

Abstract
Prostate-specific membrane antigen (PSMA) is one of the most specific cell surface markers for prostate cancer diagnosis and targeted treatment. However, achieving molecular imaging using non-invasive MRI with high resolution has yet to be achieved due to the lack of contrast agents with significantly improved relaxivity for sensitivity, targeting capabilities and metal selectivity. We have previously reported our creation of a novel class of protein Gd3+ contrast agents, ProCA32, which displayed significantly improved relaxivity while exhibiting strong Gd3+ binding selectivity over physiological metal ions. In this study, we report our effort in further developing biomarker-targeted protein MRI contrast agents for molecular imaging of PSMA. Among three PSMA targeted contrast agents engineered with addition of different molecular recognition sequences, ProCA32.PSMA exhibits a binding affinity of 1.1 ± 0.1 μM for PSMA while the metal binding affinity is maintained at 0.9 ± 0.1 × 10−22 M. In addition, ProCA32.PSMA exhibits r1 of 27.6 mM−1 s−1 and r2 of 37.9 mM−1 s−1 per Gd (55.2 and 75.8 mM−1 s−1 per molecule r1 and r2, respectively) at 1.4 T. At 7 T, ProCA32.PSMA also has r2 of 94.0 mM−1 s−1 per Gd (188.0 mM−1 s−1 per molecule) and r1 of 18.6 mM−1 s−1 per Gd (37.2 mM−1 s−1 per molecule). This contrast capability enables the first MRI enhancement dependent on PSMA expression levels in tumor bearing mice using both T1 and T2-weighted MRI at 7 T. Further development of these PSMA-targeted contrast agents are expected to be used for the precision imaging of prostate cancer at an early stage and to monitor disease progression and staging, as well as determine the effect of therapeutic treatment by non-invasive evaluation of the PSMA level using MRI.
Introduction
Prostate cancer is the most common non-skin cancer diagnosed in men and currently is the second leading cause of deaths in men in the United States.1 Current treatment methods for prostate cancer have high morbidity and a high probability for relapsing after treatment, which endangers the patient’s quality of life and survival. The growth and spread of prostate tumor are often very slow and remain undetected at the early stages of the cancer. However, if it is diagnosed early, there is a 98.9% chance of five year survival; therefore, early detection of prostate cancer becomes crucial for devising effective strategies towards curative treatment options.2
To date, biopsy is commonly used to diagnose prostate cancer recurrence or metastasis. This invasive procedure has a large sampling error especially for small tumor (<11.9 mm)3 and is associated with hemorrhaging, infections, and has the risk of spreading cancer cells. The introduction of the PSA (prostate specific antigen) test significantly decreased the mortality rate.4 However, it does not provide information about tumor location and suffers from a high false positive rate.5
Prostate specific membrane antigen (PSMA) has been shown to be one of the best diagnostic and therapeutic biomarkers for prostate cancer.6,7 PSMA is a 100 kDa glutamate carboxypeptidase II and is also a folate hydrolase and N-acetyl-α-linked acidic dipeptidase I (NAALADase I).8 It plays important roles in physiological processes such as signal transduction, receptor function, nutrient uptake and cell migration. The expression level of PSMA in normal prostate is usually 10 times higher than that in other types of organs, which is further increased ~10 folds in prostate tumors.9,10 In addition, the expression level of PSMA is increased when the androgen receptor is down regulated.11 The increasing expression level of PSMA is associated with tumor grade, pathologic stage, high Gleason score and PSA recurrence.12–14 Furthermore, there is a clear trend towards more and earlier PSA recurrence in strongly PSMA positive tumors.12 Perner et al.,13 characterized PSMA protein expression in a high-risk patient cohort by univariate Cox regression analysis to predict PSA recurrence. In their study, PSMA overexpression was significantly associated with PSA recurrence as were other prognostic markers including seminal vesicle invasion, high Gleason score, high metastatic tumor burden and the presence of extraprostatic extension. Ross et al.,14 also indicated that the incidence of PSA recurrence in patients with high PSMA expression tumors significantly increased than those with lower PSMA levels. They identified that PSMA was an independent predictor of pathologic stage by multivariate analysis for biochemical recurrence. PSMA is also expressed in tumor metastasis from prostate to spleen and bone. Studies have shown that if the cells expressing PSMA can be specifically targeted, both localized and aggressive conditions can be treated.15 Therefore, noninvasive monitoring of the PSMA expression level and three-dimensional distribution provides an attractive avenue for accurate diagnostics and staging as well as drug treatment. Targeting PSMA with a positron emission tomography (PET) imaging moiety enables more specific imaging of primary and metastasis prostate tumor than 18F-FDG-PET that has relatively low avidity of prostate cancer.2 [64Cu]6, a 64Cu labeled PSMA inhibitor, was developed by the conjugation of 64Cu chelated CB-TE2A to the lysine–glutamate urea scaffold. It shows high tumor penetration, and specific distribution to the PSMA positive tumor implanted in the mice model compared with the control PSMA negative tumor and background tissue.16
Among several clinical imaging modalities, magnetic resonance imaging (MRI) has the advantages of high resolution without depth limitation, requiring no ionized radiation and being able to image subjects repeatedly. While T2-weighted MRI has been applied to diagnose primary prostate cancer, it has limitations in differentiating between tumors since more than 70% of cancer in the peripheral zone of prostate shows decreased intensity in T2-weighted MRI. Prostatitis, scars, infarction, calcification in the peripheral zone, as well as post biopsy haemorrhage in the gland also show similar T2-weighted MRI signal decrease.17
To enhance sensitivity, contrast agents with paramagnetic metal ions are applied to generate either brighter (T1) or darker (T2) images. Clinically approved MRI contrast agents, such as Gd-DTPA, have an r1 relaxivity of 4 mM−1 s−1 and r2 relaxivity of 5 mM−1 s−1. Because of such low relaxivities, a local concentration of 0.1 mM is needed to generate a significant signal change.18 However, the expression level of biomarkers is usually relatively low (at nM or pM level).19–21 This low relaxivity limits their capability to target biomarkers such as PSMA at this level.22,23 In addition, the improper PK/PD limits their capability to capture biomarkers with high quality MRI. Therefore, there is a pressing need to develop MRI contrast agents with significantly improved relaxivity as well as PSMA-targeting capability for the early detection, disease progression, and monitoring of prostate cancer.2,15,24
In this paper, we report the development of PSMA-targeted protein MRI contrast agents for the molecular imaging of the PSMA level extending the targeting capability of our recently reported ProCA32, a protein MRI contrast agent (ProCA).25 By protein engineering, we have created a PSMA targeted MRI contrast agent, named ProCA32.PSMA, using ProCA32. ProCA32.PSMA shows high Gd3+ binding affinity and metal selectivity, relaxivity, and strong PSMA targeting capability. This is the first report to achieve molecular imaging of PSMA expression in tumor bearing mice using Gd3+-based MRI contrast agents with both T1 and T2-weighted MRI. The developed PSMA-targeted protein MRI contrast agents are expected to have applications in the molecular imaging of early stage of prostate cancer and follow drug treatment effects by noninvasive evaluation of the PSMA level using MRI.
Experimental
Protein design, expression and purification
In the contrast agent design, PSMA-targeting peptides (MAEWQPDTAHHWATLPDP for Saupw-1, SHSFSVGSGDHSPFT for 562 and LSFFSCWLRRSFSLT for 564) were linked to the C-terminal of ProCA32 by a flexible linker. These PSMA-targeted protein MRI contrast agents were named, ProCA32.wp, ProCA32.562 and ProCA32.PSMA, respectively. These PSMA-targeted MRI contrast agents were expressed according to the previously reported method.25 In brief, the E. coli competent cell strain BL21(DE3)plysS was transformed with plasmids of PSMA-targeted contrast agents. The protein expression was induced by 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) when the bacterial growth is up to the exponential phase. After IPTG induction, the culture temperature was maintained at 37 °C for 3 h, and then decreased to 25 °C overnight. Cell pellets were re-suspended in PBS buffer supplemented with benzonuclease and phenylmethanesulfonyl fluoride and were completely broken by a sonicator and cell disruptor. The supernatant of bacteria lysates was boiled at 90–95 °C for 10 min. The precipitates after boiling were removed by centrifuge. The supernatant was mixed with 3% streptomycin sulphate and placed at 4 °C overnight to precipitate DNA in the solution. On the next day, the precipitate DNA was removed by centrifuge and the supernatant was dialyzed in 10 mM HEPES buffer (pH 8.0) at 4 °C overnight. After dialysis, the protein solution was filtered by a 0.45 μm filter and further purified by fast protein liquid chromatography (FPLC) equipped with a HiTrap Q HP column. The purified protein MRI contrast agents by FPLC were confirmed by SDS-PAGE and UV spectrum. Gd3+ was loaded with these protein MRI contrast agents at a 2:1 ratio. The concentration of Gd3+ used in this paper was calibrated by Varian inductively coupled plasma optical emission spectrometers (ICP-OES) at 342.246 nm based on the standard curve generated by gadolinium standard solution (High-Purity™ Standards). 2 ppm YCl3 solution was used as an internal reference.
Cell culture
Human prostate cancer (PCa) cell lines PC3-Luc and C4-2-Luc were kindly provided by Dr Leland WK Chung (Cedars-Sinai Medical Center, Los Angeles, CA), and routinely maintained in T-medium (Invitrogen, Carlsbad, CA) with 5% fetal bovine serum (FBS). A final concentration of 400 μg ml−1 G418 was added to maintain the stable expression of luciferase. PC3, LNCaP and C4-2 cells are cultured in RPMI 1640 medium (Cellgro) supplemented with 5% FBS.
PSMA expression on LNCaP and C4-2 cells by western blot
To identify the PSMA expression in PC3, LNCaP and C4-2 cell lines, the same amount of cell lysates were applied in western blot. Proteins from the total cell lysates of PC3 and LNCaP cells were separated by 15% polyacrylamide gel electrophoresis (PAGE) and transferred to the PVDF membrane. A rabbit anti-PSMA antibody (ABcam) was applied as the primary antibody to directly interact with the membrane. The PSMA band was then visualized by using Immun-Star™ AP Chemiluminescence Kits (BioRad).
