

Summary
This Perspective delineates how redox signaling impacts activity of specific enzyme isoforms and how this property may be harnessed for rational drug design. Covalent drugs have resurged in recent years and several reports have extolled the general virtues of developing irreversible inhibitors. Indeed, many modern pharmaceuticals contain electrophilic appendages. Several invoke a warhead that hijacks active-site nucleophiles whereas others take advantage of spectator nucleophilic side-chains that do not participate in enzymatic chemistry, but are poised to bind/react with electrophiles. The latest data suggest that innate electrophile sensing—that enables rapid reaction with an endogenous signaling electrophile—is a quintessential resource for development of covalent drugs. For instance, based on recent work documenting isoform-specific electrophile sensing, isozyme non-specific drugs may be converted to isozyme-specific analogs by hijacking privileged first-responder electrophile-sensing cysteines. Because this approach targets functionally-relevant cysteines, we can simultaneously harness previously-untapped moonlighting roles of enzymes linked to redox sensing.
Cysteine is an Enigma
Cysteine is the only conventional coding residue housing a third-row element capable of forming a reactive anionic, thiolate species. This property endows cysteine with generally high nucleophilicity and polarizability (Figure 1A), although defining highly reactive cysteines within the proteome and the reasons for their reactivity are complex problems. Nonetheless, cysteine serves as a catalytic nucleophile in many classes of enzyme, such as cysteine proteases and tyrosine phosphatases, and assumes regulatory roles through post-translational covalent modifications. As the most easily-oxidized of the coding amino acids, cysteine is often exploited as a redox switch both in natural (Holmstrom and Finkel, 2014; Klomsiri, et al., 2010; Paulsen and Carroll, 2010) and engineered (Meyer and Dick, 2010) proteins, and as a redox-active catalytic residue, e.g., in thiol oxidoreductases (Dmitri, et al., 2008).
Figure 1. Electrophilic inhibitors are targeted by nucleophilic amino acid residues.
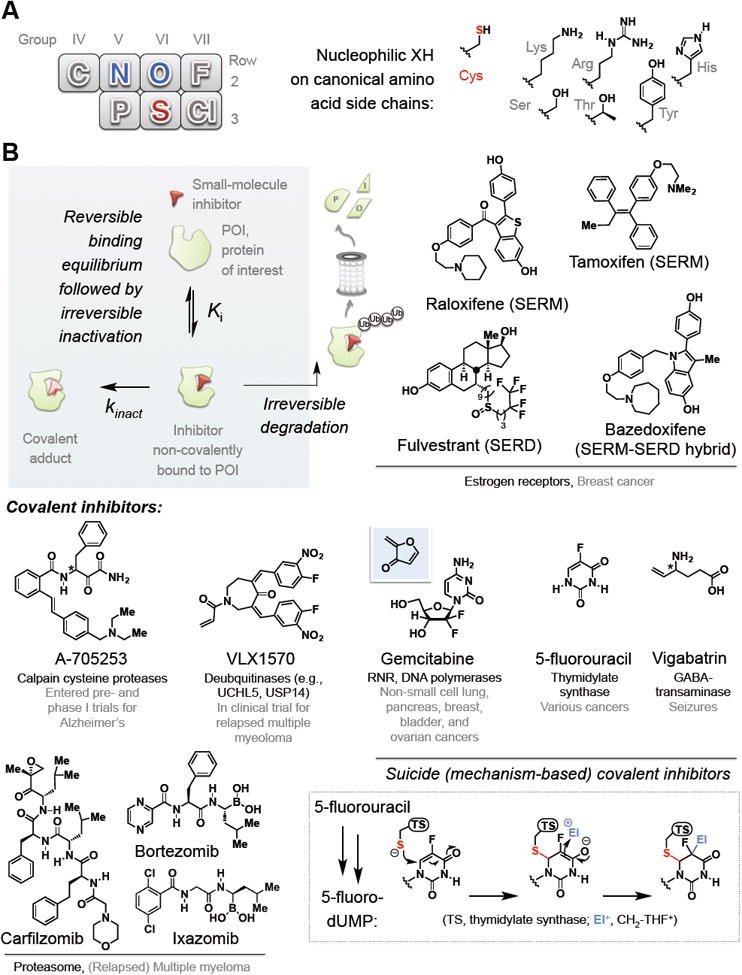
(A) (Left) Part of the periodic table showing second and third row non-metals. Blue-colored elements are components of “hard” (tend to favor carbonyl addition) nucleophilic amino acids from Row 2: sulfur (colored red) is the only primary coding residue in the third row and the only “soft” nucleophile. (Right) chemical structures of the side chains on specific residues.
(B) Clockwise from top left inset: a schematic illustration of irreversible inhibition. Top right: representative estrogen-receptors-targeting SERMs and SERDs approved for breast cancer. Middle right: examples of mechanism-based inactivators: inset at the bottom right shows currently-accepted mechanism for thymidylate synthetase (TS) inactivation by 5-FU. The active form of 5-FU (5-F-dUMP) is covalently attacked by TS, generating an enolate that traps out methylene-tetrahydrofolate (CH2-THF+) electrophile. The chemistry cannot proceed further due to the presence of the fluoride. Gemcitabine—a mechanism-based inactivator—generates the postulated reactive Michael acceptor in situ (shown above against the blue background)]. Bottom left: proteasome inhibitors present a special class of irreversible inhibition and representatives are shown. Middle left: representative covalent inhibitors of specific protein/pathway targets and disease. [Note: Stereogenic centers (*) on A-705253 and vigabatrin are undefined as they are both epimerizable in vivo].
Cysteine is underrepresented in the genome (Go, et al., 2015), indicating that Nature is discerning about cysteine's placement. In a study of 2413 mutations across 44 proteins, cysteine emerged to be one of the more frequently-mutated residues, and was the most common mutant residue (Khan and Vihinen, 2007). Thus, gain or loss of specific cysteines may be critical for maintaining fitness and disease etiology/progression.
Covalent Drug Discovery Gets its ‘Second Wind’
Several innovative drug-development strategies take advantage of cysteine and similar nucleophilic residues. This impetus is spurred on by the renewed interest in development of covalent inhibitors (Singh, et al., 2011). Although irreversible-inhibitor drugs have had a checkered history, such inhibitors offer several benefits over non-covalent counterparts. The principal win arises because unlike non-covalent inhibitors, ‘adsorption, distribution, metabolism, elimination (ADME)’ (Hodgson, 2001) does not strongly influence drug efficacy for covalent inhibitors. As irreversible inhibition (Figure 1B, inset) persists beyond drug clearance, covalent inhibitors facilitate relatively less-frequent dosing and/or lower dosing. The other class of inhibitors for which ADME is weakly linked to efficacy is protein degradation inducers (Lai and Crews, 2017) (Figure 1B). Degradation-inducing ligands far exceed efficacy of their traditional-binding counterparts: for instance, selective estrogen receptor downregulators (SERDs)— e.g., fulvestrant—are more effective than selective estrogen receptor modifiers (SERMs)—such as tamoxifen. For instance, fulvestrant and bazedoxifene are significantly more effective than the corresponding SERMS, tamoxifen and raloxifene, against hormone-independent breast cancer lines (Lewis-Wambi, et al., 2011; Wardell, et al., 2013) (Figure 1B).
This is because for a covalent inhibitor (or degradation inducer), once the target protein is inhibited, new protein synthesis is required to regain activity. This scenario is called “The Ultimate Physiological Goal” of inhibitor design (Lewandowicz, et al., 2003)—a moniker reflecting the fact that ∼50% of potential drugs in development fail due to ADME issues (Paul, et al., 2010) and a further ∼50% of approved drugs have minor ADME/toxicity problems (Hodgson, 2001). Additionally, ligand efficiency (average binding energy derived per non-hydrogen atom) of covalent binders can far exceed the maximum possible for non-covalent inhibitors. This feature allows covalent drugs to be low molecular weight, while maintaining high affinity.
Interestingly, basic science has shown examples of both covalent (Lim, et al., 2015) and non-covalent ligands causing protein degradation (Neklesa and Crews, 2012). However, since the ADME benefits and outcomes of irreversible inhibitors and protein degradation inducers overlap, there may be less benefit from converting a covalent inhibitor into a degradation inhibitor. One exception is if the protein of interest (POI) is multifunctional. This is because degradation inhibits all function and perturbs protein-protein interaction (PPI) networks: inhibition (likely) only inhibits one function. There are benefits of converting non-covalent inhibitors to degradation inducers even if there is no obvious second enzymatic function. For instance, PROTACS form a transient (non-covalent) ternary complex with a POI and an E3 ligase, leading to POI degradation. These molecules sub-stoichiometrically degrade POIs (a mode of action not available to irreversible degradation inducers) (Bondeson, et al., 2015). For this reason, their effects are significantly more persistent than non-degradation-inducing, reversible inhibitors (Lu, et al., 2015) targeting the same proteins.
