mini-gene constructs were specifically PCR amplified and Sanger sequenced. For the first splice site variant, c.760+2T>C within the 5’ splice site of exon 5, a 1,457 bp genomic fragment of the TEK gene encompassing all of intron 4, exon 5, intron 5, exon 6 and intron 6 was inserted into the construct (Figure 1A). RT-PCR and Sanger sequencing of the mini-gene expressed transcripts showed that the mutation destroyed the splice donor site, which resulted in partial intron 5 inclusion before a cryptic splice site was utilized (Figure 1C). This splicing error is predicted to result in a translational frameshift and premature termination signal, which would likely lead to transcript elimination via the nonsense-mediated decay pathway. For the second splice site variant, c.3300+2delT within the 5’ splice site of exon 22, an 831 bp genomic fragment of the TEK gene encompassing all of intron 21, exon 22 and intron 22 was inserted into the construct (Figure 1B). RT-PCR and Sanger sequencing of the mini-gene expressed transcripts revealed that the splice donor mutation led to skipping of exon 22, which is also predicted to result in a translational frameshift and premature termination signal in the genomic context of the patient (Figure 1C).
Figure 1. Exon trapping assay.

Vector exons V1 and V2, are depicted as black boxes and TEK exons 5, 6, and 22 are shown in gray. Vector exon-specific primers are indicated by half-arrows in (A) and (B). Wild-type (WT) and mutant (M) splicing products, with included exon sizes in base pairs, are indicated by dashed lines above and below the construct, respectively. The locations of the splice site mutations are shown as an asterisk (*). A. Wild-type (WT-5) and mutant (M-5) genomic fragments containing TEK exons 5 and 6 were used to model the c.760+2T>C mutation. B. Wild-type (WT-22) and mutant (M-22) genomic fragments containing TEK exon 22 were used to model the c.3300+2delT mutation. C. Gel electrophoresis of RT-PCR products from transfected COS-7 cells. ‘Empty Vector’, cells transfected with vector containing no gDNA insert; ‘TF –ve’ (transfection negative), cells transfected with QIAGEN buffer EB only; ‘PCR –ve’ (PCR negative), PCR contamination control substituting water for cDNA template. Wild-type and mutant transcript content, determined by Sanger sequencing, is depicted to the right of the gel image. The additional 21 bp of intron 5 sequence identified within the M5 transcript is shown incorporating a premature termination codon between exons 5 and 6.
Materials and Reagents
-
PCR
0.2 ml PCR tubes with caps (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12225)
1.5 ml conical base screw cap tube (USA Scientific, catalog number: 1415-8399)
Human genomic DNA samples with and without splice site variant (20 ng/μl)
TE buffer, 10 mM Tris 1 mM EDTA pH 8.0 (Sigma-Aldrich, catalog number: 93283)
-
Custom-synthesized oligonucleotide primers incorporating restriction endonuclease sites to the 5’ end (Integrated DNA Technologies; https://www.idtdna.com/site/order/oligoentry):
TEK_E5-F: 5’-ctgactgaCTCGAGCACAGCTCCAGCCTGTAACCAT-3’
TEK_E5-R: 5’-tcagtcagGGATCCTCGGAACTACTTGGGAGCCTGT-3’
TEK_E22-F: 5’-ctgactgaCTCGAGATTCCAAGGCAAATGCTGCTCT-3’
TEK_E22-R: 5’-tcagtcagGGATCCTTGACTCCCAGATCGGTACAGC-3’
Note: Genome-specific sequences are underlined, restriction sites are shown in BOLD and an extra 8 bp added to the 5’ end are shown in lowercase. 5’-CTCGAG-3’ is the recognition sequence for XhoI and 5’-GGATCC-3’ is the recognition sequence for BamHI. ‘-F’ and ‘-R’ refers to the forwards and reverse primers in a pair, respectively. Resuspend primers and make 10 μM stocks with TE buffer.
Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F549S)
Molecular grade water, nuclease-free (Dot Scientific, catalog number: DS248700)
dNTP Set (QIAGEN, catalog number: 201913)
2 mM dNTPs (see Recipes)
-
Gel electrophoresis
1 M Trizma hydrochloride solution, Tris-HCl, pH 7.8 (Sigma-Aldrich, catalog number: T2569)
3 M sodium acetate buffer solution, pH 5.2 (Sigma-Aldrich, catalog number: S7899)
0.5 M EDTA, pH 8.0 (Santa Cruz Biotechnology, catalog number: sc-203932)
Agarose (IBI Scientific, catalog number: IB70042)
SYBR safe DNA gel stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S33102)
Ficoll-400 (Dot Scientific, catalog number: DSF10400-25)
Bromophenol blue, sodium salt (MP Biomedicals, catalog number: 02152506)
Orange G (Sigma-Aldrich, catalog number: O3756)
HyperLadder 1 kb (Bioline, catalog number: BIO-33053)
1x TAE buffer (see Recipes)
1% or 1.5% agarose gel (see Recipes)
10x gel loading dye (see Recipes)
-
Cloning
1.5 ml Eppendorf tubes (VWR, catalog number: 20170-022)
Petri dishes, 100 x 15 mm (VWR, catalog number: 25384-302)
Whatman GD/X syringe filters, 0.2 μm pore size (Whatman, catalog number: 6901-2502)
BD 60 ml syringes, Luer-Lok Tip (BD, catalog number: 309653)
Pasteur glass pipettes, 230 mm (for spreading bacteria on plates) (WHEATON, catalog number: 357335)
2 ml cryovial, self-standing (Simport, catalog number: T310-2A)
50 ml polypropylene conical tube, Falcon (Corning, Falcon®, catalog number: 352070)
0.5 ml PCR tubes (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12275)
JM109 E. coli competent cells (Promega, catalog number: L2001)
pSPL3 vector (Thermo Fisher Scientific, Invitrogen)
Restriction endonuclease, XhoI (New England Biolabs, catalog number: R0146S)
Restriction endonuclease, BamHI (New England Biolabs, catalog number: R0136S)
QIAquick PCR Purification Kit (QIAGEN, catalog number: 28104)
T4 DNA ligase (New England Biolabs, catalog number: M0202S)
SOC medium (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15544034)
Luria Bertani broth (Lennox) powder microbial growth medium (Sigma-Aldrich, catalog number: L3022)
Select Agar (Thermo Fisher Scientific, InvitrogenTM, catalog number: 30391023)
Carbenicillin, disodium salt (Dot Scientific, catalog number: DSC46000-5)
Glycerol (Sigma-Aldrich, catalog number: G5516)
QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104)
Carbenicillin antibiotic, 1,000x (see Recipes)
LB (Luria-Bertani) medium with carbenicillin antibiotic (see Recipes)
LB agar with carbenicillin antibiotic plates (see Recipes)
-
Mammalian cell culture
-
Serological pipettes
5 ml (Corning, Costar®, catalog number: 4487)
10 ml (Corning, Costar®, catalog number: 4488)
25 ml (Corning, Costar®, catalog number: 4489)
Transfer pipette, polyethylene, general purpose blood bank, bulb draw 1.9 ml, sterile (Sigma-Aldrich, catalog number: Z350699)
Pasteur glass pipettes, 5.75” (for aspiration) (VWR, catalog number: 14673-010)
25 cm2 (T25) cell culture flasks with 0.2 μm vent caps (Corning, catalog number: 430639)
COS-7 mammalian cells (ATCC, catalog number: CRL-1651)
Dulbecco’s modified Eagle’s medium, DMEM, + GlutaMAX-1 cell culture medium (Thermo Fisher Scientific, GibcoTM, catalog number: 10569010)
Fetal bovine serum, FBS (Thermo Fisher Scientific, GibcoTM, catalog number: 10437028)
Phosphate-buffered saline, PBS, pH 7.4, 1x (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023)
Penicillin-streptomycin, 10,000 U/ml (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122)
Trypsin-EDTA (0.5%), no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 15400054)
FuGENE 6 Transfection Reagent (Promega, catalog number: E2691)
Opti-MEM I reduced serum medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31985062)
-
-
RNA extraction
RNeasy Mini Kit (QIAGEN, catalog number: 74104)
QIAshredder (disposable cell-lysate homogenizers) (QIAGEN, catalog number: 79654)
-
cDNA generation
High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4374966)
-
RT-PCR
0.2 ml PCR tubes with caps (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12225)
HotStarTaq DNA polymerase (QIAGEN, catalog number: 203203)
TE buffer, 10 mM Tris 1 mM EDTA pH 8.0 (Sigma-Aldrich, catalog number: 93283)
-
Vector-specific oligonucleotide primers (Integrated DNA Technologies; https://www.idtdna.com/site/order/oligoentry):
V1-F: 5’-TCTGAGTCACCTGGACAACC-3’
V2-R: 5’-ATCTCAGTGGTATTTGTGAGC-3’
Note: Resuspend primers and make 10 μM stocks with TE buffer.