Enzyme-linked immunosorbent assay (ELISA)
LNCaP, C4-2 or PC3 cell lysates from 104 cells were coated in 96-well plates at 4 °C overnight. After being blocked with 5% bovine serum albumin (BSA), a series of different concentrations of ProCA32.wp and ProCA32.PSMA were incubated with cell lysate. After incubation at 4 °C overnight, the unbounded ProCA32 variants were washed away with 1× TBST. The homemade polyclone rabbit-anti-ProCA32 antibody was applied with 1:1000 dilution as the primary antibody against protein contrast agents. A stabilized goat-anti-rabbit HRP-conjugated antibody (Pierce) was used as the secondary antibody. After a robust wash with 1× TBST, the remaining secondary antibody in the 96 well plate was visualized by using 1-Step™ Ultra TMB-ELISA Substrate Solution (Thermo Fisher Scientific). The absorbance intensity was detected by the FLUO-star OPTIMA plate reader at an absorbance wavelength of 450 nm.
Metal binding affinity
Determining Tb3+ binding affinity of ProCAs was based on the Tb3+ luminescence resonance energy transfer (LRET) experiment.25 30 μM ProCAs was prepared in 5 mM DTPA, 50 mM HEPES, 150 mM NaCl at pH 7.2. The ratio of Tb-DTPA concentration ([Tb-DTPA]) and free DTPA concentration ([DTPA]free) were controlled by titration of TbCl3 in the system. The protein-Tb3+ LRET emission spectra were collected between 520 and 580 nm using an excitation wavelength of 280 nm. The free Tb3+ concentrations ([Tb]free) in each titration point were calculated by eqn (1)
| (1) |
where is the dissociation constant between Tb3+ and DTPA based on National Institute of Standards and Technology Standard Reference Database 46. The dissociation constant between Tb3+ and ProCA is calculated by the Hill equation (2)
| (2) |
where f is the fractional change of the LRET signal at each titration point and n is the hill number.
Gd3+ binding affinities to ProCAs were measured by the LRET competition method.25 10 μM of ProCA and 20 μM Tb3+ were incubated with 0 to 200 μM of GdCl3 at room temperature overnight. The Tb3+ LRET spectra were collected between 520 and 580 nm using an excitation wavelength of 280 nm. The apparent dissociation constants were calculated by fitting the plot of LRET peak intensities over different concentrations of Gd3+ (eqn (3)) and the dissociation constants of Gd3+ to ProCAs were calculated by eqn (4),
| (3) |
| (4) |
where f is the fractional change of the LRET signal, [Tb]T is the total Tb3+ concentration, [Gd]T is the total Gd3+ concentration in each titration point, and is the dissociation constant between Gd3+ and ProCA determined by eqn (2).
The dissociation constant between Zn2+ and PSMA-targeted ProCAs was determined by the fluorescence competition method with some modifications.26 The fluorescence of 2 μM Fluozin-1 was excited at 495 nm and the emission spectra were collected between 500 and 600 nm in the presence of 2 μM Zn2+ and different concentrations of PSMA-targeted ProCAs. The apparent dissociation constant was calculated by eqn (5),
| (5) |
where f is the fractional change of the fluorescence intensity of Fluozin-1, [Zn]T is the total Zn2+ concentration, and [ProCA]T is the total concentration of protein contrast agents at each titration point.
The dissociation constants between ProCAs and Zn2+ were then calculated by eqn (6),
| (6) |
where is determined by eqn (5), is the Zn2+ affinity to Fluozin-1 and [Fluozin]T is the total Fluozin-1 concentration.
The dissociation constants between Ca2+ and ProCAs were determined based on Tsien’s method27 with some modifications. 10 μM ProCAs were added into the calcium-buffer system containing 50 mM HEPES, 100 mM NaCl, 5 mM EGTA, at pH 7.2. The system was titrated with different concentrations of CaCl2 to alter the concentration ratio between the Ca-EGTA ([Ca-EGTA]) and free EGTA ([EGTA]free). The tryptophan (Trp) fluorescence changes were monitored under the emission spectra between 300 and 390 nm as excited at 280 nm. The free calcium concentration at each titration point was calculated by eqn (7)
| (7) |
where the dissociation constant of Ca2+ to EGTA ( ) is 1.51 × 10−7 M.27 The Kd of Ca2+ to ProCAs was determined by eqn (8)
| (8) |
where f is the fractional change of Trp fluorescence intensity, [CA]free is the free Ca2+ concentration in each titration point determined by eqn (7).
Relaxivity
Different concentrations of ProCAs were mixed with GdCl3 at a 1 : 2 ratio. The T1 and T2 relaxation times of water in the presence or absence of ProCAs were measured at 37 °C by using a 1.4 T Bruker Minispec using saturation recovery and CPMG sequence, respectively. The T1 and T2 of Gd-DTPA and ProCA32.562 were also measured by using a 7 T-Agilent scanner using saturation recovery and spin echo sequence. r1 and r2 were calculated by eqn (9)
| (9) |
where Ti,S is the relaxation time of water in the presence of a contrast agent, Ti,C is the relaxation time of water in the absence of a contrast agent, and C is the concentration of Gd3+.
Animal study
All animal experiments were performed in compliance with the National Institutes of Health guidelines and Institutional Animal Care and Use Committees from Georgia Regents University, Georgia State University and Emory University. Institutional Animal Care and Use Committees from Georgia Regents University, Georgia State University and Emory University have approved the animal experiments reported in this paper. Athymic male nude mice (Hsd: Athymic Nude-nu; six-week-old; Harlan Laboratories, Indianapolis, IN) were used to inoculate PCa tumors subcutaneously. For each mouse, two sites (over the shoulder and over the flank area) on the left side were inoculated with PC3-Luc cells, and two sites on the right side were inoculated with C4-2-Luc cells, respectively. For each site, 2.0 × 106 PCa cells (in 100 μl ice-cold PBS) were mixed with Matrigel® high concentration (Corning, Inc., Corning, NY) at a ratio of 1:1 before inoculation. Blood specimens were collected at the end points for serum prostate-specific antigen (PSA) determination using an ELISA kit from United Biotech, Inc (Mountain View, CA).
Bioluminescence imaging (BLI)
Ami X optical imaging system (Spectral Instruments Imaging, LLC, Tucson, AZ) was used for BLI of PC3-Luc and C4-2-Luc tumors. Mice were anesthetized by isoflurane, and injected with D-luciferin (Caliper Life Sciences, Waltham, MA) at a dose of 100 mg per kg body weight.
MRI
MRI of tumor bearing mice was collected at Bruker 7 T MRI scanner at Georgia Regents University (GRU). T2-weighted MRI images were collected before contrast agent injection and different time points after I.V. injection of 100 μl 5 mM ProCA32 or ProCA32.PSMA using the T2-weighted RARE sequence under the following setup: repetition time (TR) = 4.4 s, echo time (TE) = 56 ms, matrix = 256 × 256, Fov = 4 cm × 3 cm and slice thickness = 1 mm. The T2-map was collected before the contrast agent injection and at different time points after I.V. injection of 5 mM ProCA32 or ProCA32.PSMA using the MSME sequence with the following parameters: TR = 3 s, different length of TE, matrix = 128 × 128, Fov = 4 cm × 3 cm, and slice thickness = 1 mm. The final T2-map images were generated by using a ImageJ plugin MRI T2 calculator.
Immunofluorescence
Immunofluorescence staining was performed on the tissues collected from the tumor bearing mice post injection of non-targeted ProCA32 or ProCA32.PSMA for 48 hours. Tissues were embedded in O.C.T cryostat sectioning medium and cut with a thickness of 5 μm. ProCA32 and ProCA32.PSMA were stained by home-made rabbit antibody against ProCA32 and Alex Fluo 555-conjugated secondary antibody against the rabbit antibody. The nuclei of the cells were stained by DAPI.
Gd3+ distribution in tissue
After injection of non-targeted ProCA32 or ProCA32.PSMA for 48 hours, the tumor-bearing mice were euthanized and tissues from these mice were digested by 70% nitric acid. These tissue digestion solutions were further diluted by 2% nitric acid before analysis. Gd3+ concentration in tissues was determined by Varian ICP-OES at 342.246 nm based on the standard curve generated by gadolinium standard solution (High-Purity™ Standards). 2 ppm YCl3 solution was used as an internal reference.
Results
Design of protein-based MRI contrast agents with PSMA targeting capability
An ideal MRI contrast agent for molecular imaging of PSMA must have the following features. First, MRI contrast agents must have targeting moieties with affinity at least in the μM range. Second, MRI contrast agents must have high relaxivity in order to bring significant contrast difference to the tumor site before and after injection of contrast agents. Third, MRI contrast agents must have strong Gd3+ affinity and high Gd3+ selectivity over the physiological metal ions to avoid free Gd3+ being released and causing toxicity. Selectivity of a contrast agent towards Gd3+ over other physiological metal ions such as Zn2+ and Ca2+ is one of the important characteristics of an ideal MRI agent since the release of Gd3+ is related with the cause of nephrogenic systemic fibrosis.28 Additionally, because PSMA is mainly expressed on prostate tumor cells that are far away from blood vessels, MRI contrast agents with strong tumor penetration are highly desired.