There are many covalent drugs, but most were discovered serendipitously (Bauer, 2015; Wagner and Schreiber, 2016). Thus, general design rules are still being written. Many different forms of covalent inhibitor exist. Many hijack inherent enzyme chemistry by binding the active-site catalytic nucleophile, forming a covalent complex (Figure 2A, Class I). These can function through direct reaction (non-suicide) or through mechanism-based behaviors (suicide). In the latter class, the enzyme chemically modifies the inhibitor to unveil a highly-reactive fragment (typically an electrophile), leading to inactivation of the enzyme prior to diffusion away from the enzyme [Figure 1B, inset (Berkowitz, et al., 2008)]. These drugs are inherently irreversible. Examples of non-suicide electrophilic drugs in clinical trials targeting catalytic cysteines include: A-705253, a calpain (cysteine-protease) inhibitor (Ono, et al., 2016), and VLX1570, a deubiquitinase inhibitor (Wang, et al., 2016) (Figure 1A).Some frontline suicide inhibitor drugs, such as gemcitabine (Aye, et al., 2015) and 5-fluorouracil (Rose, et al., 2002) target active-site cysteines. Nevertheless, specifically targeting active-site cysteines using electrophiles not generated in situ remains a challenge. Some suicide inactivators, such as the antiepilepsy drug vigabatrin (Storici, et al., 2004), target catalytic lysines, but drugs targeting catalytic serine (e.g., penicillin) (Singh, et al., 2011), and threonine (proteasome inhibitors) are arguably more common. The proteasome is now an inherently-druggable target (Esseltine and Mulligan, 2012) and a fleet of rationally-designed covalent proteasome-inhibitors has been developed (Figure 1B). Bortezomib (Velcade; boronic-acid-based electrophile), ixazomib (Ninlaro; administered as boronic-ester prodrug) and carfilzomib (Kyprolis; epoxide electrophile) form covalent bonds to the threonine nucleophile within the proteasome active site (Kisselev, et al., 2012) (Figure 2A). Because these inhibitors target residues required to perform chemically-essential functions in the catalytic cycle, mutation of these residues is an unlikely means to gain drug resistance. For bortezomib, non-active-site point mutations, overexpression of compensating factors (such as heat-shock proteins), and elevation of survival pathways are documented drug resistance mechanisms (Lü and Wang, 2013).
Figure 2. Different classes of covalent inhibitors.
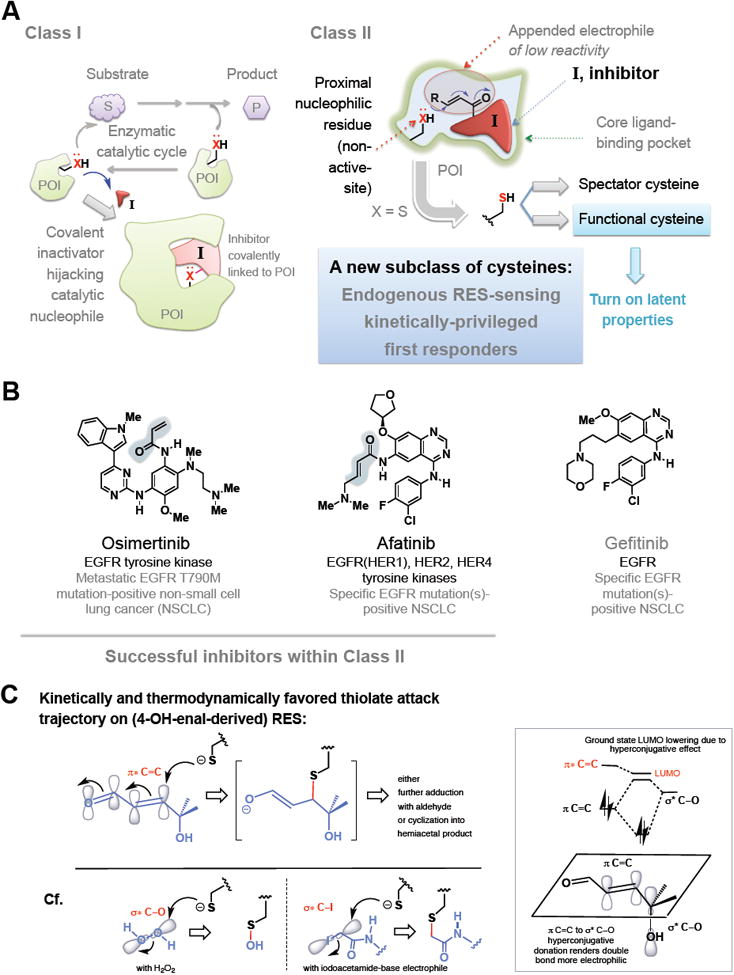
(A) Class I: active-site blockers. These electrophiles bind at the active site and engage chemically with active-site nucleophile to form an inhibited complex. Class II: These bind reversibly but are trapped out by a proximate nucleophilic residue (principally cysteine) not part of the catalytic chemistry step. This cysteine could either be a rank-and-file residue (see text), or a functional kinetically-privileged first-responding RES/ROS-sensor. Also see Figure 3.
(B) Representative inhibitors in Class II and their protein targets and disease. Blue shade marks the appended electrophile. For representative inhibitors in Class I, see carfilzomib/bortezomib in Figure 1B. Gefitinib is often considered as a non-covalent analog of afatinib.
(C) Different stereoelectronic requirements for chemical bond formation events between sp2-carbon-derived RES-based electrophiles such as HNE and sp3-based oxidants and electrophiles such as H2O2 and iodoacetamide(IA)-derivatives, respectively. With HNE, the allylic hydroxyl group enhances the partial positive charge (thus electrophilicity) at the Michael acceptor (sp2-hybridized) carbon through simultaneous hyperconjugation and inductive electron-withdrawing effects from electronegative oxygen. Inset shows hyperconjugation in ground state including molecular orbital mixing effect, resulting in overall lowering of the lowest unoccupied molecular orbital (LUMO) (π* of enal). The transition state (TS) of the reaction favors Felkin-Ahn type addition wherein σ*C—OH orients anti-periplanar to the forming thiolate carbon bond as shown. Negative charge in the TS is delocalized over what is formally an allylic anion-type intermediate: this factor contributes significantly to TS-stabilization in RES–thiolate addition, and recognition likely plays a role in kinetically-privileged RES-sensing. [The square bracket shows the first-formed intermediate, which progresses to yield indicated product(s) (see, for example, Long and Aye 2016) ]. By contrast, thiolate addition to H2O2 and IA-derived ROS and RES, respectively, has less negative-charge delocalization in the TS.
Targeting Non Active-Site Cysteines is a Growing Strategy in Drug Design
Drugs such as omeprazole and derivatives target non-active-site cysteines within the gastric H+/K+-ATPase by forming a disulfide (Sachs, et al., 2007). Some mechanism-based inhibitors, such as triflurothymidine (Santi and Sakai, 1971) and eflornithine (Poulin, et al., 1992) target spectator lysines and cysteines, respectively. In a similar vein, a new class of non-active site, nucleophilic-residue-targeting covalent inhibitors has recently arisen. These drugs feature (1) a ligand core that binds a specific pocket on targets and (2) an appended low-reactivity (typically an α,β-unsaturated carbonyl) electrophile that enables covalent binding to cysteine(s) proximal to the specific site to which the ligand core binds (Figure 2A, Class II). Because these drugs target off-active-site residues and use low-reactivity soft electrophiles (that house sp2-hybridized electrophilic carbon), cysteine is the most common residue targeted. Examples include osimertinib (Tagrisso, AZD9291) and afatinib (Gilotrif), approved to treat non-small cell lung cancer (Figure 2B). Similar to the comparison of SERDs and SERMs, covalent drugs that bind off-target cysteines are typically more effective than analogous non-covalent inhibitors. Several excellent commentaries have recently highlighted this strategy, for instance, in kinase inhibitor development (Muller, et al., 2015): afatinib is more effective than the chemically-similar, ATP-competitive reversible inhibitor gefitinib (Miller, et al., 2012; Ninomiya, et al., 2013; Park, et al., 2016) (Figure 2B). However, afatinib has an expanded target repertoire compared to gefitinib (Solca, et al., 2012) beyond the epidermal growth factor receptor (EGFR) (Figure 2B), making this comparison far from direct.
