

Abstract
Background: Although imatinib mesylate has revolutionized the management of patients with gastrointestinal stromal tumor (GIST), resistance and progression almost inevitably develop with long‐term monotherapy. To enhance imatinib‐induced cytotoxicity and overcome imatinib‐resistance in GIST cells, we examined the antitumor effects of the pro‐apoptotic Bcl‐2/Bcl‐xL inhibitor ABT‐737, alone and in combination with imatinib.
Methods: We treated imatinib‐sensitive, GIST‐T1 and GIST882, and imatinib‐resistant cells with ABT‐737 alone and with imatinib. We determined the anti‐proliferative and apoptotic effects by cell viability assay, flow cytometric apoptosis and cell cycle analysis, immunoblotting, and nuclear morphology. Synergism was determined by isobologram analysis.
Results: The IC50 of single‐agent ABT‐737 at 72 h was 10 μM in imatinib‐sensitive GIST‐T1 and GIST882 cells, and 1 μM in imatinib‐resistant GIST48IM cells. ABT‐737 and imatinib combined synergistically in a time‐ and dose‐dependent manner to inhibit the proliferation and induce apoptosis of all GIST cells, as evidenced by cell viability and apoptosis assays, caspase activation, PARP cleavage, and morphologic changes. Isobologram analyses revealed strongly synergistic drug interactions, with combination indices <0.5 for most ABT‐737/imatinib combinations. Thus, clinically relevant in vitro concentrations of ABT‐737 have single‐agent antitumor activity and are synergistic in combination with imatinib.
Conclusion: We provide the first preclinical evidence that Bcl‐2/Bcl‐xL inhibition with ABT‐737 synergistically enhances imatinib‐induced cytotoxicity via apoptosis, and that direct engagement of apoptotic cell death may be an effective approach to circumvent imatinib‐resistance in GIST.
Keywords: GIST, Imatinib, ABT-737, Apoptosis
Abbreviations
- GIST
gastrointestinal stromal tumor
- IM
imatinib mesylate
- PIIM
propidium iodide
- TUNEL
TdT-mediated dUTP Nick-End Labeling
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt
- PMS
phenazine methosulfate
- EB/AO
ethidium bromide/acridine orange
- PARP
poly-ADP-ribose polymerase
- TKI
tyrosine kinase inhibitor
- IC50
half-maximal inhibitory concentration
1. Introduction
Gastrointestinal stromal tumor (GIST) is the most common sarcoma of the digestive tract, and is a paradigm for the targeted therapy of solid tumors (Benjamin et al., 2009). GISTs share a common lineage with the pacemakers of gut peristalsis, the interstitial cell of Cajal (ICC), and are characterized by expression of the receptor tyrosine kinase KIT, homolog of the Hardy‐Zuckerman feline sarcoma viral oncogene (v‐KIT). GISTs are driven by mutations in the KIT or platelet‐derived growth factor receptor‐alpha (PDGFRA) genes, which occur in 85% and 5% of tumors, respectively (Heinrich et al., 2003; Hirota et al., 1998). These mutations trigger constitutive, ligand‐independent signaling, promoting proliferation and survival. Imatinib mesylate (imatinib, Gleevec; Novartis) is a small‐molecule tyrosine kinase inhibitor (TKI) that blocks KIT and PDGFR‐α signaling. Before imatinib, patients with recurrent or metastatic GIST had overall responses of <10% with conventional chemotherapy and radiation regimens, and experienced median overall survival (OS) of 9–12 months (DeMatteo et al., 2000; Trent et al., 2003). Imatinib revolutionized the prognosis of these patients, conferring clinical benefit in 85% and extending median OS to 57 months (Blanke et al., 2008, 2008).