2 mM dNTPs (see Recipes)
-
Sanger sequencing
Sanger sequencing (GeneWiz commercial sequencing service; https://www.genewiz.com/en/Public/Services/Sanger-Sequencing)
Sequencing primers (V1-F and V2-R oligonucleotide primers, same as for RT-PCR)
Equipment
Gel doc system (Labnet International, model: EnduroTM GDS)
Water bath (37 °C) (Fisher Scientific, model: Fisher ScientificTM IsotempTM 215)
Bacterial shaking incubator (37 °C) (GeneMate, model: Incubated Shaker Mini)
Bacterial incubator (37 °C) (Lab-Line Imperial III Incubator)
Mammalian cell incubator (37 °C, 5% CO2) (Eppendorf, model: Galaxy® 170 S)
Bunsen Burner, simple natural gas burner version (Fisher Scientific, catalog number: S95941)
NanoDrop Lite spectrophotometer (Thermo Fisher Scientific)
Eppendorf centrifuge (benchtop) (Eppendorf, model: 5415 D)
PCR thermocycler (Eppendorf, model: Mastercycler® Pro S)
Pyrex bottle/flask, 500 ml
HotPlate Stirrer (GeneMate, model: HotPlate Magnetic Stirrer)
VWR Spinbar Magnetic Stir Bar, Polygon with Pivot Ring, 6 mm (") diameter, 35 mm (1") length (VWR, catalog number: 74950-290)
Fisher Vortex Genie 2 (Thermo Fisher Scientific, catalog number: 12-812)
Household microwave oven, 1,000 W
Gel electrophoresis tank (and tray) (Bio-Rad Laboratories, model: Wide Mini-Sub GT Cell)
Electrophoresis power pack (Bio-Rad Laboratories, model: PowerPacTM Basic Power Supply)
Autoclave (Medium Healthcare Sterilizer) (Tuttnauer, model: 6690)
Biological safety cabinet (The Baker Company, model: SterilGARD e3 High Efficiency SG403A-HE)
Handheld aspirator and collector trap (Argos Technologies, model: EV514)
Software
Sequencher software (version 5.2.4, Gene Codes Corporation; http://www.genecodes.com/sequencher)
Primer3Plus (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi)
Procedure
-
Amplification of experimental genomic fragment
Custom oligonucleotide primers are designed to amplify the genomic regions of interest and incorporate restriction endonuclease sites at their 5’ ends, ready for subsequent cloning. An additional 8 bp is added to the 5’ end of each primer to increase the efficiency of post-PCR endonuclease digestion. Primer design is performed using Primer3Plus (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi). For the first construct, modeling the variant within the 5’ splice donor site of exon 5 (c.760+2T>C), a 1,457 bp genomic fragment of the TEK gene encompassing all of intron 4, exon 5, intron 5, exon 6 and intron 6 was amplified. This gDNA sequence lacked recognition sites for both XhoI and BamHI endonucleases, so these two sites were incorporated into the primers for downstream cloning. For the second construct, modeling the variant within the 5’ splice donor site of exon 22 (c.3300+2delT), an 831 bp genomic fragment of the TEK gene encompassing all of intron 21, exon 22 and intron 22 was amplified, and XhoI and BamHI recognition sites were also incorporated for subsequent cloning. Phusion Hot Start II High-Fidelity DNA polymerase is used for amplification, due to its low error rates, fast extension speed and ability to handle larger amplicon sizes. Genomic DNA from both patient and control subjects should be used as template to amplify both mutant and wild-type genomic regions, respectively.
Note: If the patient harbors a heterozygous mutation, both mutant and wild-type alleles can be sub-cloned from this single gDNA sample.
-
In 0.2 ml PCR tubes, set up 20 μl PCR reactions as follows:
gDNA template (20 ng/μl) 3 μl 5x Phusion HF buffer 4 μl 2 mM dNTPs 2 μl 10 μM forwards primer 1 μl 10 μM reverse primer 1 μl Phusion polymerase (2 U/μl) 0.2 μl Nuclease-free water 8.8 μl Include a negative control sample (add 3 μl water instead of gDNA) to test for DNA contamination.
Use the ‘Tm Calculator’ (www.thermofisher.com) to calculate the appropriate annealing temperature for PCR cycling. If the Tm values are higher than 69 °C, as was the case for the two TEK gene examples, a 2-step cycling protocol using a 72 °C combined annealing-extension step is recommended.
-
PCR cycling should be performed as follows:
Initial denaturation 98 °C 30 sec 35 cycles of 98 °C 10 sec (denaturation) 50-72 °C 30 sec (annealing temperature of primers) 72 °C 30 sec per kb amplicon (extension) Final extension 72 °C 10 min Hold temperature 4 °C -
Agarose gel electrophoresis should be performed as follows:
Prepare a 1% agarose gel in TAE buffer containing SYBR safe DNA gel stain (see Recipes).
Add 1 μl of 10x gel loading dye to 4 μl of water and 5 μl of the PCR reaction. Load all 10 μl into a well of the agarose gel. Add 5 μl of HyperLadder 1 kb marker to an adjacent lane of the gel for sizing.
Perform agarose gel electrophoresis for approximately 30 min at 120 V.
Inspect the gel using a UV lamp gel documentation system. A single distinct band should be present at the expected size (1,485 bp for the TEK exon 5 amplimer and 859 bp for the TEK exon 22 amplimer) before proceeding further (Figure 2).
-
-
Cloning of genomic fragments into pSPL3 exon trapping vector
The PCR products are now purified and inserted into the multiple cloning site (MCS) of the exon trapping vector, pSPL3. The MCS is located within an intronic region between vector exons V1 and V2. First, the PCR products are purified to remove buffers, unincorporated dNTPs and primers. Then, the vector and PCR products are digested with the relevant restriction endonucleases, in these examples XhoI and BamHI, and again purified. The ‘sticky-ended’ vector and genomic ‘insert’ fragments are then ligated together. The ligation products are transformed into chemically competent JM109 E. coli cells and plated onto LB plates containing carbenicillin antibiotic for selection of plasmid-carrying bacteria. After incubating the plates overnight, several bacterial colonies are picked, grown up in liquid culture, and their plasmid content harvested. The purified plasmid DNA is then Sanger sequenced to verify which of the plasmid clones contains an insert with the correct content and orientation.