Analysis of the mechanism unique to this non-catalytic and proximal cysteine-targeting strategy clearly spotlights an ‘Achilles heel’: loss of the targeted cysteine, such as C797 within EGFR, targeted by the covalent inhibitors renders the drug less effective, opening a route to drug resistance (Thress, et al., 2015). Indeed, osimertinib-treated patients showed that 30% had acquired the C797S mutation on EGFR. Interestingly, C797 is a potent sensor of endogenously-produced reactive-oxygen species (ROS) (Brewer, et al., 2015) (Figure 3, right panel). One important consequence of this oxidation event is a sizeable enhancement of EGFR tyrosine kinase activity (Paulsen, et al., 2012). Thus, chronic oxidative stress both stimulates EGFR and selectively renders the activated EGFR refractory to covalent drugs that target C797, like afatinib (Figure 2B) (Schwartz, et al., 2014; Truong, et al., 2016). Clearly, these data make a convincing argument that physiologic impacts of endogenous oxidation/electrophilic modification events at a specific target residue should be considered when planning to exploit ROS-sensing functional cysteines as covalent-drug targets. As we describe below, tools to perform these investigations are now available.
Figure 3. Lessons from Akt2 and Akt3.
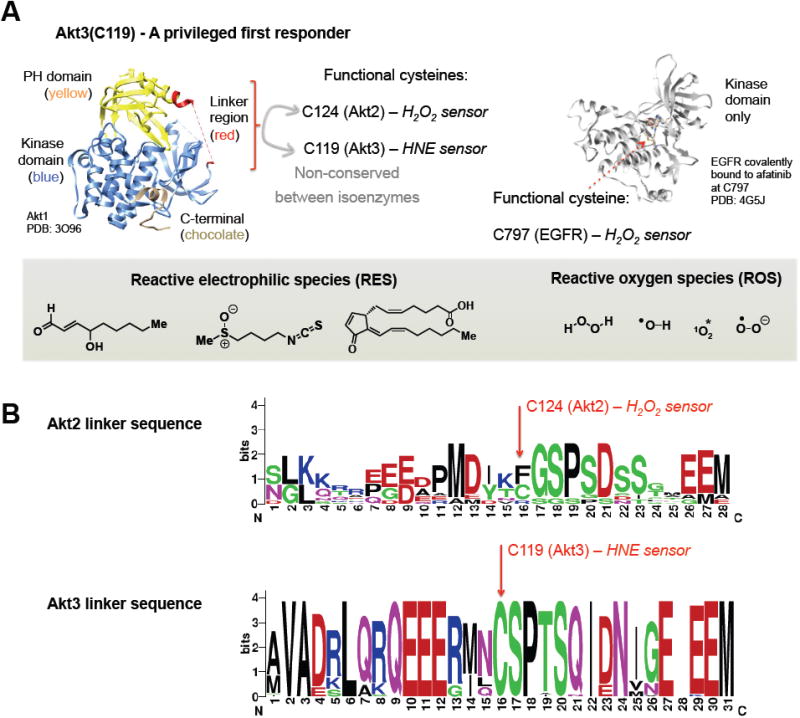
(A) (Left) X-ray crystallographic analysis of Akt1, showing specific domains and the disordered linker region that is not conserved between the specific human Akt1–3 isoforms; (right) X-ray crystallographic analysis of EGFR showing the ROS-sensitive functional cysteine. (Inset) A schematic of natural ROS and RES commonly encountered. Left to right under RES: 4-hydroxynonenal (HNE), sulforaphane, and 15-deoxy-Δ12,14-prostaglandin J2. Note the α,β-unsaturated carbonyl electrophile is a common motif in natural RES, making the basic reactivity of endogenous electrophiles similar to drug-like covalent pharmocophores.
(B) Sequence logo highlighting the amino acid sequence conservation within the linker region of either Akt2 (top) and Akt3 (bottom), across 10 and 36 species, respectively. The species are the same as those shown in Fig. 4. Relative sizes of residues reflect their frequency and the Y-axis indicates the information content of the position in bits. The sequence logo was generated using WebLogo (invented by Steven E. Brenner et al., Computational Genomics Research Group, University of California, Berkeley).
A recent innovative strategy has focused specifically on targeting acquired cysteines, i.e., cysteines that arise during disease progression (Visscher, et al., 2016). Such an approach is potentially selective for diseased cells over healthy tissue. Furthermore, because these cysteines are inherent to the disease state, mutagenesis of these residues may be an unlikely pathway to acquired resistance. However, patients with the EGFR(T790M) mutation that is responsive to osimertinib (Figure 2B) have acquired resistance through apparent reversion to wild-type (Planchard, et al., 2015), indicating that cancers can backtrack to an earlier state to evade drug toxicity. Additionally, targeting acquired cysteines requires intimate knowledge of the disease state(s) and necessitates a “personalized approach” to the specific mutations in question.
Our Perspective does not seek to change the direction of research, but rather we propose a rubric to identify ideal target cysteines, using covalent non-catalytic-residue-targeting inhibitors (Figure 2A, Class II).Where possible, acquired cysteines are exemplary targets for covalent inhibition. However, in lieu of such residues being generally available/known, we propose to focus on cysteines that are primed to react with native RES [principally α,β-unsaturated carbonyl-containing lipid-derived signaling electrophiles (LDEs)] at basal concentrations. We propose that these cysteines (dubbed “first responders”) are evolutionarily selected specifically because their low-occupancy modifications phenotypically impact enzymatic or regulatory functions. In a similar way to how reversible PROTACS improve efficacy over traditional reversible inhibitors, it is our opinion that tapping into phenotypically-dominant mechanisms will help prolong action, reduce dosing, and improve efficacy over traditional reversible compounds and covalent inhibitors that do not target privileged residues. Importantly, early findings indicate that RES and ROS sensing functions may be mutually exclusive, in at least some cases (Long, et al., 2017; Wani, et al., 2011).Thus, targeting a privileged RES-sensor cysteine—as we discuss below—may mitigate refractory issues intrinsically linked to cysteine oxidation.
Moreover, since covalent drug binding is more analogous to RES-based electrophilic adduction (as opposed to ROS-based oxidation such as sulfenylation), it is more likely that targeting privileged RES-based sensors (relative to privileged ROS-sensors) will bring translational benefits associated with phenotypically-dominant behavior and be directly applicable to electrophilic drug design, most of which is centered on the α,β-unsaturated carbonyl reactivity handle (Figure 2B). Novel ROS sensors are also important handles for covalent drug design, and impactful work has been performed in this area: at least one covalent kinase inhibitor binds a known ROS-sensor cysteine, for instance (Paulsen, et al., 2012; Truong, et al., 2016). However, the signaling mechanisms, bonding possibilities, and chemical reactivity of sulfenic acids and Michael-acceptor-alkylated cysteines are different (Figure 2C). The Michael-acceptor RES-sensing is more functionally analogous to the POI-cysteine reaction with Michael-acceptor-appendages present in several covalent drugs (Figure 2B). We thus posit that the mechanisms that usher ROS-sensing are less likely to transpose to benefits for drug design. There is, however, no reason to outright assume that a ‘privileged’ ROS-sensor cannot be a privileged RES-sensor, nor that an enzyme can have both ROS- and RES-sensing functions in different cysteines (and this is likely the case for Keap1 as we detail below).
We believe privileged sensing is important because to elicit functionally-relevant impacts from low-occupancy RES modifications, first-responder cysteines likely function through a phenotypically-dominant mechanism. This is because, due to the promiscuity of RES, high occupancy is hard to achieve without incurring many off-target modifications. Clearly, any strategy that targets genome-encoded cysteines has a risk of targeting essential proteins in healthy cells. But, our recent research (Fang, et al., 2013; Lin, et al., 2015; Long, et al., 2017; Parvez, et al., 2015; Parvez, et al., 2016) suggests that innate LDE sensitivity/responsiveness (i.e., RES sensing) can be highly enzyme- and isoform-specific, limiting collateral damage and improving tolerability.
Mother Nature Knows Best?
Endogenous RES are small-molecule by-products of poly-unsaturated-fatty-acid peroxidation events formed during basal/transient cellular stress (Schopfer, et al., 2011) (Figure 3, inset). Exposure to similar reactive electrophilic entities can occur through our diet (Ahn, et al., 2010) and from various external sources, such as smoke inhalation (Kensler, et al., 2007). Unsurprisingly, reactive electrophiles are bad for us in high doses: these molecules have many targets, including proteins, DNA, and biomolecules important for maintaining physiological redox balance. Hence these reactive entities can in principal affect manifold signaling pathways. Indeed, endogenous lipid-derived electrophiles (LDEs) such as 4-hydroxynonenal (HNE) (Figure 3, inset) (Jacobs and Marnett, 2010) are routinely discussed in toxicology journals.