Clinical evidence suggests that imatinib is unable to kill all GIST cells in a tumor efficiently. Whereas 80% of patients with metastatic disease initially benefit from imatinib, 10–20% exhibit primary resistance and immediate progression (Reichardt, 2008). In responding patients, 50% develop resistance and progression by 2 years. In these patients, quiescent tumor cells are found on pathological examination, and discontinuation of imatinib leads to rapid progression of disease (Blay et al., 2007), supporting the hypothesis that KIT inhibition is cytostatic in GIST cells and is not sufficient to eradicate tumors (Liu et al., 2008; Miselli et al., 2008). Acquired resistance to imatinib is an important clinical challenge, and various mechanisms that circumvent KIT inhibition have been characterized in GIST (Liegl et al., 2008). The most important is the development of isoallelic secondary mutations in the kinase domains of KIT, which disrupt imatinib‐binding and restore oncogenic signaling (Lasota and Miettinen, 2008; Lim et al., 2008; Nishida et al., 2008). Currently, second‐generation TKIs are used for patients with imatinib‐refractory disease, but these provide limited benefit prior to progression (Goodman et al., 2007). Given the vast heterogeneity of primary and secondary KIT and PDGFRA mutations observed in GIST, and their equally‐vast resistance profiles, TKIs as a sole therapeutic strategy may not be sufficient for cure (Agaram et al., 2008; Wardelmann et al., 2006). Thus, novel therapeutic strategies must be sought to augment the current standard of care and overcome imatinib‐resistance. In this regard, addition of a pro‐apoptotic agent may enhance cell death and prevent resistant cells from emerging.
Evasion of apoptosis is a hallmark of cancer as it promotes tumor survival and resistance to therapy (Hanahan and Weinberg, 2000). Accumulating evidence suggests that cell death in GIST is controlled by the Bcl‐2 family of proteins, which regulates intrinsic apoptosis (Gordon and Fisher, 2010; Sambol et al., 2006; Steinert et al., 2006; Yang et al., 2010). The pro‐survival members of this family, Bcl‐2, Bcl‐xL, Bcl‐w, A1, and Mcl‐1, prevent apoptosis by binding and sequestering the effectors of mitochondrial permeabilization, Bcl‐2‐associated X protein (BAX) and Bcl‐2 homologous antagonist killer (BAK). Our patient‐based investigations have found that Bcl‐2 is expressed in >80% of GISTs (Steinert et al., 2006), while amplification of Bcl‐2 and Bcl‐xL loci may be common features of GIST progression, as suggested by microarray comparative genomic hybridization (Yang et al., 2010). Further, Bcl‐2 interacting mediator of apoptosis (BIM) is a Bcl‐2 homology domain 3 (BH3)‐only protein that targets and inhibits the pro‐survival Bcl‐2 proteins. BIM was recently implicated as a mediator of imatinib‐induced apoptosis in GIST cells (Gordon and Fisher, 2010), but while BIM appears to be important for apoptosis, sufficient neutralization of pro‐survival Bcl‐2 proteins may not be achievable with imatinib alone (Sambol et al., 2006). One approach to enhance GIST eradication is to concurrently inhibit oncogenic KIT signaling while actively engaging the apoptotic pathway. We thus proposed to therapeutically modulate the BIM/Bcl‐2 axis toward apoptosis via targeted inhibition of pro‐survival Bcl‐2 proteins with ABT‐737, a small‐molecule inhibitor with high affinity (K i < 1 nM) for Bcl‐2 and Bcl‐xL (Oltersdorf et al., 2005). Studies in numerous tumor models have demonstrated that ABT‐737 acts downstream and independently of TKIs to cause time‐ and dose‐dependent activation of apoptosis (Cragg et al., 2008; Jayanthan et al., 2009; Kuroda et al., 2008). In this study, we found that ABT‐737 synergizes with imatinib at physiologically–relevant concentrations to inhibit the proliferation and induce the apoptotic cell death of GIST cells, irrespective of their underlying sensitivity or resistance to kinase inhibition.
