

Abstract
Alterations in the myelin proteolipid protein gene (PLP1) may result in rare X-linked disorders in humans such as Pelizaeus–Merzbacher disease and spastic paraplegia type 2. PLP1 expression must be tightly regulated since null mutations, as well as elevated PLP1 copy number, both lead to disease. Previous studies with Plp1-lacZ transgenic mice have demonstrated that mouse Plp1 (mPlp1) intron 1 DNA (which accounts for slightly more than half of the gene) is required for the mPlp1 promoter to drive significant levels of reporter gene expression in brain. However not much is known about the mechanisms that control expression of the human PLP1 gene (hPLP1). Therefore this review will focus on sequences in hPLP1 intron 1 DNA deemed important for hPLP1 gene activity as well as a couple of “human-specific” supplementary exons within the first intron which are utilized to generate novel splice variants, and the potential role that these sequences may play in PLP1-linked disorders.
Keywords: alternative splicing, gene expression, myelin proteolipid protein, Pelizaeus–Merzbacher disease, spastic paraplegia type 2, X-linked genetic disorders
Introduction
Myelin proteolipid protein (PLP) is the most abundant protein found in myelin from the central nervous system (CNS; Eng et al., 1968; Norton and Poduslo, 1973; Jahn et al., 2009). The tetraspan protein plays a major role in the structure and function of myelin (Klugmann et al., 1997) and is thought to be a possible immunogen for multiple sclerosis (MS; Pender and Greer, 2007; Greer and Pender, 2008). PLP is important for communication between oligodendrocytes and axons (Gruenenfelder et al., 2011). The protein serves a neuroprotective role; it is required for proper sequestration of proteins into the myelin compartment, allegedly important for axo-glial metabolism and long-term support of axons (Werner et al., 2007). Expression of the PLP1 gene is regulated in a spatiotemporal manner (Wight and Dobretsova, 2004). High levels are produced in oligodendrocytes concurrent with the active myelination period of development. Other select cells in the CNS and periphery express the gene albeit to lower levels, except for olfactory ensheathing cells (OECs) where levels are high (Dickinson et al., 1997). Its expression must be tightly regulated as evidenced by X-linked genetic diseases associated with PLP1. In humans, both segmental deletion (Raskind et al., 1991; Inoue et al., 2002) and duplication (Sistermans et al., 1998; Inoue et al., 1999; Mimault et al., 1999; Hodes et al., 2000) or higher copy number (Wolf et al., 2005) of the chromosomal region containing the PLP1 gene can lead to the dysmyelinating disorder Pelizaeus–Merzbacher disease (PMD) or the related allelic disorder, spastic paraplegia type 2 (SPG2; Inoue, 2005; Werner et al., 2007; Hobson and Kamholz, 2013). Genetically engineered mice in which the gene has been deleted, or contain added copies of PLP1 (or cDNA transgenes), demonstrate a variety of conditions ranging from late onset demyelination and axonopathy, to severe early onset demyelination (Kagawa et al., 1994; Readhead et al., 1994; Klugmann et al., 1997; Rosenbluth et al., 2006; Karim et al., 2007, 2010; Clark et al., 2013). Thus, PLP1 expression is akin to a “Goldilocks” scenario—just the right amount of protein must be produced for normal function. However, very little is known about the mechanisms that regulate expression of the human PLP1 gene (hPLP1). Previously our group has shown that the first intron of the mouse Plp1 gene (mPlp1) contains regulatory element(s), crucial for its expression (Li et al., 2002). Thus this review will focus on sequences in intron 1 that have a bearing on hPLP1 expression, and their possible significance in PLP1-related disorders.
PLP1 Gene
The PLP1 gene is located on the X chromosome in a single copy (Figure 1). The gene contains seven major exons, distributed over nearly 16 kb of DNA in human and mouse (Diehl et al., 1986; Macklin et al., 1987; Ikenaka et al., 1988). The coding sequence from these species displays 95% identity at the nucleotide level, and at least 90% identity over the promoter (proximal 150 bp) and 5′- and 3′-untranslated regions (Macklin et al., 1987; Janz and Stoffel, 1993). The first intron is relatively large (>8 kb), accounting more than half of the gene, and is moderately conserved (Figure 2); 60% identity between human and mouse.