To meet these criteria, we designed PSMA-targeted Gd3+-and protein-based MRI contrast agents based on the following considerations. We have previously developed a non-targeted protein based MRI contrast agent, ProCA32, by converting the calcium binding sites in rat α-parvalbumin to Gd3+ binding sites and further modified with PEG. ProCA32 shows high Gd3+ stability and at least 10 times greater per Gd3+ r1 and r2 relaxivities compared with that of clinical MRI contrast agents.25 We chose ProCA32 for further development based on its high relaxivity at clinical field strength and high field strength, such as 4.7 and 7 T respectively. Second, these contrast agents have small size (<3 nm) with efficient tumor penetration that is required for quantitative evaluation of the expression level of biomarkers. Third, targeting peptides such as Saupw-1, 562, and 564, have been shown to be able to interact with cancer cells with high expression of PSMA.24,29 We developed three PSMA-targeted protein contrast agents named ProCA32.wp, ProCA32.562 and ProCA32.PSMA by engineering these three different PSMA-targeting peptides at the C-terminal of ProCA32, respectively (Fig. 1). A flexible linker was inserted between ProCA32 and the targeting peptide to avoid the interference of the targeting capability of the peptide by the protein frame. Addition of the targeting moiety at the C-terminal of ProCA32 has less effect on the expression and folding of the protein variants. Fourth, we further PEGylated these protein contrast agents to improve their in vivo properties.30 We hypothesize that incorporation of PSMA targeting peptide to ProCA32 with a flexible linker equips ProCA32 with PSMA-targeting capacity while maintaining high metal affinity and selectivity as well as high relaxivities.
Fig. 1.
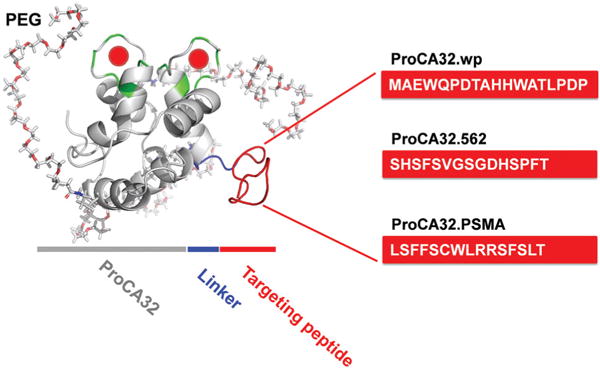
Model structure of PSMA-targeted protein MRI contrast agents for the molecular imaging of PSMA in prostate cancer. Each PSMA targeting peptide (red) was fused at the C-terminal of PEGylated ProCA32 (gray) using flexible peptide linkers (blue).
PSMA-targeted ProCAs have high relaxivities
The designed PSMA-targeted ProCAs expressed in bacteria, were purified and loaded with Gd3+ using established methods.25 Gd3+ concentration was determined by ICP-OES. A fluorescence stoichiometry study confirmed that the loading ratio of Gd3+ to ProCAs is 2:1 with the expected loading efficiency (Fig. S1†). The PSMA-targeted ProCAs were developed based on the determined crystal structure of rat α-parvalbumin (pdb code: 1RWY)31 with a determined size of ~3 nm in diameter. The molecular weight of purified ProCA32.562 determined by matrix assisted laser desorption/ionization mass spectrometry is 13568.5 Da and is in good agreement with the expected value, corresponding to the calculated size of approximately 3 nm in diameter.32 The relaxivities of ProCA32.PSMA, ProCA32.562 and ProCA32.wp were measured at 37 °C, 1.4 T. As shown in Fig. 2 and Table 1, ProCA32.PSMA, ProCA32.562 and ProCA32.wp all have about 7–8 times higher r1 and r2 relaxivities than that of Gd-DTPA. Among three PSMA-targeted contrast agent variants, ProCA32.PSMA has the highest relaxivities. The per Gd3+ r1 and r2 for ProCA32.PSMA are 27.6 ± 0.9 mM−1s−1 and 37.9 ± 1.1 mM−1 s−1, respectively. Since these contrast agents have two Gd3+ binding sites, the per particle r1 and r2 for ProCA32.PSMA are 55.2 ± 1.8 mM−1 s−1 and 75.8 ± 2.3 mM−1 s−1, respectively. Interestingly, at 7 T, ProCA32.PSMA exhibits per Gd3+ r1 and r2 of 18.6 ± 0.4 mM−1 s−1 and 94.0 ± 4.1 mM−1 s−1, respectively. The per molecule r1 and r2 for ProCA32.PSMA are 37.2 ± 0.8 mM−1 s−1 and 188.0 ± 8.2 mM−1 s−1 at 7 T, respectively. These results are consistent with our simulation of r1 and r2 at different magnetic fields based on the Solomon–Bloembergen–Morgan theory18 (Fig. S2†). To our knowledge, such high values in both r1 and r2 at both 1.4 and 7 T have not been reported before.
Fig. 2.
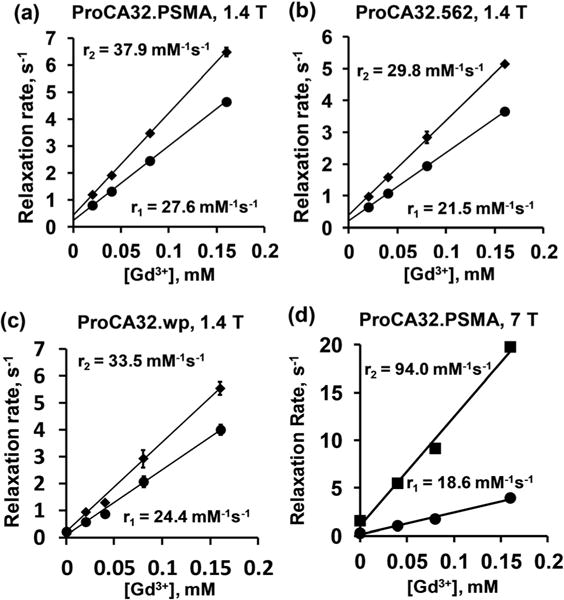
Relaxivity measurements (r1 and r2) of PSMA-targeted protein MRI contrast agents. Changes in R1 and R2 relaxation rates were plotted over various concentrations of ProCA32.PSMA (a, d), ProCA32.562 (b) and ProCA32.wp (c) at 1.4 T (a–c) and 7 T (d).
Table 1.
Summary of in vitro properties of PSMA-targeted ProCAs
| Contrast agent | ProCA32.wp | ProCA32.562 | ProCA32.PSMA |
|---|---|---|---|
| r1 (mM−1 S−1) | 24.4 ± 0.08 | 21.5 ± 0.2 | 27.6 ± 0.9 |
| r2 (mM−1 S−1) | 33.5 ± 0.1 | 29.8 ± 0.3 | 37.9 ± 1.1 |
| Kd (Tb3+) (M) | 6.8 ± 0.2 × 10−22 | 2.5 ± 0.3 × 10−22 | 3.3 ± 0.3 × 10−22 |
| Kd (Gd3+) (M) | 2.4 ± 0.3 × 10−22 | 1.3 ± 0.1 × 10−22 | 0.9 ± 0.1 × 10−22 |
| Kd (Ca2+) (M) | 1.2 ± 0.03 × 10−8 | 1.4 ± 0.01 × 10−8 | 1.7 ± 0.02 × 10−8 |
| Kd (Zn2+) (M) | 1.4 ± 0.03 × 10−6 | 2.5 ± 0.05 × 10−6 | 1.6 ± 0.04 × 10−6 |
| Kd (PSMA) (μM) | 1.1 ± 0.1 |
PSMA-targeted ProCAs have high Gd3+ binding affinity and high metal selectivity
Strong Gd3+ binding affinity is required to enable contrast agents to maintain a stable complex with Gd3+ in vivo. We applied Tb3+-DTPA buffer system and Tb3+-sensitized luminescence energy transfer (Tb3+-LRET) to determine the Tb3+ affinity to these PSMA-targeted ProCAs.25 As shown in Fig. 3a, the LRET fluorescence between Trp and Tb3+ in PSMA-targeted ProCAs increase when the free Tb3+ concentration is higher than 10−22 M, indicating an extremely strong binding between PSMA-targeted ProCAs and Tb3+. The Kd between Tb3+ and PSMA-targeted ProCAs are 3.3 ± 0.3 × 10−22, 2.5 ± 0.3 × 10−22, 6.8 ± 0.2 × 10−22 M for ProCA32.PSMA, ProCA32.562 and ProCA32.wp, respectively (Table 1). We further used a competition assay to determine the Gd3+ binding affinity to these targeted ProCAs. As shown in Fig. 3b, the Trp-Tb3+ LRET signal intensity decreased upon increase of the Gd3+ concentration, indicating that Gd3+ competes for the same metal binding sites. The Gd3+ binding affinity of ProCA32.wp, ProCA32.562 and ProCA32.PSMA are 2.4 ± 0.3 × 10−22, 1.3 ± 0.1 × 10−22, and 0.9 ± 0.1 × 10−22 M, respectively (Table 1). The Gd3+ binding affinity of PSMA-targeted ProCAs are comparable with clinical MRI contrast agents, such as Magnevist (Gd-DTPA).
Fig. 3.
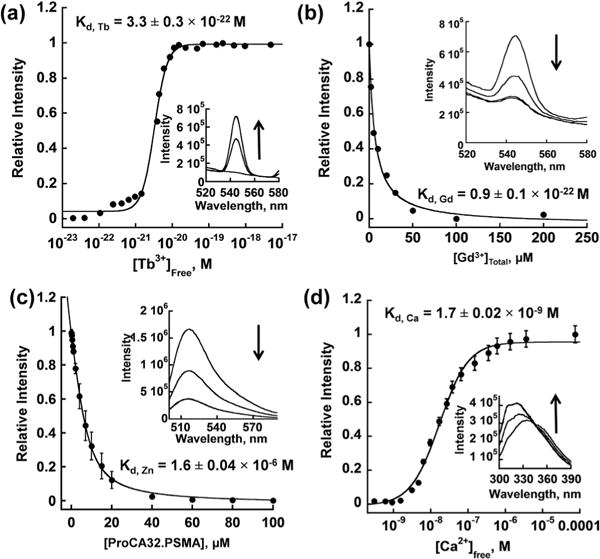
Determination of Tb3+, Gd3+, Zn2+ and Ca2+ affinity to ProCA32. PSMA. (a) Determined Tb3+ affinity to ProCA32.PSMA using the Tb-DTPA chelator buffer system. Free [Tb3+] was maintained in a range between 10−23 and 10−17 M by a tightly controlled concentration ratio between Tb-DTPA and free DTPA. The interaction between Tb3+ and ProCA32. PSMA was quantified by fluorescence intensity increase mediated by the luminescence resonance energy transfer between Trp in ProCA32.PSMA and bounded Tb3+. (b) Determined Gd3+ affinity by using Gd3+ competition assay. Different concentrations of Gd3+ were incubated with Tb3+-loaded ProCA32.PSMA. The fluorescence intensity caused by the luminescence resonance energy transfer between Trp in ProCA32.PSMA and bounded Tb3+ decrease when Gd3+ competed Tb3+ out of the metal binding pocket. (c) Determined Zn2+ affinity using Fluozin-1 competition assay. (d) Determined Ca2+ affinity to ProCA32.PSMA using the Ca2+-EGTA buffer system. Free [Ca2+] was maintained in a range between 10−10 and 10−4 M by the tightly controlled concentration of Ca2+ and EGTA. The interaction between Ca2+ and ProCA32.PSMA was monitored by the Trp fluorescence intensity increase by increasing Ca2+ concentration.