2. Materials and methods
2.1. Chemicals and antibodies
Imatinib was purchased from the M.D. Anderson Cancer Center Pharmacy. ABT‐737 and its inactive enantiomer (Compound A‐793844) were provided by Abbott (Abbott Park, IL). All three drugs were dissolved in DMSO (Fisher Bioreagents, Fair Lawn, NJ) at 10 mM, filtered through 0.22 micron filters, and stored at −20 °C, protected from light. Primary antibodies used to detect poly‐ADP‐Ribose polymerase (PARP) (rabbit polyclonal, #9542; 1:1000), caspase 3 (mouse monoclonal, #9668; 1:1000), Bcl‐2 (rabbit monoclonal, #2870; 1:1000), Bcl‐xL (rabbit monoclonal, #2764; 1:1000), and Mcl‐1 (rabbit polyclonal, #4572; 1:1000) were procured from Cell Signaling Technology (Danvers, MA). Horseradish peroxidase (HRP)‐conjugated goat anti‐mouse (sc‐2031; 1:2000) and donkey anti‐rabbit (sc‐2305; 1:2000) secondary antibodies, and primary antibody to β‐actin (mouse monoclonal, sc‐8432; 1:5000), were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2. Cell culture
The GIST‐T1 cell line was established from a patient with metastatic imatinib‐naïve GIST, and harbors an imatinib‐sensitive KIT exon 11 mutation (V560‐Y579del) (Taguchi et al., 2002). GIST882 cells were established from a patient with imatinib‐naïve GIST, and harbor imatinib‐sensitive KIT exon 13 mutations (K642E) (Tuveson et al., 2001). GIST‐T1 and GIST882 cells were kindly provided by Drs. Andrew Godwin (Fox Chase Cancer Center; Philadelphia, PA) and Jonathan Fletcher (Dana‐Farber Cancer Institute; Boston, MA), respectively, and were cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (FBS). The imatinib‐refractory cell line GIST48IM was derived, by extended culture in imatinib, from the previously described GIST48 (Rossi et al., 2006). The parental GIST48 cells, which were established from a GIST which progressed after initial response to imatinib (Bauer et al., 2006), harbor homozygous KIT exon 11 mutations (V560D) and a heterozygous secondary exon 17 mutation (D820A). GIST48IM cells were kindly provided by Dr. Anette Duensing (University of Pittsburgh Cancer Institute; Pittsburgh, PA), and cultured in Ham's F‐10 media with 15% FBS, 2 mM l‐glutamine, 1% penicillin/streptomycin, 0.1% amphotericin, 10 μg/ml gentamycin, 0.5% MITO + serum extender, and 1% bovine pituitary extract (VWR International, Roden, The Netherlands). A204 cells are derived from an unclassified sarcoma with wild‐type KIT and PDGFRA, and were purchased from the American Type Culture Collection (ATCC; Manassas, VA). A204 cells were cultured in McCoy's 5A medium supplemented with 10% heat inactivated fetal bovine serum. All cells were maintained at 37 °C in a humidified incubator, with 5% CO2.
2.3. Immunoblot analysis
Cells were harvested and washed twice with PBS, and pellets were lysed on ice for 5 min in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl, pH 7.4, containing 1% Nonidet P‐40, 150 mM NaCl, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM EDTA), with protease inhibitors 1 mM PMSF, 5 μg/ml aprotinin, and 5 μg/ml pepstatin (Sigma, St. Louis, MO), followed by sonication. Lysates were centrifuged at 14,000×g for 10 min at 4 °C, and protein concentration was measured with the Bio‐Rad Protein Assay (Bio‐Rad, Hercules, CA). Lysates were diluted 1:2 with 10 mM DTT SDS‐polyacrylamide gel electrophoresis (SDS‐PAGE) loading buffer, and heated to 70 °C for 10 min. Thirty micrograms of protein was resolved by SDS‐PAGE at 100 V for 35 min on pre‐cast 4–12% gels (Invitrogen, Carlsbad, CA), and transferred to activated polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA) by wet electrophoretic transfer (Bio‐Rad Laboratories, Hercules, CA) for 1 h at 100 V. Western blotting was performed as previously described (Dupart et al., 2009).