On the other hand, at low doses, RES serve an important, possibly essential, fine-tuning function that modulates flux through specific signaling pathways, to precondition for protection against an array of maladies. The most widely appreciated of these pathways is the Keap1/Nrf2 antioxidant response (AR). This electrophile-responsive pathway that upregulates a battery of antioxidant/detoxification genes protects against oxidative stress, carcinogen exposure, and even chemical warfare agents. The approval of dimethyl fumarate (Tecfidera), a RES-based pharmaceutical that upregulates AR, for the treatment of relapsing multiple sclerosis underscores the biomedical relevance of AR. However, recent work suggests that Nrf2 is not required for Tecfidera's function, implying other protein targets other than Keap1 are the root-cause for Tecfidera's immune-suppressive properties (Blewett, et al., 2016). Such controversies are to be expected with drugs that hit multiple targets.
Privileged First Responders are Equipped to Relay Important Signals at Low Occupancy
But if native electrophile signaling is an endogenous response pathway, there are likely some mechanisms to ensure beneficial signaling nodes are tripped and health-promoting signals are propagated, and non-specific effects are filtered out. We have proposed two non-exclusive mechanisms which are likely common to privileged sensors of RES: (1) privilege of occupancy, wherein “intended targets” trigger phenotypically-dominant downstream responses; (2) kinetic privilege, wherein functionally-impactful reactions occur at cysteines that are inherently more nucleophilic than the host of other potential nucleophiles within the cell (Long and Aye, 2016). Unfortunately, teasing out these privileged residues from the majority ‘rank-and-file’ cellular cysteines is difficult.
Conventional strategies use bolus dosing with electrophiles in a hard-to-control (dose/timing/locale/target) setting (Codreanu and Liebler, 2015). Under these conditions, the global proteome (often lysates) is exposed to prolonged treatment with the electrophile. Since electrophile– nucleophile covalent (and largely irreversible) reactions are non-equilibrium and time-dependent, targets are biased by mass action in terms of both protein expression and numbers of cysteines per protein. Some reactive electrophiles can also be converted to secondary species through reactions in situ with endogenous small-molecules (e.g., GSH–HNE adduct that still features an electrophilic aldehyde), weakening the link between the original RES and the measured response.
In an innovative ‘activity based protein profiling (ABPP)’-derived strategy, a panel of reactive electrophile proxies (affinity agents) profiles a subset of cysteine residues by mass spectrometry (MS) (Johnson, et al., 2010). This ingenious and versatile strategy continues to offer important insights into electrophile-reactive targets as well as mechanistic information into covalent drug action. Much of our current understanding of novel electrophile-regulated targets and pathways has been derived using this highly powerful profiling approach. Based on the number of unique cysteines in human genome (∼250,000)1 and the maximum number of cysteines profiled across numerous runs (∼6000), the proportion of cysteines captured from the profiling methods is modest (typically 1–5% of total cysteome). Although this number seems disappointingly low, it is important to stress that the reactive cysteome is a uniquely complex, and dynamic system that is finely balanced and responsive to stimuli. It is thus a testament to the pioneering efforts of Cravatt and other likeminded individuals that we have been able to gain the understanding we currently possess. Assuming that the 6000 identified cysteines do not reflect the whole reactive cysteome (which does appear to be the case, see below), there are several possible reasons for the low coverage. (1) Many (although not all) of these experiments are carried out on soluble lysates, created in the absence of not only reducing agents but also supplemented metals, and at relatively low protein concentrations. (2) Some reactive cysteines may be extensively modified prior to (or possibly oxidized during) the experiment, making the amount of “free”cysteines that can be detected by the affinity reagent too low to be detectable, despite their intrinsic reactivity. (3) The fraction of cysteines detected may not reflect all the cysteines labeled by iodoacetamide, as there could be technical limitations to detection/product stability/ionization. Such issues may be improved in the future by technological advancements. (4) It is unlikely that iodoacetamide is able to react with all reactive cysteines. For instance, many active-site cysteines—such as the catalytic thiol within deubiquitinating enzymes—do not show up in iodoacetamide-based ABPP screens. Ironically, these residues are detected by tailor-made affinity labels, such as ubiquitin vinyl sulfone (Borodovsky, et al., 2001) or ubiquitin propargylamide (Ekkebus, et al., 2013), that house “electrophilic” centers of much lower reactivity than iodoacetamide. Indeed, propargylamides are conventionally of poor electrophilicity. There has been some effort to develop more reactive affinity handles. One such example is ethynyl benziodoxolone (Abegg, et al., 2015). A direct comparison of this probe with an iodoacetamide analog showed that ethynyl benziodoxolone could label more proteins, although the number of enriched peptides identified by mass spectrometry was similar between iodoacetamide and ethynyl benziodoxolone (ca. 2000 each). Interestingly, around 30% of the identified cysteines were different between these two probes, indicating that no single probe will ID all reactive cysteines.
Nonetheless, the profiling method is broadly applicable, and gives precise, reliable information about cysteine reactivity using a protocol that has been widely used. Interestingly, this approach is strongly complementary to other protein-by-protein methods we will describe below. Importantly, as the profiling method measures differential loss of labeling by the proxy (e.g., iodoacetamide-derived probe) along with the requirement for a stringent threshold, the method provides an indirect measure of electrophile sensing and scores hits typically only when there is relatively high-occupancy of the specific cysteine by the electrophile. The indirect nature of the readout may render conclusions influenced by off-target and/or secondary modifications/functional coupling, and in the case of reversible RES-protein adducts (Delmastro-Greenwood, et al., 2014), RES-signal exchange between one site or protein target to another. Whether these variables are a benefit or confounding depends upon the intended use of the assay. Furthermore, the relatively low resolution of these assays may not account for the low-labeling (low-occupancy) threshold required to elicit phenotypically-dominant (either gain-of-function or dominant loss-of-function) downstream signaling, or account for compartmentalization of signaling (Hung, et al., 2013; Long, et al., 2017).
A recent extension of this assay identified fragments that bind proximal to specific cysteines using reactive fragment-based ligand discovery (Backus, et al., 2016; Parker, et al., 2017). This protocol compared loss of proxy labeling upon treatment with either an acyl chloride or an enamide. Interestingly, the number of proteins labeled by each probe set broadly correlated with reactivity. However, some proteins were labeled selectively by less reactive probes, consistent with matching between the whole probe (warhead and non-reactive fragment) and the specific cysteine. Furthermore, similar warheads gave different cysteine-labeling profiles, indicating that labeling may be dominated by binding and highlighting how binding can influence cysteine reactivity. Given this analysis, this assay could have identified some privileged sensors, but likely reports mostly on site occupancy and electrophile presentation to the cysteine in question. Either way, numerous modifications that elicited enzyme inhibition were identified, testifying to the utility of the method.
These ligandable cysteine/ligand pairs may share some mechanistic commonalities with privileged cysteine sensors, because one way to engender rapid second-order kinetics is to locate a reactive cysteine next to a ligand-specific binding site. For instance, the enzyme glutathione reductase reacts with HNE in a two-step process proceeding through a presumed non-covalent encounter complex (Ki= 0.5 μM), followed by an irreversible binding event, presumably cysteine attacking the enone. The second-order rate for the process is 500 M-1s-1 (37 °C, pH 7.0) (Vander Jagt, et al., 1997), which is respectable, but far below optimal. Nevertheless, 500 M-1s-1 represents over a >100-fold improvement on reaction of cysteine with HNE (e.g., 1.3 M-1s-1, 23 °C, pH 7.4) (Long, et al., 2016; McGrath, et al., 2011). Thus, we speculate that at least some privileged sensors may have binding sites for a subset of biological electrophiles, simultaneously predicting that these sensors will be discriminatory, and providing a mechanism for how binding can be linked to function (i.e., high-affinity interaction between POI and RES-based ligand drives conformation changes that affect function. For allosteric enzymes, this could lead to dominant loss as well as gain of function).
One potential outlier to this argument is Keap1 that we and others have shown to be a promiscuous sensor (Hayes and Dinkova-Kostova, 2014; Lin, et al., 2015; Parvez, et al., 2015; Parvez, et al., 2016). Since human Keap1 contains 27 cysteines that react with varied electrophiles, this protein likely contains multiple binding sites with privileged cysteines positioned to react with a specific electrophile class. We have found different Keap1 cysteines are labeled by different electrophile classes under T-REX conditions, consistent with this proposal (Lin, et al., 2015; Parvez, et al., 2015; Parvez, et al., 2016).
Viewing the aforementioned recent data (Backus, et al., 2016) through this specific lens, most of these sensor/ligand pairs identified are of relatively low affinity (with some notable exceptions that prove the powerful utility of the method for traditional lead drug discovery) and given the time of the experiments likely have relatively poor second-order kinetics of binding (although this was not extensively investigated). Hence, based on the available data, most ligands likely do not interact with the target (sensor residues) sufficiently to drive downstream signaling in a manner similar to native RES-orchestrated signaling through “privileged” sensors. To definitively ascertain if these residues/ligand pairs identified (Backus, et al.,2016) are kinetically- and phenotypically-privileged, the extent to which such covalent modifications could occur (semi)-selectively against the backdrop of the whole proteome and/or elicit gain of function would need to be evaluated.