A fluorescence spectroscopic method was used to investigate the selectivity of PSMA-targeted ProCAs towards Gd3+ over Zn2+ (Fig. 3c). Additionally, the Ca2+ affinities of PSMA-targeted ProCAs were determined based on the Trp fluorescence change in the tightly controlled free calcium concentration by the Ca2+-EGTA buffer system (Fig. 3d).27 As shown in Table 1, all three PSMA-targeted ProCAs have significantly lower affinities to Zn2+ and Ca2+ than to Gd3+. For example, the binding affinities of ProCA32.PSMA for Zn2+ and Ca2+ were 1.6 × 10−6 and 1.7 × 10−8 M, respectively. The Zn2+ and Ca2+ affinities for DTPA (6.3 × 10−19 M for Zn2+ and 1.5 × 10−10 M for Ca2+) are much stronger than that of ProCAs.25 These results suggest that in addition to the high Gd3+ binding affinity and high PSMA targeting capability, the developed contrast agent is highly selective towards Gd3+ and has 1013 and 102-fold higher metal selectivity than DTPA for Zn2+ and Ca2+, respectively. To the best of our knowledge, ProCA32.PSMA has the greatest metal selectivity among all Gd3+-based contrast agents. We further applied competitive binding assays to compare the metal selectivity between ProCA32 and clinical contrast agents. Transmetallation study was performed by incubating ProCA32.PSMA with phosphate and ZnCl2 at 37 °C and ProCA32.PSMA remained intact under such conditions for more than 4000 hours (Fig. S3†). In addition, we compared Gd3+ selectivity over Zn2+ by direct incubating Omniscan (containing 20 μM Gd-DTPA-BMA and 1 μM Ca-DTPA-BMA) and 20 μM of apo-ProCA32 (ProCA32 without loading Gd3+) in the presence of 100 μM ZnCl2. This mixture has per Gd r1 of 5.8 mM−1 s−1 without heating which indicates that the Omniscan held the majority of Gd3+ under such conditions. Interestingly, after incubation at 95 °C for 30 min, the per Gd r1 of this mixture increased to 21.2 mM−1 s−1 indicating that most of the Gd3+ in the Omniscan was competed out by ProCA32 and Zn2+, and the released Gd3+ bound to ProCA32 to generate high r1 (Table S1†). These two experiments also showed that high concentration of Zn2+ is not able to compete with Gd3+ in ProCA32. Thus, these results strongly demonstrated that ProCA32 has higher Gd3+ selectivity than that of clinical contrast agents, such as Omniscan.
ProCA32.PSMA targets prostate cancer cells with high PSMA expression
We chose PC3, LNCaP and C4-2 prostate cancer cell lines to investigate the interaction between ProCA32 variants and PSMA. PC3 is an androgen-independent cell line while LNCaP and C4-2 are androgen-dependent. Western blotting showed (Fig. 4 and Fig. S4†) that PSMA expression levels in C4-2 and LNCaP cell lysates are much greater than the PC3 cell. These results are consistent with the reported literature which states that LNCaP and C4-2 cells are high in PSMA expression levels with 251 900 and 204 900 molecules per cell, respectively.22 Therefore, we use the LNCaP or C4-2 cell lines as PSMA-positive cells whereas PC3 is a PSMA-negative cell line.
Fig. 4.
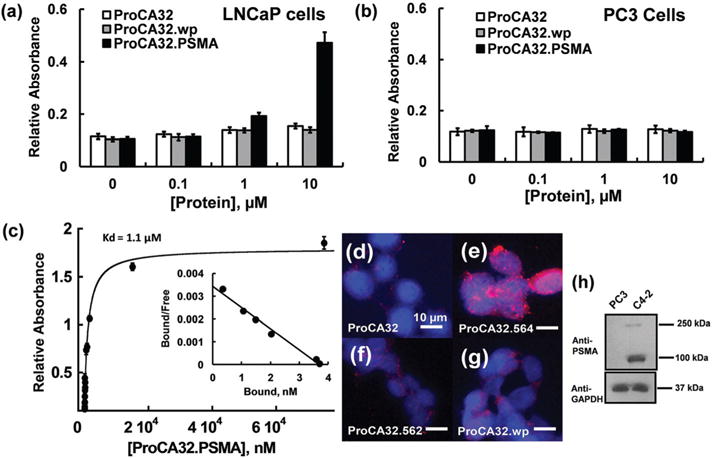
Characterization of PSMA targeting capacities of protein contrast agents at the cell level. (a, b) Comparison of the binding capability between ProCA32.wp and ProCA32.PSMA in LNCaP (a) and PC3 (b) cell lysates by ELISA. ProCA32.PSMA binds to LNCaP cell lysate at 1 and 10 μM. ProCA32.PSMA does not bind to PSMA-negative PC3 cells. (c) Determination of PSMA affinity by ELISA coupled with the Scatchard plot. (d–g) fluorescence staining of C4-2 cells incubated with ProCA32 (d), ProCA32.PSMA (e), ProCA32.562 (f), and ProCA32.wp (g). Among these PSMA-targeted ProCAs, ProCA32.PSMA shows the best PSMA targeting capacity. (h) Western blot shows the high expression of PSMA in C4-2 cells and no expression of PSMA in PC3 cells.
Next, we evaluated the PSMA targeting capacity of designed contrast agents by enzyme linked immunosorbent assay (ELISA). As shown in Fig. 4a, ProCA32.PSMA shows the highest LNCaP cell-targeting capacity among the three PSMA-targeted ProCAs when the concentration of PSMA-targeted ProCAs increased from 1 μM to 10 μM. There is no significant ELISA signal enhancement when the concentrations of these PSMA-targeted ProCAs were lower than 1 μM. Incubation of ProCA32, ProCA32.wp and ProCA32.PSMA in PSMA negative PC3 cells did not cause any significant ELISA signal increase (Fig. 4b). The affinities of PSMA-targeted ProCAs to C4-2 cells were further determined by the ELISA coupled Scatchard plot.33 As shown in Fig. 4c, ProCA32.PSMA binds to C4-2 cells with 1.1 ± 0.1 μM affinity.
The PSMA-targeting capacity of these contrast agents was further accessed by immunofluorescence imaging of C4-2 cells. As shown in Fig. 4d–g, ProCA32.PSMA has the highest fluorescence intensity among the four ProCAs, further suggesting that ProCA32.PSMA has the greatest PSMA targeting capability (Fig. 4e). On the other hand, non-targeted ProCA32 is not able to target C4-2 cells (Fig. 4d).
Taken together, ProCA32.PSMA exhibits the greatest molecular imaging capability of prostate cancer cells with high PSMA expression due to its high Gd3+ affinity, stability and high PSMA affinity.
Molecular imaging of PSMA in tumor beard mice
We then evaluate the MR imaging capacity of ProCA32.PSMA in the mice model. Luciferase labeled PC3 (PC3-Luc) and C4-2 (C4-2-Luc) cells were implanted in the left and right flank of the mice, respectively. The growth of both cell lines were confirmed by bioluminescence Imaging (Fig. 5). Compared with the C4-2 tumor without ProCA32.PSMA injection, I.V. injection of ProCA32.PSMA for 30 min–24 hours in tumor bearing mice led to approximately 50% signal decrease in the C4-2-Luc tumor at T2-weighted MRI at 7 T (Fig. 6). The percentage of signal change in C4-2-Luc cells at 48 hours post injection of ProCA32.PSMA is decreased to ~15% due to the excretion of an injected contrast agent. Tail vein injection of ProCA32. PSMA did not generate any significant SNR change in the PC3-Luc tumor without PSMA expression between 30 min and 48 hours (Fig. 6). To demonstrate that T2-weighted MRI signal change is not due to non-specific accumulation of contrast agents in tumors, we injected non-targeted ProCA32 in the mice implanted with both PC3-Luc and C4-2-Luc tumors. Injection of ProCA32 did not generate any significant signal change in PC3-Luc and C4-2-Luc tumors between 30 min and 48 hours post injection of ProCA32 (Fig. 6g). These results indicate that the T2-weighted MRI signal change in the C4-2 tumor is due to the specific interaction between PSMA and ProCA32.PSMA. However, due to 5-fold lower r1 than r2 at 7 T, the C4-2-Luc tumor has a less dramatic signal change in T1-weighted MRI (Fig. 6j and k).
Fig. 5.
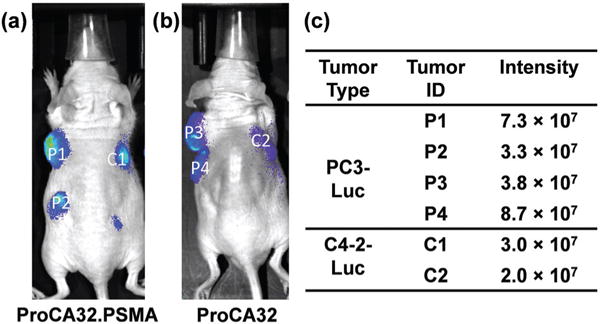
Bioluminescence imaging of nude mice implanted with PC3-Luc and C4-2-Luc tumors after injection of the substrate (a, b). Each tumor was given a different ID. The bioluminescence intensity of each tumor was listed in the table (c).