2.4. Analysis of cell proliferation and viability
Cell viability and proliferation were assessed using the CellTiter 96 AQueous Non‐Radioactive Cell Proliferation Assay (Promega Corporation, Madison, WI), which measures the bioreduction of 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt (MTS). Conversion of MTS into soluble formazan occurs in metabolically active cells, and 490 nm absorbance is directly proportional to the number of living cells in culture. For this experiment, 4000 cells per well were seeded onto 96‐well microtiter plates and incubated at 37 °C for 24 h. Vehicle‐control (DMSO), ABT‐737 or A‐793844 (0.1, 1, 10, 20 μM), as single agents or with imatinib (0.1, 1, 10 μM) were added in a checkerboard fashion to a final volume of 100 μL per well. After treatment for 24–72 h, 20 μL of 20:1 mixture of MTS and phenazine methosulfate (PMS) was added to each well and cells were incubated for 4 h at 37 °C. Absorbance at 490 nm was measured using KC Junior software and microplate reader (Bio‐Tek Instruments, Winooski, VT). Relative cell viability (%) was calculated as the mean absorbance of replicate treatment‐wells minus the mean absorbance of replicate background wells, divided by the mean absorbance of replicate DMSO‐treated wells minus the mean absorbance of replicate background wells, multiplied by 100.
2.5. Cell cycle analysis
Apoptosis is characterized in part by DNA fragmentation and loss of nuclear DNA content. Evaluation of propidium iodide (PI)‐stained cells by flow cytometry enables identification and quantification of apoptotic cells with hypodiploid DNA content (sub‐G1 phase). Cells were cultured in 100 mm dishes (Corning Life Sciences, Corning, NY) to 80% confluence, and treated with ABT‐737, singly or with imatinib. Non‐adherent cells were harvested by centrifugation (500×g for 5 min at 4 °C), and adherent cells were harvested by trypsinization and centrifugation. Cells were washed twice with PBS and permeabilized in ice‐cold 70% ethanol at −20 °C overnight. After washing with PBS, cells were incubated in the dark for 30 min in PBS containing RNAse‐A (1 μg/ml) and propidium iodide (50 μg/ml). DNA content was analyzed on a FACSCanto II flow cytometer using FACS Diva 6.1 software (BD Biosciences, San Jose, CA).
2.6. TUNEL apoptosis assay
To evaluate the induction of apoptotic DNA fragmentation in GIST cells, we used the DeadEnd Fluorometric TdT‐mediated dUTP Nick‐End Labeling (TUNEL) System (Promega Corporation, Madison, WI). TUNEL is widely used for detecting and quantifying apoptotic cells within cell populations, based on the incorporation of fluorescein‐conjugated dUTP by cells undergoing apoptosis‐induced DNA fragmentation. Cells were cultured and treated as in Section 2.5, non‐adherent and adherent cells were collected and combined, washed twice with PBS, fixed with 1% paraformaldehyde for 15 min at RT, washed twice with PBS, permeabilized in ice‐cold 70% ethanol and stored at −20 °C. Fixed, permeabilized cells were washed twice in PBS, equilibrated in commercial equilibration buffer (Promega Corporation, Madison, WI), and incubated with 50 μL of recombinant TdT‐fluorescein‐12‐dUTP cocktail for 2 h at 37 °C protected from light exposure. The reaction was terminated with 20 mM EDTA, cells were washed twice in PBS, and incubated in the dark for 30 min in PBS containing RNAse‐A at 1 μg/ml and 50 μg/ml PI. Apoptotic cells were defined as those positive for F‐dUTP (FITC fluorescence) and PI, and were quantified using the FACSCanto II flow cytometer and FACS Diva 6.1 software.
2.7. Ethidium bromide/acridine orange (EB/AO) apoptosis assay
For assessment of apoptosis‐related morphologic changes, cells were cultured and treated in 96‐well plates as described for MTS assay, and stained with ethidium bromide and acridine orange as described elsewhere (Ribble et al., 2005). Briefly, after 72 h, 20 μl of freshly‐prepared dual stain containing 10 μg/ml acridine orange and 5 μg/ml ethidium bromide was added to each well and the plates were centrifuged for 100×g for 5 min. Apoptosis was defined as the appearance of nuclear fragmentation and/or chromatin condensation; necrosis as the incorporation of ethidium bromide (orange stain) into normal‐sized nuclei (loss of plasma membrane integrity without pyknosis); and vital cells as normal‐sized, round nuclei staining positively for acridine orange (green stain). Apoptosis was calculated as the proportion of apoptotic cells in 4 replicate treatment‐wells, by counting at least 200 cells per well.