Purify the PCR products using a QIAquick PCR Purification Kit column following QIAGEN’s instructions. This is the ‘insert’ DNA and should be at a concentration of 50-150 ng/μl. To achieve this concentration, it may be necessary to pool 3 x 20 μl PCR reactions into each purification column and to elute in a final volume of 30 μl elution buffer (EB).
The pSPL3 plasmid DNA should also be pure in 1x TE or in EB from a QIAprep Spin Miniprep Kit purification. This is the ‘vector’ DNA and should be at a concentration of 200-400 ng/μl. If pSPL3 plasmid needs to be harvested from bacteria using a Miniprep Kit, please refer to step 7 of this section (‘Grow up and harvest plasmid from bacterial colonies’).
-
Set up restriction double digestions in 0.5 ml PCR tubes as follows:
Vector sample Insert sample DNA 4 μg 2 μg BamHI enzyme (20 U/μl) 4 μl 2 μl XhoI enzyme (20 U/μl) 4 μl 2 μl 10x NEBuffer 3.1 20 μl 10 μl Nuclease-free water Make up to 200 μl Make up to 100 μl Incubate reactions at 37 °C in a water bath for 2 h.
Purify the digested DNA products using another QIAquick PCR Purification Kit column following QIAGEN’s instructions. This removes the buffer salts and unwanted small digestion fragments. Elute the insert sample in 30 μl EB and the vector sample in 50 μl EB. DNA concentrations will be approximately 50-70 ng/μl for both insert and vector. At this point, 5 μl of each sample can be run out on a 1% agarose gel to check fragments are of the expected sizes (1,463 bp for the TEK exon 5 insert fragment, 837 bp for TEK exon 22 insert fragment, and 6,011 bp for the digested pSPL3 vector fragment; Figure 3).
-
Next, the insert and vector fragments will be ligated together in a 3:1 molar ratio. The NEBioCalculator tool (https://nebiocalculator.neb.com/) is helpful for calculating the amount of insert required to achieve these molar ratios. The linearized pSPL3 vector fragment is 6.011 kb, and the TEK exon 5 and exon 22 inserts are 1.463 kb and 0.837 kb, respectively. For 200 ng of vector DNA, 146 ng and 84 ng of the insert DNA will be required for each ligation, respectively. Set up ligation reactions in 0.2 ml PCR tubes as follows:
Insert + vector Vector only Vector DNA 200 ng 200 ng Insert DNA (E5/E22) 146 ng/84 ng Not added 10x T4 DNA ligase buffer 4 μl 4 μl T4 DNA ligase (400 U/μl) 2 μl 2 μl Nuclease-free water Make up to 40 μl Make up to 40 μl Notes:
Add the ligase enzyme last. ‘E5/E22’ refers to the TEK exon 5 and exon 22 inserts, respectively. The ‘vector only’ sample is a control to determine the degree to which vector DNA may be re-ligating to itself in the experimental ‘insert + vector’ sample.
Set a PCR thermocycler to incubate the ligation reactions at 16 °C overnight (~16 h), followed by a heat-inactivation step of 65 °C for 10 min, then holding at 4 °C.
-
Next, transform the ligation products into competent JM109 E. coli using a modified version of the Multiple-Use Protocol supplied by Promega (requires use of less cells):
Heat a water bath to exactly 42 °C.
For each sample, label and chill a 1.5 ml Eppendorf tube on ice.
Thaw an aliquot of frozen competent cells on ice.
When cells are just thawed (~10 min), gently flick the tube to mix the cells.
Add 50 μl of cells to chilled Eppendorf.
Add 25 ng (~5 μl) of DNA from ligation reaction to cells and gently flick to mix.
Leave tube containing cell-DNA mix on ice for 10 min.
Heat-shock cells for 50 sec in the 42 °C water bath and immediately return the tube to ice for 2 min.
Add 450 μl of SOC medium (room temperature) and flick to mix.
Secure tubes horizontally in an incubator for 1 h at 37 °C with shaking at 225 rpm.
Make 1:10 and 1:100 dilutions of the respective transformed cells with SOC medium, as follows: To a clean 1.5 ml Eppendorf tube, add 450 μl of SOC medium and 50 μl of the transformed cell suspension and mix by gently flicking the tube–this is the 1:10 dilution. To another clean 1.5 ml Eppendorf tube, add 450 μl of SOC medium and 50 μl of the 1:10 cell dilution–this is the 1:100 dilution. Mix all three cell suspensions by gently flicking the tube immediately prior to plating the cells.
-
On 3 LB plates containing 100 μg/ml carbenicillin, plate 100 μl of undiluted, 1:10 and 1:100 cell dilutions, respectively. Use a sterile cell spreader, or glass beads, to evenly distribute the cell suspension across each LB plate until all liquid is absorbed into the agar.
Note: An effective cell spreader can be created by melting the end of a Pasteur glass pipette with a Bunsen burner flame so that it forms an ‘L’ shape. Sterilize the glass spreader between cell platings by immersing it into 100% ethanol and burning off the alcohol. Let the glass cool before spreading cells onto another plate.