Figure 1.
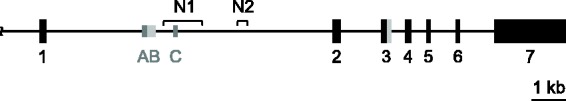
Schema of hPLP1 gene. The hPLP1 gene contains seven major exons represented by the numbered boxes; black lines indicate introns or upstream (5′-flanking) DNA. Exon 3 contains an internal splice donor site and, when utilized, results in the DM20 isoform (the PLP-specific portion of exon 3 is indicated in gray). As well, the first intron of hPLP1 contains a couple of minor exons (AB and C; symbolized by smaller gray boxes) which get incorporated separately in splice variants. Exon AB may get incorporated in its entirety, or just as select portions of the A-specific region (dark gray area). N1 and N2 indicate areas of hPLP1 intron 1 DNA orthologous to the wmN1 and wmN2 enhancer regions in mPlp1.
Figure 2.
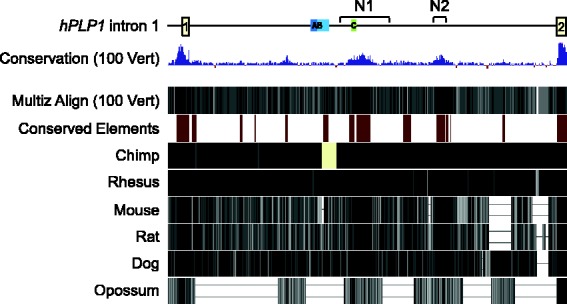
Comparison of hPLP1 intron 1 sequence with other species using the UCSC Genome Brower (GRCh38/hg38 assembly). Sequence conservation for 100 vertebrates (including human) is indicated by vertical blue lines based on Multiz Alignments (100 Vert). Conserved elements between human and six other species (chimp, Rhesus, mouse, rat, dog, and opossum) are indicated by dark red blocks. Sequence conservation of human versus chimp, Rhesus, mouse, rat, dog, or opossum is shown at the bottom.
Classic PLP1 Products (PLP/DM20) and Novel Splice Isoforms
The amino acid sequence of PLP is highly conserved in mammals; there is 100% identity among human and mouse. Two major (classic) protein products are generated due to alternative splicing in exon 3: PLP, a 276 amino acid polypeptide, and a similar protein called DM20, which lacks PLP residues 116 to 150 (Nave et al., 1987; Simons et al., 1987). DM20 is expressed prior to PLP early in development (Timsit et al., 1992), but as development proceeds, PLP becomes the predominant isoform (Kronquist et al., 1987; Gardinier and Macklin, 1988; LeVine et al., 1990; Schindler et al., 1990). In addition to the classic PLP/DM20 transcripts, which are generated in all mammals, other splice variants are produced in some species through addition of supplementary exons present within intron 1. The first intron of hPLP1 contains two such exons, exon AB and exon C, which get spliced independently of one another between hPLP1 exons 1 and 2, in the human fetal and adult CNS (Figure 1). Utilization of these supplementary exons is for the most part restricted to the human species, although exon AB or A-specific portions of it have been found in transcripts from macaque, while exon C-containing transcripts are present in bovine (Sarret et al., 2010). These novel splice variants are expressed preferentially in neurons (Sarret et al., 2010), unlike their classic (PLP/DM20) counterparts. Splice variants that incorporate only A-specific regions of exon AB (A and A′) encode proteins containing an additional nine amino acids at the N-terminus of PLP or DM20. These isoforms are trafficked to the plasma membrane as shown by transfection analysis with applicable expression constructs (Sarret et al., 2010). Thus these novel proteins could pose as targets for immune-mediated degeneration of axons/neurons in MS patients. In contrast, it is not known whether the splice variants that contain exon AB (in its entirety) or exon C encode protein or function solely as noncoding RNAs. If they encode a protein product, the start of translation is predicted to be located near the end of hPLP1 exon 4, due to introduction of multiple in-frame stop codons downstream of the traditional start site in exon 1 used to make the classic (PLP or DM20) products. This in turn would yield a peptide corresponding to the last 72 amino acids of PLP (Sarret et al., 2010). Interestingly, a secreted C-terminal PLP product has been shown to increase proliferation of oligodendrocyte lineage cells in vitro (Yamada et al., 1999). Thus, it is conceivable that exon AB- or C-containing mRNAs may encode a protein that acts as a growth factor.