Fig. 6.
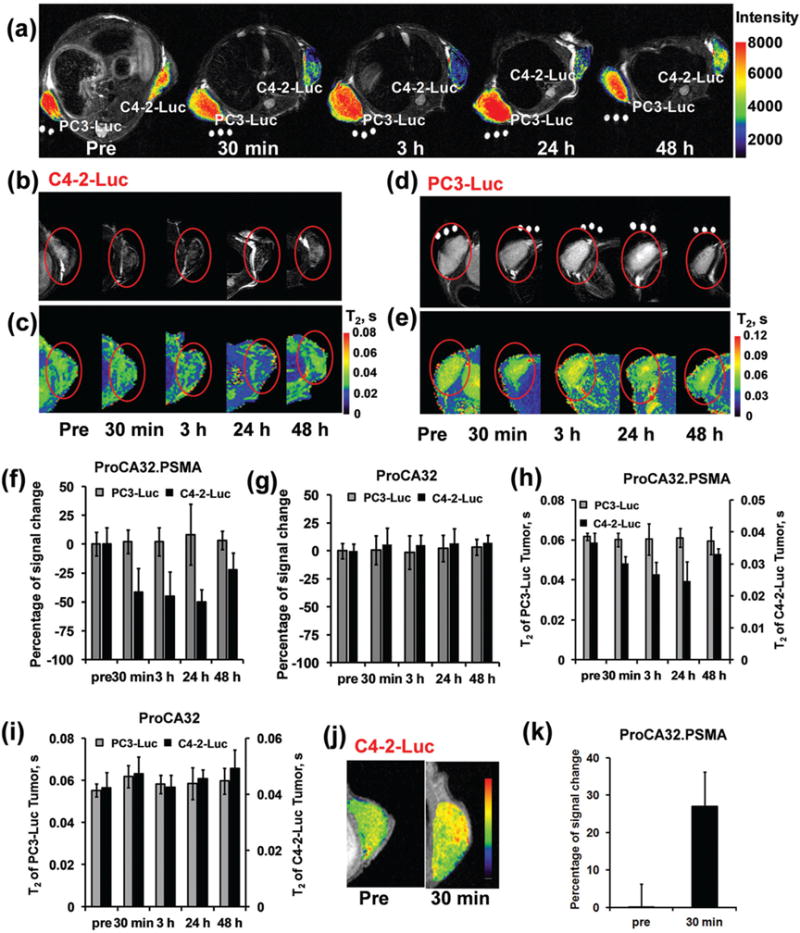
MR imaging of PSMA in xenografted mice tumors by protein based MRI contrast agents. (a) T2-weighted MRI of the mice implanted with both PC3-Luc and C4-2-Luc tumors before and after injection of ProCA32.PSMA. The PSMA-positive tumor, C4-2-Luc, exhibited decreased MRI signal intensity at 30 min–48 hours post injection of ProCA32.PSMA. After injection of ProCA32.PSMA, the PSMA-negative tumor, PC3-Luc, did not show any significant MRI signal change. (b) T2-weighted MRI of the C4-2-Luc tumor implanted mice before and after injection of ProCA32.PSMA. (c) T2 map of C4-2-Luc tumor implanted mice before and after injection of ProCA32.PSMA. (d) T2-weighted MRI of the PC3-Luc tumor implanted mice before and after injection of ProCA32.PSMA. (e) T2 map of PC3-Luc tumor implanted mice before and after injection of ProCA32.PSMA. (f) After injection of ProCA32.PSMA, the PSMA positive tumor (C4-2-Luc) shows a dramatically decreased signal between 30 min and 24 hours in T2-weighted MRI at 7 T. The PSMA negative tumor, PC-3-Luc, did not show any significant MRI signal change before and after injection of ProCA32. PSMA. (g) After injection of non-targeted ProCA32, the T2-weighted MRI signals in both PC3-Luc and C4-2-Luc were not changed due to the lack of PSMA targeting moieties in ProCA32. (h) After injection of ProCA32.PSMA, the C4-2-Luc tumor shows a decreased T2 value at 30 min, 3 hours and 24 hours. T2 of the C4-2-Luc returned to 33 ms at 48 hours post injection of ProCA32.PSMA. As a PSMA negative tumor, the T2 of PC-3-Luc did not have a significant signal change before and after injection of ProCA32.PSMA. (i) T2 of both C4-2-Luc and PC3-Luc tumors did not have significant change before and after injection of non-targeted ProCA32. (j) After I.V. injection of ProCA32.PSMA, the C4-2-Luc tumor in the xenografted mice showed increased MRI signal in the T1-weighted MRI. (k) The percentage signal increase plot of C4-2-Luc tumor in xenografted mice after injection of ProCA32.PSMA.
The percentage of MRI signal change in T2-weighted MRI is mainly due to the change of T2 relaxation time by the accumulation of MRI contrast agents. The T2 map of the same MRI slices was generated based on a set of MRI collected under different echo times. The T2 relaxation time of C4-2-Luc tumor is 37 ms at 7 T without injection of MRI contrast agents. Consistent with T2-weighted MRI, the T2 relaxation time of the C4-2-Luc tumor decreased to 30, 26, 23, and 33 ms at 30 min, 3 hours, 24 hours and 48 hours post injection of ProCA32. PSMA (Fig. 6h). Injection of non-targeted ProCA32 did not cause any significant T2 change in the C4-2-Luc tumor (Fig. 6i). Additionally, due to the lack of PSMA expression, the T2 relaxation time of PC3-Luc tumor does not change before and after injection of ProCA32.PSMA or ProCA32 (Fig. 6h and i). Three dimensional T1-weighted MRI further showed that ProCA32 is mainly distributed in the liver, kidney and blood at 45 min and 3.5 hours post injection (Fig. S5†). These MRI results were further confirmed by immunofluorescence staining and ICP-OES analysis of tissue after the injection of contrast agents for 48 hours. As shown in Fig. 7, the C4-2-Luc tumor from mice with I.V. injection of ProCA32.PSMA shows strong fluorescence staining in fluorescence imaging, indicating that ProCA32.PSMA accumulated in the C4-2-Luc tumor. As a PSMA-negative tissue, PC3-Luc tumor and heart from the same mouse did not have strong contrast agent staining. Consistent with the MRI of the mice injected with non-targeted ProCA32, C4-2-Luc, PC3-Luc and heart tissue from these mice after injection of non-targeted ProCA32 did not have strong fluorescence staining (Fig. 7). Additionally, ICP-OES analysis demonstrates that ProCA32.PSMA has three times higher Gd3+ distribution in the C4-2-Luc tumor than that of the PC3 tumor (Table 2). These results strongly support that ProCA32.PSMA has the capacity to target PSMA in C4-2-Luc tumor and efficiently change its relaxation properties for in vivo MRI.
Fig. 7.
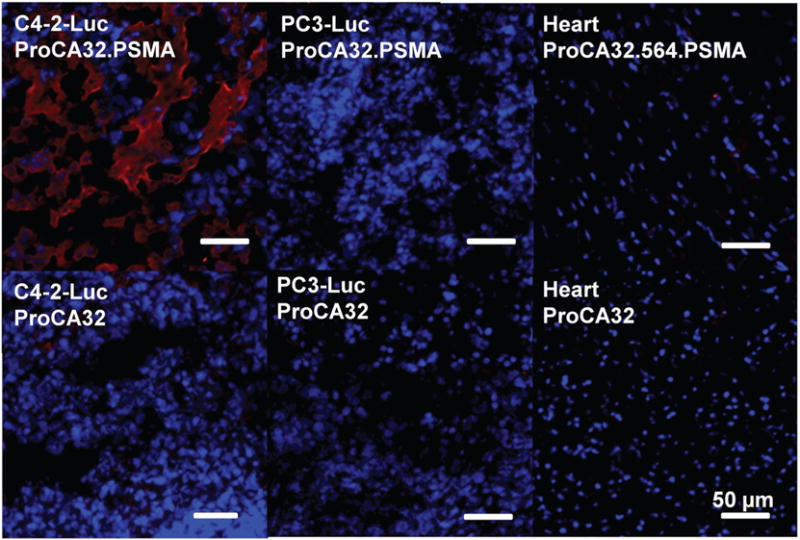
Immunofluorescence imaging of tumor implanted mice tissues (C4-2-Luc tumor, PC3-Luc tumor and heart) after injection of ProCA32. PSMA or ProCA32 for 48 hours. Red color stains ProCA32.PSMA or ProCA32. Consistent with MRI results, only C4-2-Luc tumor tissue from mice injected with ProCA32.PSMA can be positively stained, indicating ProCA32.PSMA targets the C4-2-Luc tumor by the specific interaction with PSMA.
Table 2.
Gd distribution of PC3 and C4-2 tumor implanted mice before and after injection of ProCA32.PSMA or ProCA32 for 48 hours
| Tissue | ProCA32 μg g−1 tissue | ProCA32.PSMA μg g−1 tissue |
|---|---|---|
| Muscle | 0.24 ± 0.05 | 0.26 ± 0.00 |
| Heart | 0.80 ± 0.01 | 0.76 ± 0.00 |
| Kidney | 1.55 ± 0.01 | 1.43 ± 0.01 |
| PC3-Luc | 0.44 ± 0.01 | 0.51 ± 0.01 |
| C4-2-Luc | 0.38 ± 0.00 | 1.29 ± 0.02 |
Discussion
PSMA as a prostate specific biomarker
PSMA with its surface expression has been proven to be a valuable and specific target to detect primary and metastatic prostate cancer. The expression level of PSMA in prostate cancer tissues is about 10 times higher than that in the normal prostate which is also much greater than that of other types of tissues.9,10 In addition, the PSMA level is increased when the androgen receptor is down regulated. Furthermore, PSMA is also expressed in the secondary tumors metastasized from prostate cancer.9 This specific prostate cancer property is very different from GRPR that is expressed from many types of cancers such as breast, colon, and lung. With these unique properties, PSMA has become one of the best recognized targets for the specific diagnosis and treatment of prostate cancer. For instance, a PSMA antibody-based SPECT contrast agent, named Prostascint, has been approved by the FDA. An inhibitor-based PSMA targeting molecule has also been robustly developed in the preclinical field for PET and SPET/CT imaging.34–36 Thus, the development of PSMA targeted imaging reagents is expected to enable most specific and accurate imaging for prostate cancer and its progression or treatment.