Incubate plates at 37 °C overnight.
-
Examine plates for bacterial colonies. There should be more colonies on the ‘insert + vector’ plates than the ‘vector only’ plates (Figure 4).
Note: No bacterial colonies were observed on the ‘vector only’ plates.
The number of colonies on the ‘vector only’ plate will indicate the proportion of colonies on the ‘insert + vector’ plate that has resulted from vector re-ligation. This should be taken into consideration when deciding upon how many clones to pick in the following steps. If there are twice as many colonies on the ‘insert + vector’ plate compared to the ‘vector only’ plate, then half of the colonies on the ‘insert + vector’ plate are expected to have no insert present. Also, if the genomic DNA sample used to PCR amplify the insert sequence harbored a heterozygous mutation, half of the clones with an insert will contain wild-type sequence and half will contain the mutant sequence. If there are relatively few colonies on the ‘vector only’ plate, picking 5 colonies from the ‘insert + vector’ plate should suffice to obtain both wild-type and mutant insert-harboring plasmids.
-
Grow up and harvest plasmid from several (5 to 10) bacterial colonies as follows:
Label 50 ml Falcon tubes.
To each tube add 10 ml of LB media containing carbenicillin (100 μg/ml final concentration).
Using a sterile pipette tip, transfer a single bacterial colony from a plate to a liquid media tube. Note: Colonies will be spaced further apart on the serial dilution plates.
Vortex the tubes briefly to transfer the bacteria from the submerged pipette tips into the liquid media.
Place the tubes into a rack and incubate at 37 °C overnight with shaking at 225 rpm.
-
Before harvesting the plasmid content, make glycerol stocks of the bacterial cultures for long-term storage:
-
To a 2 ml cryovial, add:
500 μl 50% glycerol (diluted with dH2O).
500 μl overnight bacterial culture.
Gently mix contents until homogeneous.
-
Freeze at -80 °C (viable for years).
Note: When recovering at a future date, scrape some of the frozen bacteria from the top of the stock and transfer to an LB plate containing antibiotic (thawing the stock is unnecessary and will reduce the lifetime of the sample).
-
Harvest the plasmid content from the remainder of the overnight culture using a QIAprep Spin Miniprep Kit, following QIAGEN’s instructions.
NanoDrop the plasmid DNA sample to quantify yield.
Sanger sequence the purified plasmid DNA to verify which of the plasmid clones contains an insert with the correct content and orientation. Once validated, these are the final mini-gene constructs–a wild-type and mutant mini-gene for each splice variant being tested.
-
Transfection of mini-gene constructs into COS-7 cells
Next, COS-7 mammalian cells (adherent, fibroblast-like cells from the kidney of the African Green Monkey) are grown in culture and the wild-type and mutant mini-gene constructs are transfected into the cells using FuGene 6 transfection reagent following Promega’s instructions:
-
COS-7 cells are seeded into 25 cm2 (T25) flasks at 25-40% confluence (6.25 x 105-1 x 106 cells) the day before transfection, so that they are 50-80% confluent the next day.
Note: COS-7 cells have a doubling time of ~18 h.
-
Prepare the transfection complex.
Note: Include control transfections of only the pSPL3 vector with no gDNA insert, and another with no plasmid (QIAGEN buffer EB only).
Allow FuGENE 6 and OptiMEM medium to equilibrate to room temperature.
-
To a 1.5 ml Eppendorf tube, add:
475 μl OptiMEM medium
15 μl of FuGENE 6 (without touching the sides of tube with the tip, pipette into the center of the medium)
Gently pipette up and down 10 times to mix
Let stand for 5 min
5 μg (10 μl) of the respective wild-type or mutant pure plasmid DNA (at 500 ng/μl in QIAGEN buffer EB) or EB buffer alone in the control transfection (without touching the sides of tube with the tip, pipette into the center of the medium)
Gently pipette up and down 10 times to mix
Let stand for 15 min
Dropwise, using a sterile plastic transfer pipette, add all 500 μl of the transfection mixture to the center of a T25 flask of cells, swirling gently with each drop added.
Incubate the cells at 37 °C with 5% CO2 for 24 h.
-
-
Total RNA isolation
Next, total RNA is isolated from the COS-7 cells using an RNeasy Mini Kit following QIAGEN’s instructions:
Note: A T25 cell culture flask containing COS-7 cells at 100% confluence (~2.5 x 106 cells) should yield ~80 μg RNA. Perform all steps including centrifugation at room temperature (21 °C). Add 10 μl 2-mercaptoethanol (2-ME) to each 1 ml of buffer RLT.
Aspirate T25 flask culture medium and wash the cells in 5 ml PBS; aspirate all PBS.
Add 3 ml trypsin-EDTA, swirl to coat the cells and quickly aspirate the solution.
Incubate cells at 37 °C until they start to detach from the flask (~5 min).
Add 5 ml medium containing 10% FBS to inactivate trypsin.
Pipette the medium up and down within the flask to resuspend the cells and transfer the mixture to a 50 ml Falcon tube.
Pellet the cells by centrifugation at 300 × g for 5 min; aspirate the supernatant.