Exons AB and C are not utilized in mouse even though the sequences are highly conserved (≥95% identity with human; Sarret et al., 2010). This is not due to a paucity of human-specific splicing components since the entire spectrum of splice variants were produced in a mouse cell line transfected with hPLP1 constructs (Hamdan et al., 2015). Thus lack of these “human-specific” isoforms in PLP1-null patients may have unique consequences in man. An altogether different pair of supplementary exons are present in intron 1 from mouse; mPlp1 exons 1.1 and 1.2 (Bongarzone et al., 1999; Li et al., 2009). Exon 1.1-containing splice variants encode an isoform with an additional 12 amino acid leader sequence (counting the initiator methionine residue), which causes the protein to be retained within the cell soma (Bongarzone et al., 1999).
PLP1-Related Disorders
As mentioned earlier, the essential role that PLP1 plays is illustrated by mutations in the gene that lead to PMD or SPG2; conditions characterized by delayed motor and cognitive development, followed by a gradual decline, with variation in the age of onset and rate of deterioration between patients (Inoue, 2005; Gruenenfelder et al., 2011; Hobson and Kamholz, 2013). There is variable deficiency of myelin and reduced numbers of oligodendrocytes, and some axonal loss may occur. Underlying PLP1 mutations include duplications (or higher copy number), deletions, and point (missense) mutations. More than 50% of PMD cases are caused by genomic duplications involving hPLP1, with point mutations and indels (small insertions and deletions) accounting for approximately 25% of PMD or SPG2 cases (Mimault et al., 1999). Increased PLP1 gene dosage results in elevated amounts of PLP (Ikenaka et al., 1988; Regis et al., 2009). Overexpression of the gene has been shown to cause accumulation of cholesterol and PLP in endosomes or lysosomes and perturb myelin protein composition and myelination in transgenic mice (Simons et al., 2002; Karim et al., 2007, 2010). Plp1 copy number in transgenic mouse models seems to roughly correlate with disease severity (Anderson et al., 1998, 1999; Karim et al., 2007). Hemizygous mice that harbor a Plp1 transgene show late onset demyelination and axonal degeneration, whereas dysmyelination and early death occur in homozygous mice (Readhead et al., 1994; Anderson et al., 1998, 1999). However, the cause of premature death in animals with significant levels of PLP overexpression (which is primarily evident during the early postnatal stage) remains unclear. Moreover, defects observed in Plp1 overexpressing mice are not limited solely to the myelin compartment, or even to oligodendrocytes. Plp1 overexpressing mice exhibit major oxidative phosphorylation deficits that include a 50% reduction in brain ATP levels and decreased mitochondrial membrane potentials in oligodendrocytes, as well as neurons (Hüttemann et al., 2009). Axonal damage has been observed in PMD patients with hPLP1 duplications (Laukka et al., 2016). In addition, massive microglial cell activation and inflammation throughout the brain has been observed in Plp1 overexpressing mice (Tatar et al., 2010).
In contrast, genomic deletion of PLP1 leads to the formation of compact myelin devoid of PLP. The resulting myelin sheath is physically fragile, making it susceptible to subsequent demyelination. The lack of PLP/DM20 may lead to disruption of myelin-axon interactions, resulting in axonal degeneration (Inoue, 2005; Gruenenfelder et al., 2011). In addition, patients lacking hPLP1 (as well as Plp1 null mice) develop length-dependent axonal degeneration, in the absence of demyelination and inflammation (Garbern et al., 2002). All told, these results suggest that the level of PLP1 gene expression must be tightly regulated; either too much or too little of the protein yield detrimental effects.