Approaches for targeting PSMA
Among several strategies available for PSMA targeting, imaging agents can be conjugated to antibody, peptide, aptamer, or inhibitors to enable the binding between PSMA and imaging agents. Aptamers and inhibitors require additional steps before conjugation with imaging agents, such modification could change their binding affinity to receptors.16,37,38 Antibodies usually have high binding affinity and specificity, but their large size limits their tumor penetration. Several antibodies against PSMA have been applied for the diagnosis of prostate cancer. However, some of them, such as 7E11,39 target the intracellular region of PSMA impairing the application of these antibodies for in vivo imaging. A monoclonal antibody in a clinical developmental phase, named J591, was designed to target the extracellular domain of PSMA for specifically delivering imaging agents or drugs to PSMA-positive prostate cells.8 Currently, PSMA antibodies have been linked to radioactive isotopes and quantum dots for the preclinical and clinical images of prostate cancer.40
Using PSMA-specific targeting peptides is an alternative way of molecular imaging PSMA with high tissue penetration without the limitation of large size. The first PSMA-targeting peptide, WQPDTAHHWATL, was screened by the phage-display technique. The monomer form of this peptide, however, lost its PSMA targeting capability.41 Wu et al. further made an improved version of the PSMA-targeting peptide by adding an additional 3 amino acids at both flanks of the WQPDTAHHWATL peptide. The improved peptide, named Saupw-1, was shown to be able to bind to PSMA of LNCaP cells.29 Additionally, the newly screened three promising peptides, named 562, 563, and 564 peptides, show strong PSMA-targeting properties at a micromolar (μM) level.24 However, the in vivo targeting capabilities of these peptides have not been fully evaluated. Our studies reported here demonstrate that we can extend ProCA32’s targeting capability by engineering the targeting peptide 564 (denoted as ProCA32. PSMA) with the strongest biomarker targeting capability. ProCA32.PSMA with 1.1 μM binding affinity to PSMA is able to monitor the biomarker expression level in tumor bearing mice by MRI.
Novel strategy to increase relaxivity for molecular MRI contrast agents
Evaluating cancer biomarkers using non-invasive imaging methods enables efficient and repetitive evaluation of disease stage and treatment. MRI, with non-invasive, non-radioactive and high soft tissue penetration properties, has become one of the leading methods for disease diagnosis. The molecular imaging of disease biomarkers using MRI, however, is underdeveloped mainly because of the lack of MRI contrast agents with high relaxivity, low toxicity, and specific targeting capacity for biomarkers. Robust progress has been made in the past several decades for the development of MRI contrast agents for the in vivo diagnosis of the disease. Clinical MRI contrast agents, such as Gd-DTPA, have low relaxivity and no targeting moieties for the surface disease biomarkers, which limits their application for molecular imaging in MRI. Increasing relaxivity often results in reduction of the relaxivity of small chelators. Improving relaxivity of MRI contrast agents while maintaining high Gd3+ binding affinity and metal selectivity is the prerequisite for achieving molecular imaging of biomarkers such as PSMA.
To overcome such limitations, various strategies have been explored by conjugating Gd3+-chelates to polymers, dendrimers, nanoparticles and proteins.42,43 An albumin based multi-modality imaging and photothermal therapy agent, HSA-Gd-IR825, was developed for the imaging and treatment of cancer cells. This agent can serve as a T1 contrast agent for imaging as well as a near infrared probe for both imaging and treatment.44 Alternatively, the modification of natural metal binding proteins to contrast agents has been demonstrated as a feasible way to improve the sensitivity of MRI contrast agents. Proteins and protein complexes with size in the nanoscale are good scaffolds for creating MRI contrast agents. Gas vesicles (typically 50–500 nm in size) formed entirely from proteins has been applied for the hyperpolarized 129Xe MRI.45 A manganese-nanocomposite with high r2 has been developed by the modification of recombinant ferritin.46 Genetically modified paramagnetic iron binding proteins, such as BM3h, with altered T1-weighted MRI signals in the presence of dopamine, have been applied for noninvasive imaging of dopaminergic signaling.47,48
In addition to its required tissue and tumor penetration, we have shown that small PEGylated protein (about 3 nm) can be used to create MRI contrast agents with optimized rotational correlation time (about 3–10 ns) to achieve both high relaxivity at medically related field strength as well as at higher field strength. By either creating Gd3+ binding sites with site-directed mutagenesis or engineering calcium binding sites in proteins to Gd3+ binding, we have developed several protein contrast agents such as ProCA1 and ProCA32 with significantly improved relaxivity.
Our previous work shows that ProCA32 has comparable Gd3+ stability to Gd-DTPA, while ProCA32 has about 10 times higher per Gd3+ relaxivity and about 20 times higher per particle relaxivity than that of Gd-DTPA while achieving unprecedented metal binding affinity and selectivities.25 Importantly, this approach also enables us to optimize relaxivity and metal binding affinity simultaneously. This overcomes the major limitations in the trading of relaxivity with metal binding stability and selectivity observed in small chelators.20,21 ProCA32 enables the detection of small metastatic liver tumors from the current size 1–2 cm to 0.24 mm.25 In this paper, we report a sensitive, specific Gd3+-based protein MRI contrast agent for the molecular imaging of PSMA in both T1 and T2-weighted MRI at high magnetic field. This is the first report of a Gd3+-based PSMA targeted contrast agent which efficiently decreases T2 of the PSMA-tumor and changes the signal in the T2-weighted MRI. We further determined the relaxivity of ProCA32.562, ProCA32.wp and ProCA32.PSMA, as summarized in Table 1. ProCA32.PSMA has high per Gd3+ relaxivities (r1 = 27.6 mM−1 s−1, r2 = 37.9 mM−1 s−1 at 1.4 T), which are 7 times higher than that of Gd-DTPA. We have shown that PSMA-targeted ProCAs have comparable Gd3+ binding affinity to Gd-DTPA. ProCA32.562 has a Gd3+ binding affinity of 1.3 × 10−22 M while ProCA32.PSMA has a Gd3+ binding affinity of 0.9 × 10−22 M. Both of them have comparable Gd3+ affinity to the clinical MRI contrast agent, Gd-DTPA (1.86 × 10−21 M). Our previous work has shown that the protein contrast agent is stable in serum for at least 12 days with high resistance for protease cleavage.25 No organ toxicity nor acute toxicity were detected for ProCAs. Additionally, the protein contrast agent had reduced Gd3+ accumulation in vivo compared with Gd-DTPA at its clinical injection dosage at 14 days post injection.25 In this paper, we further show that the protein contrast agent has higher Gd3+ selectivity over Zn2+ by a direct competitive binding assay (Table S1†). Omniscan is a clinical MRI contrast agent reported to have the best metal selectivity among all Gd3+-based clinically approved MRI contrast agents. However, incubation of apo-ProCA32 (ProCA32 without loading Gd3+) with Omniscan in the presence of Zn2+ at 95 °C for 30 minutes caused the increase of r1 from 5.8 mM−1 s−1 to 21.2 mM−1 s−1. This result suggests that our ProCA32 is able to chelate Gd3+ released from Omniscan and is consistent with its stronger Gd3+ binding affinity and metal selectivity. High stability and relaxivity of ProCAs support their promise in in vivo applications. These studies along with our previous studies further demonstrate the strategy of extending the protein contrast agent to biomarker targeting by the addition of peptide, glycans, or synthesized small ligands without sacrificing metal binding affinity and relaxivity.19–21,30
Implication for clinical molecular imaging of prostate cancer
Non-invasive molecular imaging reagents with high sensitivity, specificity and resolution are highly desired for the diagnosis and stage determination of prostate cancer. Targeting PSMA with a positron emission tomography (PET) imaging moiety enables specific imaging of the primary and metastasis prostate tumor rather than 18F-FDG-PET which has relatively low avidity for prostate cancer.2 [64Cu]6, a 64Cu labeled PSMA inhibitor developed by the conjugation of 64Cu chelated CB-TE2A to the lysine–glutamate urea scaffold, shows high tumor penetration, and specific distribution to the PSMA positive tumor implanted in the mice model compared with the control PSMA negative tumor and background tissue.16,49–52
Although MRI has superior resolution, and no radiation compared with other imaging techniques, the PSMA-targeted MRI contrast agents are less developed compared with the robust development of imaging reagents for other imaging techniques. Currently, PSMA-targeted MRI contrast agents are mainly developed by the iron oxide nanoparticle conjugated with PSMA-targeting moieties.15,53–59 Given the fact that larger size in the nanoparticle hampers efficient tumor tissue penetration, it would be imperative to develop PSMA-targeted MRI contrast agents with smaller size and higher tumor penetration efficiency. PSMA-targeted MRI contrast agents derived from high payload Gd-DOTA are also under development for T1-weighted MRI.60 Gd3+ based MRI contrast agents are routinely applied as T1-weighted MRI contrast agents. For the clinical MRI contrast agent, due to its low r2, it is not efficient to significantly change T2 of the tissue at a physiologically achievable concentration. We have previously demonstrated that non-targeted ProCA32 with increased r2 relaxivity is able to decrease the liver signal in T2-weighted MRI.25
Different from other contrast agents targeting PSMA, ProCA32.PSMA is the first Gd3+-based MRI contrast agent capable of altering both the T1 and T2-weighted MRI signal, especially T2-weighted, for the molecular imaging of PSMA. We further demonstrate that PSMA-targeted contrast agents can decrease T2 of C4-2 tumor from 37 ms to 23 ms, which causes the decrease of the MRI signal in a typical T2-weighted MRI. ProCA32.PSMA only targets the PSMA positive tumor rather than the PSMA negative tumor. Thus, ProCA32.PSMA not only has high sensitivity for the molecular imaging in T2-weighted MRI but also has high specificity for targeting the PSMA positive tumor. Additionally, ProCA32.PSMA also enhances the C4-2-Luc tumor in the T1-weighted MRI at 7 T. However, because of the relatively low r1 relaxivity at 7 T, the MRI signal change of the C4-2-Luc tumor in the T1-weighted MRI is less dramatic than that in the T2-weighted MRI. Although decrease of r1 is less desired, increased r2 at high field could improve molecular imaging by the T2-weighted MRI. In this paper, we report the first Gd3+-based MRI contrast agents for molecular imaging of PSMA by T2-weighted MRI.