Specific Isoforms, such as Akt3, Function as First Responders
We recently used a medium-throughput screen of several redox-sensitive kinases to establish a novel electrophile-sensing/signaling role of Akt3 (Long, et al., 2017). The live-cell-based screen used a recent innovation, called T-REX (targetable reactive electrophiles and oxidants) (Parvez, et al., 2016), with which select proteins in a panel are screened for sensitivity to HNE [a number of other LDEs can be evaluated using the “T-REX electrophile toolbox” (Lin, et al., 2015)]. In contrast to bolus profiling methods above, the electrophile is only made available in amounts sub-stoichiometric to the target protein, within its proximity (Long, et al., 2016; Long, et al., 2017), and at/for a user-defined time/duration in an otherwise unperturbed native environment/redox state in live cells (Fang, et al., 2013; Parvez, et al., 2016).
A benefit of T-REX that sets it apart from most other electrophile-sensitivity screens is that T-REX can directly evaluate downstream signaling responses that are specific to the ‘electrophile-modified state’ of the privileged sensor protein (Long and Aye, 2016; Long, et al., 2017; Parvez, et al., 2015; Parvez, et al., 2016). This capacity is achievable because (1) the electrophile is delivered selectively to that protein in intact cells or animals, and (2) occupancy of the electrophile on the target POI is limited to maximally 100% but practically is 10–30% for a functional privileged sensor (Long, et al., 2017; Parvez, et al., 2016).
This method has proven a powerful means to link target-specific RES-modification to function and assess the mechanism through which signaling occurs. The method is equally functional in cells and fish. Although we have written at length about the limitations of this method (Long, et al., 2017; Parvez, et al., 2016), we stress here that T-REX is a protein-by-protein protocol that requires genetic fusion of each POI to a HaloTag (a protein handle to allow recognition for POI-specific LDE-delivery) and photouncaging of a latent electrophile. We are currently working on removing reliance on HaloTag through all small-molecule T-REX delivery to native POIs. An extension of this concept that we are also focusing on is targeting privileged cysteines through drug-LDE chimeras that we propose in this Perspective.
In the case of Akt3, this sensing function occurs through HNE-modification of a single cysteine (C119)–in preference to seven other spectator cysteines within human Akt3. C119 is conserved across vertebrates and loss of this sensing cysteine selectively ablates the electrophile-modification-dependent signaling function in human cells and in zebrafish, even though the C119S/A-mutant enzyme retains its canonical kinase activity. Other Akt isoforms (1 and 2) do not manifest potent electrophile-sensing like Akt3. Comparison of the amino-acid sequences reveals that Akt1 and Akt2 do not contain C119. No perturbation of the downstream function occurs upon T-REX-redox-targeting on Akt1/2 (Long, et al., 2017).
Interestingly, the sensing residue (C119) within Akt3 resides within a loop region bridging the plekstrin-homology (PH) and kinase domains that are both highly conserved across human isoforms. The loop sequence displays poor conservation across isoforms, explaining the divergent cysteine-sensing abilities. Intriguingly, a cysteine in the analogous loop region in Akt2 senses and responds to endogenously-produced H2O2 (Wani, et al., 2011). The cysteine within this loop region of Akt2 is also conserved from humans to reptiles (Anolis carolinensis) but is not found in fish (Figure 3B). Akt1 harbors no analogous loop cysteine. Interspecies conservation of the isoform-specific sensor cysteines has allowed us to propose that Nature has evolved isozymes to differentially sense and respond to chemically-distinct RES versus ROS signals. Because at least in the case of Akt3, this sensor residue is conserved across a wide range of species (Figure 4), it is likely that these functions are critical for fitness.
Figure 4. Evolutionary analyses [with emphasis on conservation of privileged cysteine(s) ] of Akt and Akt-like isoenzymes based on protein sequence data.
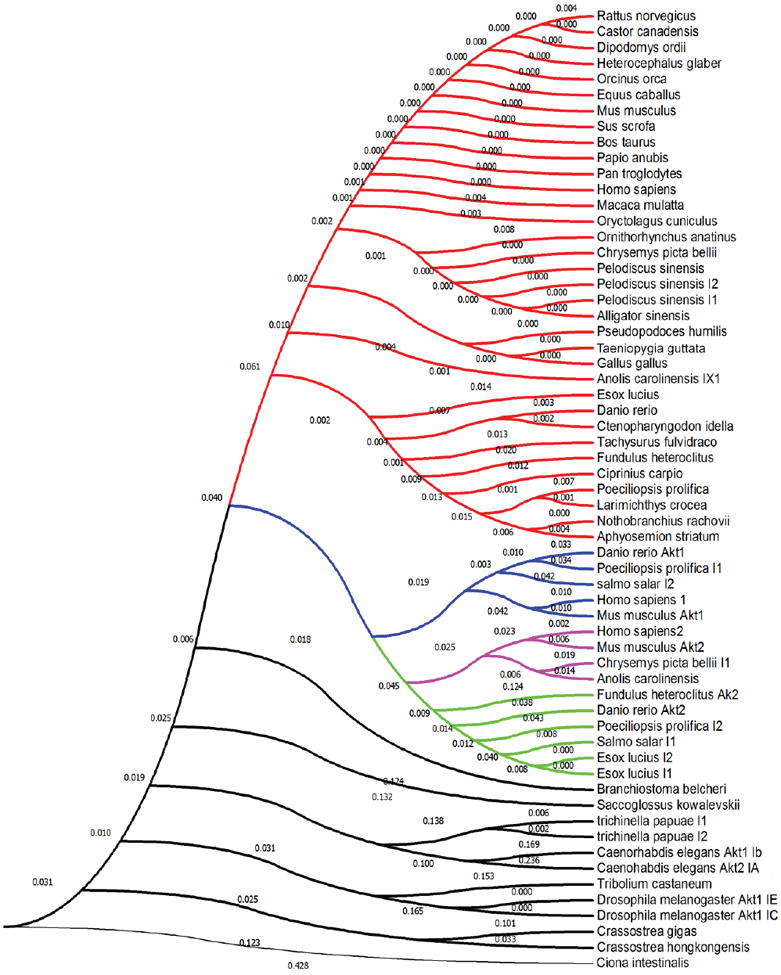
The amino acid conservation tree was constructed in MEGA-7 (Kumar, et al., 2016) using the Neighbor-Joining method (Saitou and Nei, 1987). The analysis involved 61 Akt sequences (whole-protein amino acid sequence) spanning Akt1–3 from indicated species, with the emphasis on Akt3. Red indicates Akt3-like proteins all of which contain the privileged cysteine analogous to C119. Blue indicates Akt1-like proteins that bear no cysteine(s) analogous to C119 and/or C124. Magenta indicates Akt2-like proteins with ROS-sensor cysteine (C124). Green indicates Akt2-like proteins that lack the ROS-sensor cysteine (C124). Black indicates Akt-like proteins from lower eukaryotes. Note: No protein aligning with Akt3 but lacking C119 was found in this set (see Fig. 3B for sequence logo). (The optimal tree with the sum of branch length = 2.99180282 is shown). The evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling, 1965) and are in the units of the number of amino acid substitutions per site.
First Responders are Ideal Targets for Covalent Drug Design
Based on these observations, we propose several reasons why privileged electrophile sensors may be ideal for covalent drug design.
These privileged sensor residues have intrinsic function that is retained (at least in the case of RES-sensor Akt3, and to a lesser extent ROS-sensor Akt2) across vertebrates and this function is dialed into specific enzymes, and even specific isoenzymes. Thus, it is less likely that these privileged sensor cysteines will be lost through drug-induced selection compared to rank-and-file cysteines, rendering drugs targeting these privileged sensor cysteines inherently less prone to resistance through cysteine mutation.
Sensing residues are inherently reactive to electrophiles. In the case of Akt3, 15–20% of photo-liberated HNE labels the specific cysteine (Long, et al., 2017) under what we believe to be the time required for HNE to escape from the coordination shell(s) of the protein to the bulk solvent [likely 100's of nanoseconds (Elber, 2010)]. This compares favorably to kinact values calculated for various irreversible kinase inhibitors that are on the order of 1 ms-1 (Schwartz, et al., 2014). Alternatively, success of T-REX-redox-targeting indicates a rapid second-order rate constant, possibly approaching diffusion control: (kinact/Ki)'s for several tyrosine kinase inhibitors bearing similar α,β-unsaturated carbonyl pharmacophores in clinical use or trials are several orders of magnitude below this. Thus, drugs that target such residues are likely to have excellent irreversible binding kinetics and/or be able to interact with weakly-electrophilic motifs appended to the inhibitor (mitigating off-target effects). Importantly, Akt3 is not the only protein we have discovered that can undergo appreciable extent of modification (i.e., high delivery efficiency) via T-REX: Keap1, small heat-shock protein 7 (hspb7), and the large subunit of ribonucleotide reductase (RNR-α) (Fang, et al., 2013; Parvez, et al., 2016) have all been similarly successful.