Transcriptional Regulatory Elements in PLP1 Intron 1
Enhancer trap studies in transgenic mice have identified a couple of conserved regions within mPlp1 intron 1 that are capable of augmenting expression of a reporter gene driven by a minimal (heat shock protein) promoter (Tuason et al., 2008). The wmN1 region encompasses 1171 bp, which appears to be important for directing high levels of expression in myelinating oligodendrocytes and OECs. The other region, called wmN2 (313 bp), conferred high level expression in Schwann cells and their progenitors, dorsal root ganglia, and OECs, and weak expression in oligodendrocyte lineage cells. In vitro studies, with hPLP1 constructs, indicate that the orthologous human wmN2 region is active in an immature oligodendrocyte (Oli-neu) cell line (Hamdan et al., 2015). Results from our laboratory indicate that the wmN1 region is essential for broad spatiotemporal expression of an hPLP1-lacZ transgene driven by its native (hPLP1) promoter (unpublished results by Hamdan et al.). Thus it is possible that mutations within these regions that disrupt enhancer function may be the underlying cause of PMD or SPG2 in patients with normal hPLP1 gene dosage, whom lack mutations in their coding sequence.
Conclusions
In summary, the first intron of hPLP1 contains several sequences important for its expression. Utilization of supplementary exons (exon AB and exon C), located in what is classically thought of as hPLP1 intron 1, leads to generation of splice variants that are primarily restricted to the human species. Some of the splice variants contain only A-specific regions of exon AB and encode a novel isoform, having an additional nine amino acids at the N-terminus of PLP or DM20. Others are predicted to encode a truncated product that potentially could act as a growth factor or may function simply as noncoding RNAs. In addition, other regions (wmN1 and wmN2) within the intron seem to behave as enhancers necessary for hPLP1 gene activity. These sequences may have special relevance for hPLP1-related disorders. For instance, mutations that disrupt enhancer function would lead to little or no expression of hPLP1, resulting in PMD or SPG2. Furthermore, splice variants generated through the addition of supplementary exons present in hPLP1 intron 1 yield novel isoforms which are primarily restricted to the human species and preferentially expressed in neurons. A lack of these isoforms in hPLP1 null patients or alternations in their levels may have unique implications in humans. Demyelinating peripheral neuropathy has been observed in humans (but not animals) that are null for PLP1 (Garbern et al., 1997). Whereas mutations in hPLP1 that alter the ratio of PLP to DM20 have been shown to cause the X-linked disorder, Hypomyelination of Early Myelinating Structures (HEMS; Kevelam et al., 2015). Thus it is conceivable that deviations from the conventional amounts of “human-specific” hPLP1 splice variants may also lead to complications.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by National Multiple Sclerosis Society (RG2705) and NIH/NINDS (R01 NS037821).
References
- Anderson T. J., Klugmann M., Thomson C. E., Schneider A., Readhead C., Nave K. A., Griffiths I. R. (1999) Distinct phenotypes associated with increasing dosage of the PLP gene: Implications for CMT1A due to PMP22 gene duplication. Ann N Y Acad Sci 883: 234–246. [PubMed] [Google Scholar]
- Anderson T. J., Schneider A., Barrie J. A., Klugmann M., McCulloch M. C., Kirkham D., Kyriakides E., Nave K. A., Griffiths I. R. (1998) Late-onset neurodegeneration in mice with increased dosage of the proteolipid protein gene. J Comp Neurol 394: 506–519. [DOI] [PubMed] [Google Scholar]