Add 350 μl buffer RLT (containing 2-ME), pipet to mix and add lysate directly into a QIAshredder spin column placed in a 2 ml collection tube.
Centrifuge for 2 min at max speed in a benchtop centrifuge.
Add 350 μl of 70% ethanol to the homogenized lysate; mix well by pipetting. Do not centrifuge at this point.
Transfer up to 700 μl of sample (including any precipitate) to an RNeasy spin column placed in a 2 ml collection tube.
Centrifuge for 15 sec at 9,000 × g; discard the flow-through.
Add 700 μl buffer RW1 to the column.
Centrifuge for 15 sec at 9,000 × g to wash the column membrane; discard the flow-through.
Add 500 μl buffer RPE (containing ethanol) to the column.
Centrifuge for 15 sec at 9,000 × g to wash the membrane; discard the flow-through.
Add another 500 μl buffer RPE to the column.
Centrifuge for 2 min at 9,000 × g to wash the membrane.
Transfer the column to a new 2 ml collection tube and centrifuge at maximum speed for 1 min to remove any residual ethanol.
Place the column in a new 1.5 ml collection tube.
Add 50 μl RNase-free water directly to the membrane.
Centrifuge for 1 min at 9,000 × g to elute the RNA sample.
Add a further 30 μl RNase-free water directly to membrane.
Centrifuge for 1 min at 9,000 × g to elute the remaining RNA.
NanoDrop the RNA sample to quantify yield (expect ~70 μl total RNA, 400-700 ng/μl).
Store the remaining RNA at -80 °C.
-
cDNA generation
Next, a High Capacity cDNA Reverse Transcription Kit is used with random primers to reverse-transcribe RNA into cDNA. To maximize the reaction, RNA samples should first be diluted to 200 ng/μl with RNase-free water. The reverse transcription reaction is then performed to convert 2 μg of total RNA to cDNA in a 20 μl volume, following Applied Biosystems’ instructions:
-
On ice, for each sample, make 10 μl of a 2x RT Master mix:
10x RT buffer 2 μl 25x dNTPs (100 mM) 0.8 μl 10x RT random primers 2 μl RNase inhibitor at 1 U/μl 1 μl Nuclease-free H2O 3.2 μl MultiScribe reverse transcriptase 1 μl On ice, mix the components gently.
Pipette 10 μl of the 2x RT Master mix into ice-cold 0.2 ml PCR tubes.
Add 10 μl RNA (200 ng/μl) and gently flick to mix.
-
Keep the reaction on ice until loaded into a PCR thermocycler programmed as follows:
25 °C 10 min 37 °C 120 min 85 °C 5 min 4 °C hold Store the resulting cDNA products at -20 °C.
-
-
RT-PCR analysis of pSPL3-derived transcripts
Next, the splicing vector-transcribed cDNA species are specifically amplified by PCR using primer pairs corresponding to sequences in the vector exons, V1 and V2 (Figures 1A and 1B). The resulting RT-PCR products from the wild-type and mutant mini-genes are visualized by gel electrophoresis and their sequence composition determined by Sanger sequencing (Figure 1C).
Note: There is no need to remove the RNA component of the cDNA sample before PCR amplification. Use 4 μl of the cDNA sample as template in a 40 μl PCR reaction. More than 4 μl (10% of the PCR reaction volume) cannot be used, as carry-over of the buffer salts can inhibit the reaction.
-
In 0.2 ml PCR tubes, set up 40 μl PCR reactions as follows:
cDNA template 4 μl 10x HotStarTaq buffer 4 μl 2 mM dNTPs 4 μl 10 μM V1-F primer 2 μl 10 μM V2-R primer 2 μl HotStarTaq polymerase (5 U/μl) 0.4 μl Nuclease-free water 23.6 μl Note: Include a negative control sample (add 4 μl water instead of cDNA) to test for reaction contamination.
-
Perform PCR amplification using the following cycling conditions:
Initial denaturation 95 °C 5 min 35 cycles of 94 °C 30 sec (denaturation) 55 °C 30 sec (annealing temperature of primers) 72 °C 3 min (30 sec per kilobase extension) Final extension 72 °C 10 min Hold temperature 4 °C -
Analyze the PCR amplification products by electrophoresis on a 1% agarose gel (Figure 1C).
Note: The pSPL3 vector with no gDNA insert will generate a single RT-PCR product of 257 bp from vector exons V1 and V2 (Figure 1C). Inclusion of TEK exons 5 and 6 will generate a single RT-PCR product of 530 bp (Figure 1C). Inclusion of TEK exon 22 will generate a single RT-PCR product of 357 bp (Figure 1C). From the mutant exon 5 and exon 22 splice site constructs, single bands of 551 bp and 357 bp are observed, respectively (Figure 1C).
-
Where a single distinct band is present, the sample should be sent for direct Sanger sequencing using primers V1-F and V2-R (Figure 1C). If multiple bands are present, each band should be separately gel extracted (QIAquick Gel Extraction Kit) prior to Sanger sequencing.