2.8. Data analysis: statistics and synergy
Statistical analyses were carried out using GraphPad Prism 5 software (San Diego, CA). To assess differences among three or more experimental groups, we used one‐ and two‐way analysis of variance (ANOVA). Bonferroni's multiple comparisons post‐tests were used, as needed, to compare two individual groups under different experimental conditions. To determine whether the cytotoxic interactions of ABT‐737 and imatinib in GIST cells were synergistic, additive, or antagonistic, drug effects were examined using the combination index (CI) method of Chou and Talalay (Chou, 2008; Chou and Talalay, 1981; Reynolds and Maurer, 2005). Briefly, the fraction affected (Fa) was calculated from cell viability and apoptosis assays, and CIs were generated using CalcuSyn software (Biosoft, Cambridge, UK).
3. Results
3.1. ABT‐737, but not its enantiomer A‐793844, results in significant growth inhibition of GIST cells
ABT‐737 has been shown to bind with high affinity (Ki < 1 nM), and inhibit the function of Bcl‐2 and Bcl‐xL in vivo and in vitro, whereas its enantiomer, compound A‐793844, binds these proteins with limited affinity (Ki > 100 nM for Bcl‐2 and Bcl‐xL) (Oltersdorf et al., 2005). We first determined whether the protein targets of ABT‐737, Bcl‐2 and Bcl‐xL, were expressed in GIST‐T1 and GIST882 cells, examining their protein levels, and potential imatinib‐induced alterations. Consistent with published data (Gordon and Fisher, 2010; Sambol et al., 2006), we found that GIST‐T1 and GIST882 expressed Bcl‐2 and Bcl‐xL, as well as Mcl‐1 (Figure 1A). The expression of these proteins was not affected by treatment with 1 μM imatinib for 24–72 h. We next asked whether single‐agent ABT‐737 exhibited cytotoxicity in GIST cells. To explore the antiproliferative activity of ABT‐737 and A‐793844, and determine a range of effective concentrations in GIST cells, we evaluated the viability of GIST‐T1 and GIST882 cells after treatment with incremental concentrations of ABT‐737 or A‐793844 as single agents for 24–72 h (Figure 1B). The concentrations used (0.1–20 μM) were comparable to those that have been used in preclinical studies of ABT‐737 (Tse et al., 2008). Limited anti‐proliferative activity in GIST‐T1 and GIST882 was observed for single‐agent ABT‐737 at concentrations below 1 μM. However, we found that ABT‐737 caused significant dose‐ and time‐dependent inhibition of viability at concentrations above this threshold (Two‐way ANOVA, p < 0.0001). Specifically, 10 μM and 20 μM ABT‐737 achieved approximately 50 and 95% inhibition in both cell lines, whereas 1 μM ABT‐737 reduced the viability of GIST‐T1 and GIST882 by less than 20%. Overall, the IC50 of ABT‐737 at 72 h was 10 μM for both GIST‐T1 and GIST882. Enantiomer A‐793844 did not affect the viability of either cell line, consistent with its decreased affinity for pro‐survival Bcl‐2 proteins.
Figure 1.