The pharmacokinetics profile of ProCA32 was previously reported.25 ProCA32 has a distribution half-life of 0.15 h, and elimination half-life of 2.86 h. The volume distribution for ProCA32 is 0.08 l kg−1 and 0.13 l kg−1 for the initial state and steady state, respectively. The values of volume distribution of ProCA32 indicate that ProCA32 is distributed in the blood stream, and extracellular extravascular space of the liver and kidney. According to the volume distribution values, it is less likely for ProCA32 to have major non-specific uptake in the cells. We will perform detailed studies to examine the pharmacokinetics of ProCA32.PSMA with the addition of the 15-amino acid targeting moiety in the future.
Potential advantages of ProCA32.PSMA over PSMA-targeted low molecular weight imaging agents in PET, SPECT, and PET-MR
Comparing with reported studies of PET, SPECT and PET-MR, our developed MRI contrast agents for imaging PSMA have the main advantage of not using harmful radioactive labeling. With more than 10 times improved per particle r1 and r2 compared with small molecular MRI contrast agents, ProCA32. PSMA greatly improved the sensitivity for MRI for molecular imaging of PSMA. The small size (about 3 nm) of ProCA32. PSMA allows high tumor penetration. In addition, owing to the unique properties of MRI over other imaging modalities, ProCA32.PSMA has much higher spatial resolution for imaging PSMA in prostate cancer compared with PET/SPECT agents. Moreover, clinical MRI scanning has much lower cost compared with that of clinical PET/SPECT, making ProCA32.PSMA more cost-effective for imaging PSMA. Besides, the molecular imaging of PSMA by ProCA32.PSMA in MRI could be easily coupled with other MRI imaging techniques to assess the physiological parameters of tumors, such as tumor blood vasculature, tumor pH and oxygen level, water diffusion, and concentration of various metabolites in the tumor. Finally, since MRI mediated interventional therapy is routinely performed for the treatment of the tumor, ProCA32.PSMA-assisted molecular imaging can be easily adapted for MRI mediated tumor interventional therapy.
Advances in molecular diagnostics and staging
We have reported several protein based contrast agents with targeting capabilities to HER2 and gastric releasing peptide receptor (GRPR) using the first generation of ProCAs (ProCA1).19,26,33,61,62 Our current studies illuminate several additional advances in molecular imaging especially for prostate cancer. First, GRPR and HER-2 are expressed in multiple tumors, therefore, these GRPR and HER-2 targeted contrast agents can be applied for the imaging of tumor originating from different tissues. Because of the unique feature of PSMA expression, ProCA32.PSMA is expected to specifically target cancer originating from the prostate, including primary prostate cancer and metastasis from the prostate. Nevertheless, our development extends our imaging capability towards different types of prostate cancers. Second, ProCA32.PSMA has significantly improved metal binding affinity and selectivity compared with ProCA1.GRPR and the ProCA1.Affibody.26,33,61 Third, here we have systemically studied both T1 and T2 effects at a high field strength 7 T for protein-based MRI contrast agents. T2-weighted MRI for the molecular imaging of biomarkers has not been well studied for Gd3+-based MRI contrast agents. The capability in a broad range of field strengths from the medically related low field strength to high field strength is very unique and important. It enables us to correlate human imaging results with mice studies with the required resolution. In addition, we demonstrate a new avenue to achieve the quantitative imaging of biomarkers and monitor their temporal and spatial changes using non-invasive whole body imaging.33 Further development of these PSMA-targeted MRI contrast agents are expected to be used for the early detection of primary and metastatic prostate cancer with the desired sensitivity and specificity and for following disease progression and therapeutic treatment effects by non-invasive determination of the PSMA level using MRI.
Conclusions
We have developed a PSMA-targeted MRI contrast agent by engineering a biomarker binding peptide into a protein contrast agent ProCA32. The newly constructed PSMA-targeted MRI contrast agent exhibits PSMA binding affinity (Kd = 1.1 ± 0.1 μM) while maintaining its high r1 and r2 values, strong metal binding affinity and selectivity. Intravenous injection of ProCA32.PSMA in tumor bearing mice results in PSMA expression level dependent MRI signal change in both T1 and T2-weighted MRI at 7 T. Further development of this PSMA targeted MRI contrast agent will advance our capability for early detection of primary and metastatic prostate cancer, and for following disease progression and treatment and facilitating image-guided interventional therapy.
Supplementary Material
Acknowledgments
We thank Christopher Middleton, Drs. Roxan Ara, Nathan Yanasak and Ali Arbab for their help in MRI data collection. Drs. Mittal Pardeep, Zhi-Ren Liu for helpful discussions and Kenneth Huang, Oluwatosin Yetunde Odubade, Cassie Miller, Jesse Liu for their critical reviews of the manuscript. This work was supported by the Molecular Basis of Disease (MBD) Fellowship to M. Salarian, NIH Research Grants EB007268, GM62999, CA118113 to J. Yang, NIH Grants 1R21CA164612-01A1 and 1R41CA186498-01 to D. Wu, 1R41CA183376 and Georgia Research Alliance VentureLab to J. Yang and Inlighta Biosciences LLC, S10RR023706 (instrumentation grant) for the University of Georgia BioImaging Research Center.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr09071g
Author contribution
F. P., M. S., S. X., A. P., S. T. performed the in vitro experiments; F. P., M. S., S. X., J. Q. and K. H. were involved in the MRI data collection, imaging analysis and biodistribution studies; F. P. and S. X. performed the immunohistochemistry; F. P., S. X., J. F., X. L. K. M., D. W. were involved in tumor model development in bioluminescence imaging; F. P., M. S., S. X., M. S., D. W. and J. J. Y. analyzed the data; F. P. and J. Z. performed the immunoblot study of the PSMA expression level in tumor cells, F. P., S. X., D. W. and J. J. Y. designed the experiments, F. P., S. X., M. S., D. W. and J. J. Y. wrote the paper. All authors reviewed the manuscript.
References
- 1.Brawley OW. World J Urol. 2012;30:195–200. doi: 10.1007/s00345-012-0824-2. [DOI] [PubMed] [Google Scholar]
- 2.Lutje S, van Rij CM, Franssen GM, Fracasso G, Helfrich W, Eek A, Oyen WJ, Colombatti M, Boerman OC. Contrast Media Mol Imaging. 2015;10:28–36. doi: 10.1002/cmmi.1596. [DOI] [PubMed] [Google Scholar]
- 3.Obek C, Doganca T, Erdal S, Erdogan S, Durak H. J Urol. 2012;187:2051–2055. doi: 10.1016/j.juro.2012.01.075. [DOI] [PubMed] [Google Scholar]
- 4.Simmons MN, Berglund RK, Jones JS. Cleveland Clin J Med. 2011;78:321–331. doi: 10.3949/ccjm.78a.10104. [DOI] [PubMed] [Google Scholar]
- 5.Selley S, Donovan J, Faulkner A, Coast J, Gillatt D. Health Technol Assess. 1997;1(i):1–96. [PubMed] [Google Scholar]
- 6.Dimakakos A, Armakolas A, Koutsilieris M. BioMed Res Int. 2014;2014:890697. doi: 10.1155/2014/890697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nogueira L, Corradi R, Eastham JA. BJU Int. 2010;105:166–169. doi: 10.1111/j.1464-410X.2009.09088.x. [DOI] [PubMed] [Google Scholar]
- 8.Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Proc Natl Acad Sci U S A. 2005;102:5981–5986. doi: 10.1073/pnas.0502101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Clin Cancer Res. 1997;3:81–85. [PubMed] [Google Scholar]
- 10.Chang SS. Rev Urol. 2004;6(suppl 10):S13–S18. [PMC free article] [PubMed] [Google Scholar]
- 11.Evans MJ, Smith-Jones PM, Wongvipat J, Navarro V, Kim S, Bander NH, Larson SM, Sawyers CL. Proc Natl Acad Sci U S A. 2011;108:9578–9582. doi: 10.1073/pnas.1106383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minner S, Wittmer C, Graefen M, Salomon G, Steuber T, Haese A, Huland H, Bokemeyer C, Yekebas E, Dierlamm J, Balabanov S, Kilic E, Wilczak W, Simon R, Sauter G, Schlomm T. Prostate. 2011;71:281–288. doi: 10.1002/pros.21241. [DOI] [PubMed] [Google Scholar]
- 13.Perner S, Hofer MD, Kim R, Shah RB, Li H, Moller P, Hautmann RE, Gschwend JE, Kuefer R, Rubin MA. Hum Pathol. 2007;38:696–701. doi: 10.1016/j.humpath.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Ross JS, Sheehan CE, Fisher HA, Kaufman RP, Jr, Kaur P, Gray K, Webb I, Gray GS, Mosher R, Kallakury BV. Clin Cancer Res. 2003;9:6357–6362. [PubMed] [Google Scholar]