Note: From the mutant TEK exon 5 construct, Sanger sequencing determined the additional transcript content to result from inclusion of 21 bp of intron 5 (Figure 1C). From the mutant TEK exon 22 construct, Sanger sequencing showed a lack of any TEK content in the transcript, with content from vector exons V1 and V2 only, indicating exon 22 is skipped entirely during RNA splicing (Figure 1C).
-
Figure 2. PCR amplification of genomic fragments.
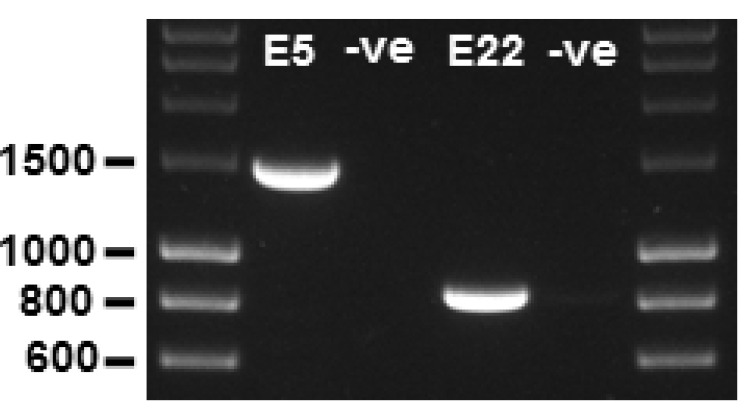
The TEK exon 5 (E5) and exon 22 (E22) amplimers are observed at 1,485 bp and 859 bp, respectively. –ve, PCR negative controls (water substituted for gDNA template) for each amplimer reaction; 1% agarose gel; molecular weight markers are 5 μl of ‘HyperLadder 1 kb’ per lane.
Figure 3. Fragments of PCR products and pSPL3 vector following restriction digestion with BamHI and XhoI endonucleases.
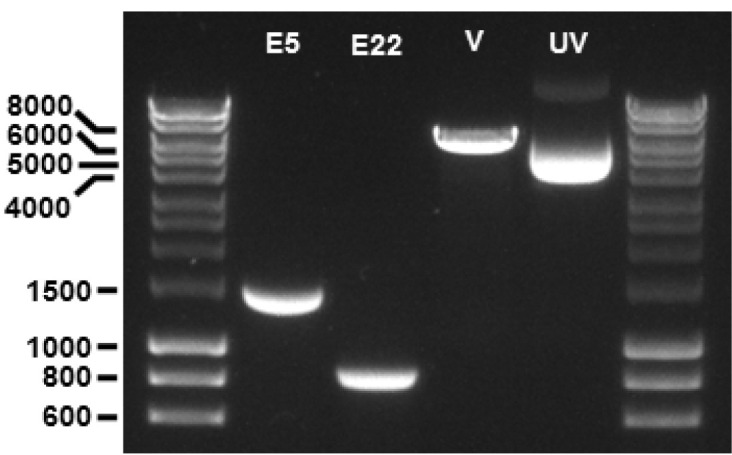
The TEK exon 5 (E5) and exon 22 (E22) fragments are observed at 1,463 bp and 837 bp, respectively. The pSPL3 vector (V) fragment is observed at 6,011 bp. A sample of undigested supercoiled pSPL3 vector (UV) is also included on the gel, migrating at ~4,000 bp. 1% agarose gel; molecular weight markers are 5 μl of ‘HyperLadder 1 kb’ per lane.
Figure 4. Bacterial colony growth on LB plates containing carbenicillin to select for circular pSPL3 plasmid-carrying bacteria.
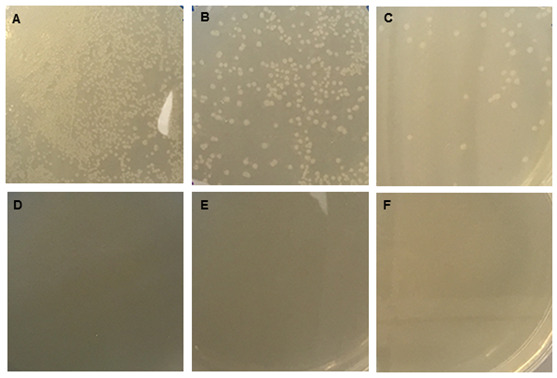
JM109 bacterial cells were transformed with either ‘insert + vector’ (panels A-C) or ‘vector only’ (panels D-F) ligation products and 100 μl was plated undiluted (panels A and D), at a 1:10 dilution (panels B and E), or at a 1:100 dilution (panels C and F).
Data analysis
Sequencher software (Gene Codes Corporation) can be used to analyze Sanger sequencing chromatogram files to determine the DNA sequence composition of the mini-gene transcripts.
Notes
The Invitrogen website supplies an unconfirmed sequence of the 6,031 bp pSPL3 vector, assembled from the known sequence of fragments used to construct it: http://tools.thermofisher.com/content/sfs/vectors/pspl3_seq.htm.
We re-sequenced our in-house pSPL3 vector (sequence given in Supplemental file 1) and found 3 bp lacking when compared to the sequence provided by Invitrogen. The 3 bases missing from our sequence were 2 bp in vector exon 1 and 1 bp in vector exon 2 (shown within square brackets). These sequence discrepancies are not proximal to exon-intron junctions and, as demonstrated by our experimental data, do not impact splicing mechanisms.