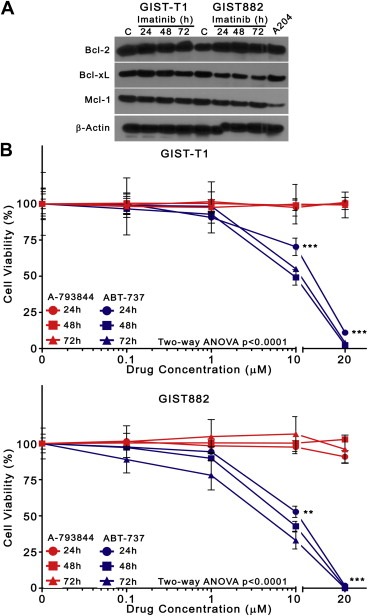
ABT‐737, but not its enantiomer A‐793844, significantly inhibits the viability of GIST cells. (A) GIST‐T1 and GIST882 cells were incubated with 1 μM imatinib or with DMSO vehicle‐control for 24–72 h, and lysates were analyzed by immunoblotting for expression of Bcl‐2, Bcl‐xL, and Mcl‐1. A lysate of untreated A204 sarcoma cells was used as a positive control to demonstrate pro‐survival Bcl‐2 protein expression (rightmost lane). β‐actin was used to demonstrate equal protein loading. (B) GIST‐T1 and GIST882 cells were incubated with increasing concentrations (0, 0.1, 1, 10, 20 μM) of ABT‐737 (blue) or enantiomer A‐793844 (red) as single agents for 24, 48, and 72 h. Inhibition of viability was determined by the MTS cell viability assay with absorbance measured at 490 nm. Symbols represent the mean of duplicate experiments; error bars represent standard deviation (SD). Three asterisks (***) represent Bonferroni's post‐test p‐value<0.0001 versus enantiomer A‐793844 at identical timepoints.
3.2. ABT‐737 and imatinib synergistically reduce the viability of GIST cells
Because single‐agent ABT‐737 proved to be a potent inhibitor of GIST viability, albeit at higher concentrations than in other tumor models (Paoluzzi et al., 2008; Shoemaker et al., 2006), we investigated its effect in combination with imatinib, hypothesizing that this rational combination would exhibit superior antiproliferative activity compared to ABT‐737 or imatinib alone. We treated cells with ABT‐737 (0, 0.1, 1, 10, 20 μM) and imatinib (0, 0.1, 1, 10 μM) in a checkerboard fashion, followed by cell viability assays at 72 h. Combined treatment resulted in significantly greater viability reductions compared with either agent alone (Figure 2A). The effect of single‐agent imatinib can be observed in the first column of each group (white), whereas the effect of increasing ABT‐737 can be observed in the second through fifth columns (shades of green). Whereas maximum growth inhibition with 0.1, 1, and 10 μM imatinib did not surpass 80% in GIST‐T1, or 60% in GIST882, the addition of ABT‐737 (>10 μM) enhanced the antiproliferative effect of imatinib, causing >90% growth inhibition in both cell lines (One‐way ANOVA, p < 0.0001). Importantly, combining imatinib with seemingly ineffective single‐agent doses of ABT‐737 (0.1 and 1 μM) appeared to potentiate the effect of ABT‐737. We thus determined whether ABT‐737 and imatinib interactions were additive or synergistic. Isobologram analysis revealed that the growth‐inhibitory effect of these drugs was strongly synergistic, with CI < 0.5 for most combinations tested (Supplementary table). The synergy results generated for GIST882 cells are depicted graphically in the Normalized Isobologram (Figure 2B, top), and Fraction affected‐Combination Index (Fa‐CI) plot (Figure 2B, bottom). Similar results are available for GIST‐T1 cells (Supplementary figure 1).
Figure 2.