- 15.Bates D, Abraham S, Campbell M, Zehbe I, Curiel L. PLoS One. 2014;9:e97220. doi: 10.1371/journal.pone.0097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banerjee SR, Pullambhatla M, Foss CA, Nimmagadda S, Ferdani R, Anderson CJ, Mease RC, Pomper MG. J Med Chem. 2014;57:2657–2669. doi: 10.1021/jm401921j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heenan SD. Prostate Cancer Prostatic Dis. 2004;7:282–288. doi: 10.1038/sj.pcan.4500767. [DOI] [PubMed] [Google Scholar]
- 18.Caravan P. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 19.Qiao J, Xue S, Pu F, White N, Jiang J, Liu ZR, Yang JJ. JBIC, J Biol Inorg Chem. 2014;19:259–270. doi: 10.1007/s00775-013-1076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue S, Qiao J, Jiang J, Hubbard K, White N, Wei L, Li S, Liu ZR, Yang JJ. Med Res Rev. 2014;34:1070–1099. doi: 10.1002/med.21313. [DOI] [PubMed] [Google Scholar]
- 21.Xue S, Qiao J, Pu F, Cameron M, Yang JJ. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2013;5:163–179. doi: 10.1002/wnan.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Ma D, Olson WC, Heston WD. Mol Cancer Ther. 2011;10:1728–1739. doi: 10.1158/1535-7163.MCT-11-0191. [DOI] [PubMed] [Google Scholar]
- 23.Casneuf VF, Delrue L, Van Damme N, Demetter P, Robert P, Corot C, Duyck P, Ceelen W, Boterberg T, Peeters M. Radiat Res. 2011;175:10–20. doi: 10.1667/RR2068.1. [DOI] [PubMed] [Google Scholar]
- 24.Shen D, Xie F, Edwards WB. PLoS One. 2013;8:e68339. doi: 10.1371/journal.pone.0068339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue S, Yang H, Qiao J, Pu F, Jiang J, Hubbard K, Hekmatyar K, Langley J, Salarian M, Long RC, Bryant RG, Hu XP, Grossniklaus HE, Liu ZR, Yang JJ. Proc Natl Acad Sci U S A. 2015;112:6607–6612. doi: 10.1073/pnas.1423021112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang JJ, Yang J, Wei L, Zurkiya O, Yang W, Li S, Zou J, Zhou Y, Maniccia AL, Mao H, Zhao F, Malchow R, Zhao S, Johnson J, Hu X, Krogstad E, Liu ZR. J Am Chem Soc. 2008;130:9260–9267. doi: 10.1021/ja800736h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grynkiewicz G, Poenie M, Tsien RY. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 28.Penfield JG, Reilly RF. Semin Dial. 2008;21:129–134. doi: 10.1111/j.1525-139X.2007.00408.x. [DOI] [PubMed] [Google Scholar]
- 29.Wu P, Kudrolli TA, Chowdhury WH, Liu MM, Rodriguez R, Lupold SE. Cancer Res. 2010;70:9549–9553. doi: 10.1158/0008-5472.CAN-10-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Jiang J, Zou J, Qiao J, Xue S, Wei L, Long R, Wang L, Castiblanco A, White N, Ngo J, Mao H, Liu ZR, Yang JJ. J Inorg Biochem. 2012;107:111–118. doi: 10.1016/j.jinorgbio.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bottoms CA, Schuermann JP, Agah S, Henzl MT, Tanner JJ. Protein Sci. 2004;13:1724–1734. doi: 10.1110/ps.03571004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erickson HP. Biol Proced Online. 2009;11:32–51. doi: 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pu F, Qiao J, Xue S, Yang H, Patel A, Wei L, Hekmatyar K, Salarian M, Grossniklaus HE, Liu ZR, Yang JJ. Sci Rep. 2015;5:16214. doi: 10.1038/srep16214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hillier SM, Maresca KP, Femia FJ, Marquis JC, Foss CA, Nguyen N, Zimmerman CN, Barrett JA, Eckelman WC, Pomper MG, Joyal JL, Babich JW. Cancer Res. 2009;69:6932–6940. doi: 10.1158/0008-5472.CAN-09-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banerjee SR, Pullambhatla M, Shallal H, Lisok A, Mease RC, Pomper MG. Oncotarget. 2011;2:1244–1253. doi: 10.18632/oncotarget.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pu F, Xue S, Qiao J, Patel A, Yang JJ. Curr Protein Pept Sci. 2016;17 doi: 10.2174/1389203717666160101123725. [DOI] [PubMed] [Google Scholar]
- 37.Pavlicek J, Ptacek J, Cerny J, Byun Y, Skultetyova L, Pomper MG, Lubkowski J, Barinka C. Bioorg Med Chem Lett. 2014;24:2340–2345. doi: 10.1016/j.bmcl.2014.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian N, Sreemanthula JB, Balaji B, Kanwar JR, Biswas J, Krishnakumar S. Chem Commun. 2014;50:11810–11813. doi: 10.1039/c4cc02996h. [DOI] [PubMed] [Google Scholar]
- 39.Wynant GE, Murphy GP, Horoszewicz JS, Neal CE, Collier BD, Mitchell E, Purnell G, Tyson I, Heal A, Abdel-Nabi H, et al. Prostate. 1991;18:229–241. doi: 10.1002/pros.2990180305. [DOI] [PubMed] [Google Scholar]
- 40.Gao X, Cui Y, Levenson RM, Chung LW, Nie S. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 41.Aggarwal S, Singh P, Topaloglu O, Isaacs JT, Denmeade SR. Cancer Res. 2006;66:9171–9177. doi: 10.1158/0008-5472.CAN-06-1520. [DOI] [PubMed] [Google Scholar]
- 42.Huang CH, Tsourkas A. Curr Top Med Chem. 2013;13:411–421. doi: 10.2174/1568026611313040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Z, Lu ZR. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2013;5:1–18. doi: 10.1002/wnan.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Q, Liang C, Wang X, He J, Li Y, Liu Z. Biomaterials. 2014;35:9355–9362. doi: 10.1016/j.biomaterials.2014.07.062. [DOI] [PubMed] [Google Scholar]
- 45.Shapiro MG, Ramirez RM, Sperling LJ, Sun G, Sun J, Pines A, Schaffer DV, Bajaj VS. Nat Chem. 2014;6:629–634. doi: 10.1038/nchem.1934. [DOI] [PubMed] [Google Scholar]
- 46.Sana B, Poh CL, Lim S. Chem Commun. 2012;48:862–864. doi: 10.1039/c1cc15189d. [DOI] [PubMed] [Google Scholar]
- 47.Lee T, Cai LX, Lelyveld VS, Hai A, Jasanoff A. Science. 2014;344:533–535. doi: 10.1126/science.1249380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapiro MG, Westmeyer GG, Romero PA, Szablowski JO, Kuster B, Shah A, Otey CR, Langer R, Arnold FH, Jasanoff A. Nat Biotechnol. 2010;28:264–270. doi: 10.1038/nbt.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben Jemaa A, Bouraoui Y, Sallami S, Banasr A, Ben Rais N, Ouertani L, Nouira Y, Horchani A, Oueslati R. J Exp Clin Cancer Res. 2010;29:171. doi: 10.1186/1756-9966-29-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang X, Mease RC, Pullambhatla M, Lisok A, Chen Y, Foss CA, Wang Y, Shallal H, Edelman H, Hoye AT, Attardo G, Nimmagadda S, Pomper MG. J Med Chem. 2016;59:206–218. doi: 10.1021/acs.jmedchem.5b01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nedrow JR, Latoche JD, Day KE, Modi J, Ganguly T, Zeng D, Kurland BF, Berkman CE, Anderson CJ. Mol Imaging Biol. 2015 doi: 10.1007/s11307-015-0908-7. [DOI] [PubMed] [Google Scholar]
- 52.Ganguly T, Dannoon S, Hopkins MR, Murphy S, Cahaya H, Blecha JE, Jivan S, Drake CR, Barinka C, Jones EF, VanBrocklin HF, Berkman CE. Nucl Med Biol. 2015;42:780–787. doi: 10.1016/j.nucmedbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Y, Sun Y, Chen Y, Liu W, Jiang J, Guan W, Zhang Z, Duan Y. Int J Mol Sci. 2015;16:9573–9587. doi: 10.3390/ijms16059573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor RM, Sillerud LO. Int J Nanomed. 2012;7:4341–4352. doi: 10.2147/IJN.S34381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abdolahi M, Shahbazi-Gahrouei D, Laurent S, Sermeus C, Firozian F, Allen BJ, Boutry S, Muller RN. Contrast Media Mol Imaging. 2013;8:175–184. doi: 10.1002/cmmi.1514. [DOI] [PubMed] [Google Scholar]
- 56.Taylor RM, Huber DL, Monson TC, Ali AM, Bisoffi M, Sillerud LO. J Nanopart Res. 2011;13:4717–4729. doi: 10.1007/s11051-011-0439-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang HW, Hua MY, Liu HL, Tsai RY, Chuang CK, Chu PC, Wu PY, Chang YH, Chuang HC, Yu KJ, Pang ST. ACS Nano. 2012;6:1795–1805. doi: 10.1021/nn2048526. [DOI] [PubMed] [Google Scholar]
- 58.Yu MK, Kim D, Lee IH, So JS, Jeong YY, Jon S. Small. 2011;7:2241–2249. doi: 10.1002/smll.201100472. [DOI] [PubMed] [Google Scholar]
- 59.Zhang F, Shan L, Liu Y, Neville D, Woo JH, Chen Y, Korotcov A, Lin S, Huang S, Sridhar R, Liang W, Wang PC. Adv Healthcare Mater. 2013;2:736–744. doi: 10.1002/adhm.201200254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Banerjee SR, Ngen EJ, Rotz MW, Kakkad S, Lisok A, Pracitto R, Pullambhatla M, Chen Z, Shah T, Artemov D, Meade TJ, Bhujwalla ZM, Pomper MG. Angew Chem, Int Ed. 2015;54:10778–10782. doi: 10.1002/anie.201503417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiao J, Li S, Wei L, Jiang J, Long R, Mao H, Wei L, Wang L, Yang H, Grossniklaus HE, Liu ZR, Yang JJ. PLoS One. 2011;6:e18103. doi: 10.1371/journal.pone.0018103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei L, Li S, Yang J, Ye Y, Zou J, Wang L, Long R, Zurkiya O, Zhao T, Johnson J, Qiao J, Zhou W, Castiblanco A, Maor N, Chen Y, Mao H, Hu X, Yang JJ, Liu ZR. Mol Imaging Biol. 2011;13:416–423. doi: 10.1007/s11307-010-0342-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.