- Bongarzone E. R., Campagnoni C. W., Kampf K., Jacobs E. C., Handley V. W., Schonmann V., Campagnoni A. T. (1999) Identification of a new exon in the myelin proteolipid protein gene encoding novel protein isoforms that are restricted to the somata of oligodendrocytes and neurons. J Neurosci 19: 8349–8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Sakowski L., Sperle K., Banser L., Landel C. P., Bessert D. A., Skoff R. P., Hobson G. M. (2013) Gait abnormalities and progressive myelin degeneration in a new murine model of Pelizaeus-Merzbacher disease with tandem genomic duplication. J Neurosci 33: 11788–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson P. J., Griffiths I. R., Barrie J. M., Kyriakides E., Pollock G. F., & Barnett S. C. (1997) Expression of the dm-20 isoform of the plp gene in olfactory nerve ensheathing cells: Evidence from developmental studies. J Neurocytol 26: 181–189. [DOI] [PubMed] [Google Scholar]
- Diehl H. J., Schaich M., Budzinski R. M., Stoffel W. (1986) Individual exons encode the integral membrane domains of human myelin proteolipid protein. Proc Natl Acad Sci USA 83: 9807–9811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng L. F., Chao R. C., Gerstl B., Pratt D., Tavastsjerna M. G. (1968) The maturation of human white matter myelin: Fractionation of the myelin membrane proteins. Biochemistry 7: 4455–4465. [DOI] [PubMed] [Google Scholar]
- Garbern J. Y., Cambi F., Tang X. M., Sima A. A., Vallat J. M., Bosch E. P., Lewis R., Shy M., Sohi J., Kraft G., Chen K. L., Joshi I., Leonard D. G., Johnson W., Raskind W., Dlouhy S. R., Pratt V., Hodes M. E., Bird T., Kamholz J. (1997) Proteolipid protein is necessary in peripheral as well as central myelin. Neuron 19: 205–218. [DOI] [PubMed] [Google Scholar]
- Garbern J. Y., Yool D. A., Moore G. J., Wilds I. B., Faulk M. W., Klugmann M., Nave K. A., Sistermans E. A., van der Knapp M. S., Bird T. D., Shy M. E., Kamholz J. A., Griffiths I. R. (2002) Patients lacking the major CNS myelin protein, proteolipid 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain 125: 551–561. [DOI] [PubMed] [Google Scholar]
- Gardinier M. V., Macklin W. B. (1988) Myelin proteolipid protein gene expression in jimpy and jimpymsd mice. J Neurochem 51: 360–369. [DOI] [PubMed] [Google Scholar]
- Greer J. M., Pender M. P. (2008) Myelin proteolipid protein: An effective autoantigen and target of autoimmunity in multiple sclerosis. J Autoimmun 31: 281–287. [DOI] [PubMed] [Google Scholar]
- Gruenenfelder F. I., Thomson G., Penderis J., Edgar J. M. (2011) Axon-glial interaction in the CNS: What we have learned from mouse models of Pelizaeus-Merzbacher disease. J Anat 219: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan H., Kockara N. T., Jolly L. A., Haun S., Wight P. A. (2015) Control of human PLP1 expression through transcriptional regulatory elements and alternatively spliced exons in intron 1. ASN Neuro 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson G. M., Kamholz J. (2013) PLP1-related disorders. In: Pagon R. A., Adam M. P., Ardinger H. H., Bird T. D., Dolan C. R., Fong C. T., Smith R. J. H., Stephens K. (eds) GeneReviews® (Internet), Seattle, WA: University of Washington. [Google Scholar]
- Hodes M. E., Woodward K., Spinner N. B., Emanuel B. S., Enrico-Simon A., Kamholz J., Stambolian D., Zackai E. H., Pratt V. M., Thomas I. T., Crandall K., Dlouhy S. R., Malcolm S. (2000) Additional copies of the proteolipid protein gene causing Pelizaeus-Merzbacher disease arise by separate integration into the X chromosome. Am J Hum Genet 67: 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M., Zhang Z., Mullins C., Bessert D., Lee I., Nave K. A., Appikatla S., Skoff R. P. (2009) Different proteolipid protein mutants exhibit unique metabolic defects. ASN Neuro 1: e00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenaka K., Furuichi T., Iwasaki Y., Moriguchi A., Okano H., & Mikoshiba K. (1988) Myelin proteolipid protein gene structure and its regulation of expression in normal and jimpy mutant mice. J Mol Biol 199: 587–596. [DOI] [PubMed] [Google Scholar]
- Inoue K. (2005) PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 6: 1–16. [DOI] [PubMed] [Google Scholar]