Simultaneous engagement with the native-electrophile-sensor residues and docking to the specific binding pocket ushers changes on the enzyme that can be harnessed for medicinal benefit. Whereas it is typical to target rank-and-file cysteines adjacent to a drug-binding site, modification of these spectator cysteines typically has no phenotypically-dominant effects on the enzyme. [EGFR(C797) may be an exception, although how RES affects C797 has not been studied in detail]. By contrast, kinetically-privileged sensor residues are intrinsically linked to enzyme function. In the case of Akt3, HNEylation of C119 at low occupancy elicits dominant-negative loss of kinase activity and suppressed downstream signaling at a magnitude that is functionally coupled to the extent of sub-stoichiometric alkylation at C119 (Long, et al., 2017). Clearly, endowing a small molecule with the ability to functionally downregulate oncogenic targets at low-occupancy imparts benefits in terms of drug efficacy and duration of action in a way similar to the longer latency of PROTACS (gain-of-function inducers) relative to analogous non-degradation inducers (Lai and Crews, 2017; Ottis and Crews, 2017; Salami and Crews, 2017).
For Akt2(ROS) and Akt3(RES), privileged sensor cysteines reside on mobile loop regions (Figure 3). This conformational plasticity will assist productive covalent bond formation between a (rigid) bound ligand and the proximal reactive privileged cysteine.
Because privileged sensors function through amplifying (either dominant-negative or gain-of-function) mechanisms that can elicit functional signaling events at low occupancy, it is likely that target occupancy by electrophiles at any time is low. Thus a substantial proportion of the sensor residue on the target is still present at its non-modified state, likely even under chronic electrophilic stress. This tenet potentially alleviates refractory responses to pharmacological treatment harnessing these cysteines. We discuss below how different phenotypic outputs factor into this analysis.
Identifying Bona Fide Privileged Sensors and their Mechanism of Action may Help Develop Better Covalent Drugs and/or Identify Cysteine Targets
To assist in drug discovery, more bona fide first responders must be identified. Although not enough information exists to date,it is our hope that as more examples of cysteines—validated for both sensing and downstream signaling—appear, we will better predict these kinetically-distinctive residues a priori: from our limited investigations, cysteine conservation (possibly in unstructured loops) may be an important clue (Figure 4), but more research is required.
Until then, it is in our opinion that the search for sensor residues, and specifically functional validations, would profit from transient release of close-to-physiological concentrations of native signaling electrophiles within the microenvironment of a target of interest or subcellular locale in vivo. It is likely these low-occupancy conditions will identify only the most reactive residues, reducing noise by maximizing stringency. We are currently developing general methods that will fulfill the need for unbiased ID of privileged sensors in a high-throughput manner while functioning within an unperturbed proteome.
Using methods available to date, assuming that (a) putative drug candidate protein(s) is(are) known, we suggest that it(they) be screened using T-REX or a similar method to evaluate kinetically-privileged, first-responsive sensing capability including interrogation for precision downstream signaling. Following T-REX in vivo, simple enrichment methods can identify the specific residue(s) modified by RES using MS (Lin, et al., 2015; Long, et al., 2017; Parvez, et al., 2015; Parvez, et al., 2016). Hypomorphic sensor point mutants where applicable validate the residue and unambiguously document signaling arises from on-target modification. Targeted covalent inhibitors could be established subsequently, by synthetically modifying known ligands that bind in the vicinity to the first-responder cysteine. Of importance, novel small-molecule ligands that bind near the sensor-residue site could also be screened by ability to block T-REX-mediated HNEylation and/or signaling output, or (more likely) using established and upcoming high-throughput readouts.
Although hydrophobic moieties—such as n-pentyl chain within HNE—are not unheard of within approved drugs (Liu, et al., 2011), there are several challenges to developing drugs around HNE-type functionalization. The transition state for addition of cysteine to an α,β-unsaturated carbonyl is reasonably invariant across different RES, and thus kinetically privileged sensors likely show elevated rates of reaction to many α,β-unsaturated carbonyl-derived RES (Figure 2C). Phenotypic dominance can arise from numerous parameters that are likely to be a function of the reaction pathway and contacts the small-molecule electrophile makes with the enzyme during this reaction landscape.
General drawbacks include: (1) lipid/HNE-interacting proteins/structures in vivo may non-specifically associate with the appended HNE on the drug, limiting bioavailability and potentially allowing unintended reaction with the enone moiety through proximity enhancement; (2) atom economy is reduced relative to acrylamide-based inhibitors; and (3) metabolism may also be increased (Alavijeh, et al., 2005). Since T-REX is suited to deliver a number of different cyclic and acyclic enones/enals to a specific POI (Lin, et al., 2015; Parvez, et al., 2016), one simple way to circumvent the hydrophobicity problem is to exploit short-chain electrophilic motifs out of T-REX toolbox, or employ user-designed electrophilic appendages to the enone during pilot tests, as opposed to n-pentyl. The latest data reveal that Keap1, an established promiscuous electrophile sensor (Hayes and Dinkova-Kostova, 2014), is a privileged sensor of numerous electrophiles, independent of alkyl chain length/topology, for example (Lin, et al., 2015; Parvez, et al., 2016).
Once a specific framework is identified, traditional SAR approaches could also be harnessed to improve drug-like properties while retaining downstream signaling behavior. In addition, use of chimeric drugs, like PROTACS, is increasing within the pharmaceutical industry (Corson, et al., 2008; Ottis and Crews, 2017). These molecules employ traditional chemical biology “linker” regions (such as polyethylene glycols) to fuse two pharmacophores together (Corson, et al., 2008). Thus, the hydrophobic chain within HNE could be substituted with a (poly)ethylene unit, and/or potentially be used as a linker region itself. Since the mechanisms leading to privileged sensing are likely multifaceted, different privileged sensors will likely respond to modification differently. Thus general rules may not be derivable, making mechanistic interrogation of each sensor at a time all the more important.
Critically, the mechanism through which first-responder-modification-initiated downstream signaling occurs is paramount. Dominant-negative (inhibition-reinforcing) signaling modes are a good basis for drug discovery. Obviously, unlike the case of EGFR(C797) oxidation above (Paulsen, et al., 2012), target cells would not benefit from upregulation of RES to block inhibitor binding, as this event would also cause inhibition. One such hub is Akt3(C119) (Figure 3–4). Using the conceptual framework proposed above, we are presently striving to repurpose known isozyme non-specific inhibitors to target this privileged residue. The ultimate goal is to create isoform-specific, covalent inhibitors. However, one can also envision that electrophile modification of the target cysteine could stimulate canonical enzymatic activity (‘inhibition antagonizing’, potentially like electrophile modification of C797 in EGFR), or even that modification would elicit gain of function. Such a gain of function could be, from the purposes of drug efficacy, (1) “null”, (2) “antagonizing”, or (3) “reinforcing”. Neither 1 nor 2 are optimal.
On the other hand, triggering two separate, yet mutually-reinforcing phenotypes—one from occupancy of the ligand-binding site; the other from modification of the cysteine—certainly is appealing. This idea is consistent with poly-pharmacology (Knight, et al., 2010) and/or combination therapies (Webster, 2016); but benefits from simplicity, atom economy, and the other wins associated with covalent binders discussed above. Finally, there is a possibility that disease states acquire privileged sensors. Such residues could occur either through elevation of nucleophilicity of an existing cysteine (via mutations of neighboring amino-acid side chains), or through mutation of any residue to cysteine (Visscher, et al., 2016). Similar to endogenous first responders, these acquired reactive cysteine residues would be coupled to downstream signaling processes likely essential to the diseased state. Such residues are ideal targets for covalent drug discovery and could be relatively easily profiled using the transient electrophile release method or cysteine-reactive fragment profiling we discuss above.
Interestingly, some key genes involved in the oncogenic program are known to have acquired cysteines. Oncogenic acquired cysteines are found in 12% of oncogenic KRAS mutations, 9% of β-catenin mutations and an astounding 88% of fibroblast growth factor receptor 3 (FGRF3) mutations (Visscher, et al., 2016; Zhou, et al., 2016). To date, none of these mutations have documented acquired RES-sensing roles. In the case of the cysteines involved in disulfide formation, there may be a privileged ROS-sensing role (one way to form a disulfide) and modification could block oncogenic function. For β-catenin proto-oncogene, the mutation occurs in its N-terminus that houses the recognition element for β-TrCP E3-ligase, driving heightened Wnt signaling (Liu, et al., 2002). It is possible that modification of this cysteine with a β-TrCP-binding ligand may recruit β-TrCP to β-catenin, rescuing degradation through gain of function (a “mutant allele-specific” PROTAC).