In our 6,028 bp pSPL3 sequence (Supplemental file 1), the two vector exons are shown in bold and italics, with the V1-F and V2-R primers underlined. The multiple cloning site (MCS) recognition sequences for XhoI (CTCGAG) and BamHI (GGATCC) are also shown in bold and underlined.
Recipes
-
2 mM dNTPs
Thaw the 4 individual 100 mM dNTPs from the dNTP Set at room temperature
Add 460 μl dH2O to a 1.5 ml screw-cap tube
Add 10 μl of each dNTP
Vortex to mix and store at -20 °C
-
1x TAE (Tris, acetate, EDTA) buffer
40 mM Tris-HCl (pH 7.8)
5 mM sodium acetate
1 mM EDTA
-
1% or 1.5% agarose gel
Add 1% or 1.5% (w/v) agarose to 1x TAE buffer in a 500 ml Pyrex bottle/flask
Heat in a microwave oven, frequently swirling the bottle carefully until all agarose has been dissolved into solution
Cool the agarose solution to ‘hand hot’ by flowing tap water over the side of the bottle whilst gently swirling
Add 10 μl of SYBR safe DNA gel stain per 100 ml of gel solution and swirl to mix
Immediately pour solution into an electrophoresis tray and allow to set at room temperature
-
10x gel loading dye
-
To a 50 ml Pyrex bottle, add
2.5 g Ficoll-400 (25% final conc.)
1 ml of 1 M Tris-HCl (pH 7.8, 100 mM final conc.)
2 ml 0.5 M EDTA, pH 8.0 (0.1 M final conc.)
Make up to 10 ml with nuclease-free water
Heat solution to 65 °C with a magnetic stir bar to dissolve Ficoll
Add ~15 mg each of bromophenol blue and Orange G dyes (add up to 25 mg if a stronger dye intensity preferred)
Mix well to dissolve dyes
Store at room temperature
-
-
Carbenicillin antibiotic, 1,000x
Add 10 ml dH2O to a 50 ml Falcon tube
Add 1 g carbenicillin disodium salt
Vortex until salt has fully dissolved
Sterilize the solution by passing it through a 0.2 μm-gage filter using a syringe
Keep the 100 mg/ml stock solution frozen in 1 ml aliquots at -20 °C
-
LB (Luria-Bertani) medium with carbenicillin antibiotic
In a 1 L autoclavable bottle, add 20 g LB broth (Lennox) powder microbial growth medium to 1 L of dH2O
Autoclave for 15 min at 121 °C to sterilize
Allow to cool to room temperature before adding 1 μl per ml carbenicillin antibiotic
-
LB agar with carbenicillin antibiotic plates
Follow the recipe for LB medium, but add 15 g Select Agar before autoclaving
Allow contents to cool to ‘hand-hot’ before adding 1 μl per ml carbenicillin antibiotic
Pour ~25 ml into each plate (burst surface bubbles with a Bunsen burner flame)
Allow to solidify at room temperature
Bag plates upside-down in a stack and store at 4 °C until needed
Acknowledgments
This protocol was used for the work previously published in The Journal of Clinical Investigation ( Souma et al., 2016 ). We thank Sean M. Martin for his careful reading of the manuscript and helpful comments. This study was funded by NIH R01 EY014685, the Research to Prevent Blindness Inc. Lew R. Wasserman Award, and the University of Wisconsin Centennial Scholars Award to Terri L. Young. The authors declare no conflict of interest.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Church D. M., Stotler C. J., Rutter J. L., Murrell J. R., Trofatter J. A. and Buckler A. J.(1994). Isolation of genes from complex sources of mammalian genomic DNA using exon amplification. Nat Genet 6(1): 98-105. [DOI] [PubMed] [Google Scholar]
- 2.Duyk G. M., Kim S. W., Myers R. M. and Cox D. R.(1990). Exon trapping: a genetic screen to identify candidate transcribed sequences in cloned mammalian genomic DNA. Proc Natl Acad Sci U S A 87(22): 8995-8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nisson P. E., Ally A., Watkins P. C.(1994). Protocols for trapping internal and 3’-terminal exons. PCR Methods Appl 4(1): S24-39. [DOI] [PubMed] [Google Scholar]
- 4.Souma T., Tompson S. W., Thomson B. R., Siggs O. M., Kizhatil K., Yamaguchi S., Feng L., Limviphuvadh V., Whisenhunt K. N., Maurer-Stroh S., Yanovitch T. L., Kalaydjieva L., Azmanov D. N., Finzi S., Mauri L., Javadiyan S., Souzeau E., Zhou T., Hewitt A. W., Kloss B., Burdon K. P., Mackey D. A., Allen K. F., Ruddle J. B., Lim S. H., Rozen S., Tran-Viet K. N., Liu X., John S., Wiggs J. L., Pasutto F., Craig J. E., Jin J., Quaggin S. E. and Young T. L.(2016). Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest 126(7): 2575-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]