- Inoue K., Osaka H., Imaizumi K., Nezu A., Takanashi J., Arii J., Murayama K., Ono J., Kikawa Y., Mito T., Shaffer L. G., Lupski J. R. (1999) Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: Molecular mechanism and phenotypic manifestations. Ann Neurol 45: 624–632. [PubMed] [Google Scholar]
- Inoue K., Osaka H., Thurston V. C., Clarke J. T., Yoneyama A., Rosenbarker L., Bird T. D., Hodes M. E., Shaffer L. G., Lupski J. R. (2002) Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet 71: 838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn O., Tenzer S., Werner H. B. (2009) Myelin proteomics: Molecular anatomy of an insulating sheath. Mol Neurobiol 40: 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz R., Stoffel W. (1993) Characterization of a brain-specific Sp1-like activity interacting with an unusual binding site within the myelin proteolipid protein promoter. Biol Chem Hoppe Seyler 374: 507–517. [DOI] [PubMed] [Google Scholar]
- Kagawa T., Ikenaka K., Inoue Y., Kuriyama S., Tsujii T., Nakao J., Nakajima K., Aruga J., Okano H., Mikoshiba K. (1994) Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron 13: 427–442. [DOI] [PubMed] [Google Scholar]
- Karim S. A., Barrie J. A., McCulloch M. C., Montague P., Edgar J. M., Iden D. L., Anderson T. J., Nave K. A., Griffiths I. R., McLaughlin M. (2010) PLP/DM20 expression and turnover in a transgenic mouse model of Pelizaeus-Merzbacher disease. Glia 58: 1727–1738. [DOI] [PubMed] [Google Scholar]
- Karim S. A., Barrie J. A., McCulloch M. C., Montague P., Edgar J. M., Kirkham D., Anderson T. J., Nave K. A., Griffiths I. R., McLaughlin M. (2007) PLP overexpression perturbs myelin protein composition and myelination in a mouse model of Pelizaeus-Merzbacher disease. Glia 55: 341–351. [DOI] [PubMed] [Google Scholar]
- Kevelam S. H., et al. (2015) Altered PLP1 splicing causes hypomyelination of early myelinating structures. Ann Clin Transl Neurol 2: 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugmann M., Schwab M. H., Pühlhofer A., Schneider A., Zimmermann F., Griffiths I. R., Nave K. A. (1997) Assembly of CNS myelin in the absence of proteolipid protein. Neuron 18: 59–70. [DOI] [PubMed] [Google Scholar]
- Kronquist K. E., Crandall B. F., Macklin W. B., Campagnoni A. T. (1987) Expression of myelin proteins in the developing human spinal cord: Cloning and sequencing of human proteolipid protein cDNA. J Neurosci Res 18: 395–401. [DOI] [PubMed] [Google Scholar]
- Laukka J. J., Kamholz J., Bessert D., Skoff R. P. (2016) Novel pathologic findings in patients with Pelizaeus-Merzbacher disease. Neurosci Lett 627: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine S. M., Wong D., Macklin W. B. (1990) Developmental expression of proteolipid protein and DM-20 mRNAs and proteins in the rat brain. Dev Neurosci 12: 235–250. [DOI] [PubMed] [Google Scholar]
- Li S., Greuel B. T., Meng F., Pereira G. B., Pitts A., Dobretsova A., Wight P. A. (2009) Leydig cells express the myelin proteolipid protein gene and incorporate a new alternatively spliced exon. Gene 436: 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Moore C. L., Dobretsova A., Wight P. A. (2002) Myelin proteolipid protein (Plp) intron 1 DNA is required to temporally regulate Plp gene expression in the brain. J Neurochem 83: 193–201. [DOI] [PubMed] [Google Scholar]
- Macklin W. B., Campagnoni C. W., Deininger P. L., Gardinier M. V. (1987) Structure and expression of the mouse myelin proteolipid protein gene. J Neurosci Res 18: 383–394. [DOI] [PubMed] [Google Scholar]
- Mimault C., Giraud G., Courtois V., Cailloux F., Boire J. Y., Dastugue B., Boespflug-Tanguy O. (1999) Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher disease: Duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The Clinical European Network on Brain Dysmyelinating Disease. Am J Hum Genet 65: 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave K. A., Lai C., Bloom F. E., Milner R. J. (1987) Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc Natl Acad Sci USA 84: 5665–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton W. T., Poduslo S. E. (1973) Myelination in rat brain: Changes in myelin composition during brain maturation. J Neurochem 21: 759–773. [DOI] [PubMed] [Google Scholar]