Acquired cysteines are also present in both dominant and recessive heritable diseases. We describe some interesting examples below where a RES-based strategy may be useful and how the genetics of the disease may help assess which diseases will respond best. However, we state upfront that to our knowledge, none of these cysteines have been shown to be privileged RES-sensors and a therapy based around RES would need to be administered likely for life. In multiple mitochondrial dysfunctions syndrome 1 (MMDS1—a disease that often causes death in perinatal stages), a recessive cysteine mutation in Nfu1 (G208C) promotes dimerization and perturbs secondary structure, likely impairing iron-sulfur-cluster biogenesis (Wachnowsky, et al., 2017). Because this mutation is recessive (i.e., the gene is haplosufficient), it is possibly an ideal candidate for a molecular intervention approach. This is because even partial blocking of dimerization via RES-modification of the acquired cysteine may alleviate disease symptoms, provided (sufficient partial) activity can be restored. Thus, it would certainly be interesting to investigate whether a specific RES could target this cysteine, and in turn prevent dimerization/rescue activity. On the other hand, a rare genetic condition, autosomal dominant sensory ataxia, is linked to an R119C mutation in the ubiquitin ligase Rnf170 (Wright, et al., 2015). This mutation destabilizes the enzyme and may result in aberrant ubiquitination, either through a compensatory mechanism, or through directly changing the client profile of Rnf170. Possibly modification of this cysteine may help to stabilize the protein, although because this is a dominant mutation, assuming equal effect on stability between Rnf170 and Nfu1, RES-occupancy may have to be higher for Rnf170(R119C) than in the case of Nfu1(G208C).
Conclusion
In sum, we hope that the redox signaling field can be of some use to drug design. It is our opinion that by harnessing kinetically-privileged cysteines that are defined by heightened RES susceptibility,we can gain extra selectivity and/or efficacy from covalent drugs.
Acknowledgments
We thank Mr. Jesse R. Poganik and Dr. Yi Zhao for helpful feedback and proofreading assistance. NSF CAREER (CHE-1351400), Beckman Young Investigator, NIH-New-Innovator (1DP2GM114850), Office of Naval Research (ONR) Young Investigator (N00014-17-1-2529), Burroughs Wellcome CRTG, and the Sloan fellowship (FG-2016-6379) programs (to Y.A.) for research; zebrafish husbandry and microinjection/imaging facility (NIH R01 NS026539, PI: J. Fetcho), Cornell NMR facility (NSF MRI: CHE-1531632, PI: Y. Aye), Cornell Imaging Center (NIH 1S10RR025502, PI: R. M. Williams), Cornell proteomics facility (NIH SIG grant 1S10RR025449-01) for resources and instrumentation, are acknowledged for supporting the RES signaling research program in the Aye lab.
Footnotes
Percentage of amino acids that are cysteines in eukaryotic proteome ∼2.2% (see, for instance, MolBiolEvol. 2000 17:1232–1239).Average MW of a protein ∼55 kDa and amino acid ∼110 Da. Total number of genes in humans ∼25,000.Thus, the estimated total number of unique cysteines in human genome ∼275,000. Also note, the reported number of cysteines was 214,000 (see, for instance, Free RadicBiol Med. 2015 84:227–245). We have taken the average of these two figures and rounded up as ∼250,000.
Conflict of interest disclosure: The authors have no conflicting financial interests. A provisional patent application relating to the inhibitors resulting from the concepts proposed in this manuscript is being pursued.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abegg D, Frei R, Cerato L, Prasad Hari D, Wang C, Waser J, Adibekian A. Proteome-Wide Profiling of Targets of Cysteine reactive Small Molecules by Using EthynylBenziodoxolone Reagents. Angew Chem Int Ed Engl. 2015;54:10852–10857. doi: 10.1002/anie.201505641. [DOI] [PubMed] [Google Scholar]
- Ahn YH, Hwang Y, Liu H, Wang XJ, Zhang Y, Stephenson KK, Boronina TN, Cole RN, Dinkova-Kostova AT, Talalay P, et al. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proc Natl Acad Sci U S A. 2010;107:9590–9595. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug Metabolism and Pharmacokinetics, the Blood-Brain Barrier, and Central Nervous System Drug Discovery. NeuroRx. 2005;2:554–571. doi: 10.1602/neurorx.2.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y, Li M, Long MJ, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34:2011–2021. doi: 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Berkowitz DB, Karukurichi KR, de la Salud-Bea R, Nelson DL, McCune CD. Use of Fluorinated Functionality in Enzyme Inhibitor Development: Mechanistic and Analytical Advantages. J Fluor Chem. 2008;129:731–742. doi: 10.1016/j.jfluchem.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, Cravatt BF. Chemical proteomic map of dimethyl fumarate–sensitive cysteines in primary human T cells. Science Signaling. 2016;9:rs10–rs10. doi: 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A, Kessler BM, Casagrande R, Overkleeft HS, Wilkinson KD, Ploegh HL. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. The EMBO Journal. 2001;20:5187–5196. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu Rev Biochem. 2015;84:765–790. doi: 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codreanu SG, Liebler DC. Novel approaches to identify protein adducts produced by lipid peroxidation. Free Radic Res. 2015;49:881–887. doi: 10.3109/10715762.2015.1019348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson TW, Aberle N, Crews CM. Design and Applications of Bifunctional Small Molecules: Why Two Heads Are Better Than One. ACS Chemical Biology. 2008;3:677–692. doi: 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmastro-Greenwood M, Freeman BA, Wendell SG. Redox-dependentanti-inflammatory signaling actions of unsaturated fatty acids. Annu Rev Physiol. 2014;76:79–105. doi: 10.1146/annurev-physiol-021113-170341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitri EF, Stefano MM, Vadim NG. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol Cells. 2008;26:228–235. [PMC free article] [PubMed] [Google Scholar]
- Ekkebus R, van Kasteren SI, Kulathu Y, Scholten A, Berlin I, Geurink PP, de Jong A, Goerdayal S, Neefjes J, Heck AJ, et al. On terminal alkynes that can react with active-site cysteine nucleophiles in proteases. J Am Chem Soc. 2013;135:2867–2870. doi: 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elber R. Ligand diffusion in globins: simulations versus experiment. Curr Opin Struct Biol. 2010;20:162–167. doi: 10.1016/j.sbi.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseltine DL, Mulligan G. An historic perspective of proteasome inhibition. Semin Hematol. 2012;49:196–206. doi: 10.1053/j.seminhematol.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Fang X, Fu Y, Long MJ, Haegele JA, Ge EJ, Parvez S, Aye Y. Temporally controlled targeting of 4-hydroxynonenal to specific proteins in living cells. J Am Chem Soc. 2013;135:14496–14499. doi: 10.1021/ja405400k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Hodgson J. ADMET—turning chemicals into drugs. Nat Biotech. 2001;19:722–726. doi: 10.1038/90761. [DOI] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Hung RJ, Spaeth CS, Yesilyurt HG, Terman JR. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat Cell Biol. 2013;15:1445–1454. doi: 10.1038/ncb2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. Systems Analysis of Protein Modification and Cellular Responses Induced by Electrophile Stress. Accounts of Chemical Research. 2010;43:673–683. doi: 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future medicinal chemistry. 2010;2:949–964. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Khan S, Vihinen M. Spectrum of disease-causing mutations in protein secondary structures. BMC Struct Biol. 2007;7:56–73. doi: 10.1186/1472-6807-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomsiri C, Karplus PA, Poole LB. Cysteine-Based Redox Switches in Enzymes. Antioxidants & Redox Signaling. 2010;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowicz A, Tyler PC, Evans GB, Furneaux RH, Schramm VL. Achieving the ultimate physiological goal in transition state analogue inhibitors for purine nucleoside phosphorylase. J BiolChem. 2003;278:31465–31468. doi: 10.1074/jbc.C300259200. [DOI] [PubMed] [Google Scholar]