- Pender M. P., Greer J. M. (2007) Immunology of multiple sclerosis. Curr Allergy Asthma Rep 7: 285–292. [DOI] [PubMed] [Google Scholar]
- Raskind W. H., Williams C. A., Hudson L. D., Bird T. D. (1991) Complete deletion of the proteolipid protein gene (PLP) in a family with X-linked Pelizaeus-Merzbacher disease. Am J Hum Genet 49: 1355–1360. [PMC free article] [PubMed] [Google Scholar]
- Readhead C., Schneider A., Griffiths I., Nave K. A. (1994) Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron 12: 583–595. [DOI] [PubMed] [Google Scholar]
- Regis S., Grossi S., Corsolini F., Biancheri R., Filocamo M. (2009) PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts from Pelizaeus-Merzbacher disease patients. Biochim Biophys Acta 1792: 548–554. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J., Nave K. A., Mierzwa A., Schiff R. (2006) Subtle myelin defects in PLP-null mice. Glia 54: 172–182. [DOI] [PubMed] [Google Scholar]
- Sarret C., Combes P., Micheau P., Gelot A., Boespflug-Tanguy O., & Vaurs-Barriere C. (2010) Novel neuronal proteolipid protein isoforms encoded by the human myelin proteolipid protein 1 gene. Neuroscience 166: 522–538. [DOI] [PubMed] [Google Scholar]
- Schindler P., Luu B., Sorokine O., Trifilieff E., Van Dorsselaer A. (1990) Developmental study of proteolipids in bovine brain: A novel proteolipid and DM-20 appear before proteolipid protein during myelination. J Neurochem 55: 2079–2085. [DOI] [PubMed] [Google Scholar]
- Simons M., Kramer E. M., Macchi P., Rathke-Hartlieb S., Trotter J., Nave K. A., Schulz J. B. (2002) Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: Implications for Pelizaeus-Merzbacher disease. J Cell Biol 157: 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons R., Alon N., Riordan J. R. (1987) Human myelin DM-20 proteolipid protein deletion defined by cDNA sequence. Biochem Biophys Res Commun 146: 666–671. [DOI] [PubMed] [Google Scholar]
- Sistermans E. A., de Coo R. F., De Wijs I. J., Van Oost B. A. (1998) Duplication of the proteolipid protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology 50: 1749–1754. [DOI] [PubMed] [Google Scholar]
- Tatar C. L., Appikatla S., Bessert D. A., Paintlia A. S., Singh I., & Skoff R. P. (2010) Increased Plp1 gene expression leads to massive microglial cell activation and inflammation throughout the brain. ASN Neuro 2: e00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timsit S. G., Bally-Cuif L., Colman D. R., Zalc B. (1992) DM-20 mRNA is expressed during the embryonic development of the nervous system of the mouse. J Neurochem 58: 1172–1175. [DOI] [PubMed] [Google Scholar]
- Tuason M. C., Rastikerdar A., Kuhlmann T., Goujet-Zalc C., Zalc B., Dib S., Friedman H., Peterson A. (2008) Separate proteolipid protein/DM20 enhancers serve different lineages and stages of development. J Neurosci 28: 6895–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner H. B., Kuhlmann K., Shen S., Uecker M., Schardt A., Dimova K., Orfaniotou F., Dhaunchak A., Brinkmann B. G., Möbius W., Guarente L., Casaccia-Bonnefil P., Jahn O., Nave K. A. (2007) Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J Neurosci 27: 7717–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight P. A., Dobretsova A. (2004) Where, when and how much: Regulation of myelin proteolipid protein gene expression. Cell Mol Life Sci 61: 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf N. I., Sistermans E. A., Cundall M., Hobson G. M., Davis-Williams A. P., Palmer R., Stubbs P., Davies S., Endziniene M., Wu Y., Chong W. K., Malcolm S., Surtees R., Garbern J. Y., Woodward K. J. (2005) Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease. Brain 128: 743–751. [DOI] [PubMed] [Google Scholar]
- Yamada M., Ivanova A., Yamaguchi Y., Lees M. B., Ikenaka K. (1999) Proteolipid protein gene product can be secreted and exhibit biological activity during early development. J Neurosci 19: 2143–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]