- Lewis-Wambi JS, Kim H, Curpan R, Grigg R, Sarker MA, Jordan VC. The selective estrogen receptor modulator bazedoxifene inhibits hormone-independent breast cancer cell growth and down-regulates estrogen receptor alpha and cyclin D1. Mol Pharmacol. 2011;80:610–620. doi: 10.1124/mol.111.072249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SM, Xie T, Westover KD, Ficarro SB, Tae HS, Gurbani D, Sim T, Marto JA, Janne PA, Crews CM, et al. Development of small molecules targeting the pseudokinase Her3. Bioorg Med Chem Lett. 2015;25:3382–3389. doi: 10.1016/j.bmcl.2015.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HY, Haegele JA, Disare MT, Lin Q, Aye Y. A generalizable platform for interrogating target- and signal-specific consequences of electrophilic modifications in redox-dependent cell signaling. J Am Chem Soc. 2015;137:6232–6244. doi: 10.1021/ja5132648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Liu J, Obando D, Liao V, Lifa T, Codd R. The many faces of the adamantyl group in drug design. Eur J Med Chem. 2011;46:1949–1963. doi: 10.1016/j.ejmech.2011.01.047. [DOI] [PubMed] [Google Scholar]
- Long MJ, Aye Y. The Die Is Cast: Precision Electrophilic Modifications Contribute to Cellular Decision Making. Chem Res Toxicol. 2016;29:1575–1582. doi: 10.1021/acs.chemrestox.6b00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MJ, Parvez S, Zhao Y, Surya SL, Wang Y, Zhang S, Aye Y. Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat Chem Biol. 2017 doi: 10.1038/nchembio.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MJ, Poganik JR, Aye Y. On-Demand Targeting: Investigating Biology with Proximity-Directed Chemistry. J Am Chem Soc. 2016;138:3610–3622. doi: 10.1021/jacs.5b12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MJ, Poganik JR, Ghosh S, Aye Y. Subcellular Redox Targeting: Bridging in Vitro and in Vivo Chemical Biology. ACS Chem Biol. 2017;12:586–600. doi: 10.1021/acschembio.6b01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomarker Research. 2013;1:13. doi: 10.1186/2050-7771-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CE, Tallman KA, Porter NA, Marnett LJ. Structure-activity analysis of diffusible lipid electrophiles associated with phospholipid peroxidation: 4-hydroxynonenal and 4-oxononenal analogues. Chem Res Toxicol. 2011;24:357–370. doi: 10.1021/tx100323m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AJ, Dick TP. Fluorescent Protein-Based Redox Probes. Antioxidants & Redox Signaling; 2010. p. 13. [DOI] [PubMed] [Google Scholar]
- Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. The Lancet Oncology. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- Muller S, Chaikuad A, Gray NS, Knapp S. The ins and outs of selective kinase inhibitor development. Nat Chem Biol. 2015;11:818–821. doi: 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- Neklesa TK, Crews CM. Chemical biology: Greasy tags for protein removal. Nature. 2012;487:308–309. doi: 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- Ninomiya T, Takigawa N, Ichihara E, Ochi N, Murakami T, Honda Y, Kubo T, Minami D, Kudo K, Tanimoto M, et al. Afatinib prolongs survival compared with gefitinib in an epidermal growth factor receptor-driven lung cancer model. Mol Cancer Ther. 2013;12:589–597. doi: 10.1158/1535-7163.MCT-12-0885. [DOI] [PubMed] [Google Scholar]
- Ono Y, Saido TC, Sorimachi H. Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov. 2016;15:854–876. doi: 10.1038/nrd.2016.212. [DOI] [PubMed] [Google Scholar]
- Ottis P, Crews CM. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem Biol. 2017;12:892–898. doi: 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- Park K, Tan EH, O'Byrne K, Zhang L, Boyer M, Mok T, Hirsh V, Yang JCH, Lee KH, Lu S, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. The Lancet Oncology. 2016;17:577–589. doi: 10.1016/S1470-2045(16)30033-X. [DOI] [PubMed] [Google Scholar]
- Parker CG, Galmozzi A, Wang Y, Correia BE, Sasaki K, Joslyn CM, Kim AS, Cavallaro CL, Lawrence RM, Johnson SR, et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell. 2017;168:527–541, e529. doi: 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S, Fu Y, Li J, Long MJ, Lin HY, Lee DK, Hu GS, Aye Y. Substoichiometric hydroxynonenylation of a single protein recapitulates whole-cell-stimulated antioxidant response. J Am ChemSoc. 2015;137:10–13. doi: 10.1021/ja5084249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S, Long MJ, Lin HY, Zhao Y, Haegele JA, Pham VN, Lee DK, Aye Y. T-REX on-demand redox targeting in live cells. Nat Protoc. 2016;11:2328–2356. doi: 10.1038/nprot.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Carroll KS. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chemical Biology. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat ChemBiol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, Soria JC. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015;26:2073–2078. doi: 10.1093/annonc/mdv319. [DOI] [PubMed] [Google Scholar]
- Poulin R, Lu L, Ackermann B, Bey P, Pegg AE. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. Journal of Biological Chemistry. 1992;267:150–158. [PubMed] [Google Scholar]
- Rose MG, Farrell MP, Schmitz JC. Thymidylate synthase: a critical target for cancer chemotherapy. Clin Colorectal Cancer. 2002;1:220–229. doi: 10.3816/CCC.2002.n.003. [DOI] [PubMed] [Google Scholar]
- Sachs G, Shin JM, Vagin O, Lambrecht N, Yakubov I, Munson K. The gastric H,K ATPase as a drug target: past, present, and future. J Clin Gastroenterol. 2007;41(Suppl 2):S226–242. doi: 10.1097/MCG.0b013e31803233b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Salami J, Crews CM. Waste disposal—An attractive strategy for cancer therapy. Science. 2017;355:1163–1167. doi: 10.1126/science.aam7340. [DOI] [PubMed] [Google Scholar]
- Santi DV, Sakai TT. Thymidylate synthetase. Model studies of inhibition by 5-trifluoromethyl-2'-deoxyuridylic acid. Biochemistry. 1971;10:3598–3607. doi: 10.1021/bi00795a018. [DOI] [PubMed] [Google Scholar]
- Schopfer FJ, Cipollina C, Freeman BA. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Natl Acad Sci U S A. 2014;111:173–178. doi: 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, Silverman RB, Schirmer T. Structures of gamma-aminobutyric acid (GABA) aminotransferase, a pyridoxal 5′-phosphate, and [2Fe-2S] cluster-containing enzyme, complexed with gamma-ethynyl-GABA and with the antiepilepsy drug vigabatrin. J Biol Chem. 2004;279:363–373. doi: 10.1074/jbc.M305884200. [DOI] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong TH, Ung PM, Palde PB, Paulsen CE, Schlessinger A, Carroll KS. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem Biol. 2016;23:837–848. doi: 10.1016/j.chembiol.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Jagt DL, Hunsaker LA, Vander Jagt TJ, Gomez MS, Gonzales DM, Deck LM, Royer RE. Inactivation of glutathione reductase by 4-hydroxynonenal and other endogenous aldehydes. Biochemical Pharmacology. 1997;53:1133–1140. doi: 10.1016/s0006-2952(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Visscher M, Arkin MR, Dansen TB. Covalent targeting of acquired cysteines in cancer. Curr Opin Chem Biol. 2016;30:61–67. doi: 10.1016/j.cbpa.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachnowsky C, Wesley NA, Fidai I, Cowan JA. Understanding the Molecular Basis of Multiple Mitochondrial Dysfunctions Syndrome 1 (MMDS1)-Impact of a Disease-Causing Gly208Cys Substitution on Structure and Activity of NFU1 in the Fe/S Cluster Biosynthetic Pathway. J Mol Biol. 2017;429:790–807. doi: 10.1016/j.jmb.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BK, Schreiber SL. The Power of Sophisticated Phenotypic Screening and ModernMechanism-of-Action Methods. Cell Chem Biol. 2016;23:3–9. doi: 10.1016/j.chembiol.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivityfor ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci Rep. 2016;6:26979. doi: 10.1038/srep26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, Furdui CM. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proceedings of theNational Academy of Sciences. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardell SE, Nelson ER, Chao CA, McDonnell DP. Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res. 2013;19:2420–2431. doi: 10.1158/1078-0432.CCR-12-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RM. Combination therapies in oncology. Nat Rev Drug Discov. 2016;15:81–82. doi: 10.1038/nrd.2016.3. [DOI] [PubMed] [Google Scholar]
- Wright FA, Lu JP, Sliter DA, Dupre N, Rouleau GA, Wojcikiewicz RJ. A Point Mutation in the Ubiquitin Ligase RNF170 That Causes Autosomal Dominant Sensory Ataxia Destabilizes the Protein and Impairs Inositol 1,4,5-Trisphosphate Receptor-mediated Ca2+ Signaling. J Biol Chem. 2015;290:3948–13957. doi: 10.1074/jbc.M115.655043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou WY, Zheng H, Du XL, Yang JL. Characterization of FGFR signaling pathway as therapeutic targets for sarcoma patients. Cancer Biol Med. 2016;13:260–268. doi: 10.20892/j.issn.2095-3941.2015.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, Pauling L. Evolutionary divergence and convergence in proteins. New York, NY, USA: Academic Press); 1965. [Google Scholar]