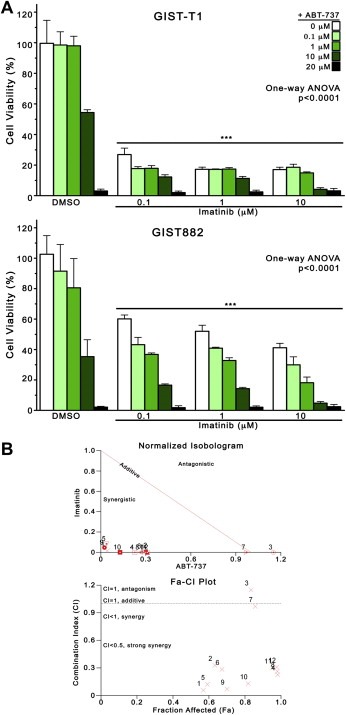
ABT‐737 and imatinib synergistically inhibit the viability of GIST cells. (A) GIST‐T1 and GIST882 cells were treated in a checkerboard fashion with increasing concentrations of imatinib (0, 0.1, 1, 10 μM) and ABT‐737 (0, 0.1, 1, 10, 20 μM) and analyzed at 72 h by MTS cell viability assay with absorbance measured at 490 nm. Columns, mean of duplicate experiments; error bars, SD. Results were analyzed by one‐way ANOVA, and three asterisks (***) represent p < 0.0001 versus DMSO control (0 μM ABT‐737 + 0 μM imatinib) by Bonferroni's post‐test comparison. (B) The Combination Indices (CI) corresponding to the Imatinib/ABT‐737 combinations tested in Figure 2A was determined by isobologram analysis (see Materials and methods). A representative normalized isobologram generated for GIST882 cells (Figure 2B, top) and a Fraction affected (Fa)‐CI plot (Figure 2B, bottom), graphically depict the interaction between imatinib and ABT‐737. Similar results for GIST‐T1 cells are available in Supplementary figure 1. Overall results of isobologram (synergy) analyses for all three cell lines are available in the Supplementary table.
3.3. ABT‐737 and imatinib induce apoptosis synergistically in imatinib‐sensitive cells
We next determined whether the potent growth‐inhibitory effects exhibited by the combination of ABT‐737 and imatinib were due to apoptosis. We treated GIST‐T1 and GIST882 cells with ABT‐737 and/or imatinib for 48 h, and quantified DNA fragmentation by cell cycle analysis (Figure 3A), and by TUNEL (Figure 3B). Overall, both methodologies revealed that combined ABT‐737 and imatinib induced greater apoptosis, compared with DMSO and with either agent alone (One‐way ANOVA, p < 0.0001). Specifically, in GIST‐T1 cells examined for sub‐G1 DNA content, there was 3% apoptosis in DMSO‐treated cells, compared with 19% with 10 μM ABT‐737 (Figure 3A, left). In combination, 10 μM ABT + 0.1 μM IM and 10 μM ABT + 1 μM IM induced 28% and 41% apoptosis, respectively. Similarly, TUNEL revealed 3% apoptosis in control GIST‐T1 cells, 13% in 10 μM ABT‐737‐treated cells, and 15% and 22% with 10 μM ABT + 0.1 μM IM and 10 μM ABT + 1 μM IM, respectively (Figure 3B, left). In GIST882 cells, there was 4% apoptosis in the control group by TUNEL, which increased to 55% and 68% with 10 μM ABT + 0.1 μM IM and 10 μM ABT + 1 μM IM, respectively (Figure 3B, right). Interestingly, we observed a substantial proportion (19%) of sub‐G1 phase GIST882 control cells (DMSO‐treated), 29% with 10 μM ABT‐737 (Figure 3A, right), and 50% with both 10 μM ABT + 0.1 μM IM and 10 μM ABT + 1 μM IM. We further confirmed that the synergy exhibited with regard to viability extended to apoptosis. As for growth inhibition, isobologram analyses revealed CI < 0.5 for most combinations with regard to apoptosis (TUNEL) (Supplementary figure 2 and Supplementary table). Apoptosis was further assessed in GIST882 cells by immunoblot analysis of caspase 3 and PARP after 72 h of treatment with ABT‐737 and imatinib as single agents (Figure 3C, left) and in combination (Figure 3C, right). As a single agent, ABT‐737 caused dose‐dependent cleavage of the inactive proform of caspase 3 (37‐kDa), and appearance of the active 19‐kDa fragment. PARP was likewise cleaved with single‐agent ABT‐737, but not imatinib, which induced minimal caspase 3 cleavage, and no PARP cleavage in GIST882 cells. The combinations 10 μM ABT + 0.1, 1, and 10 μM IM caused cleavage of both caspase 3 and PARP, beyond the effect of 10 μM ABT‐737 alone (Figure 3C, right). Notably, the levels of cleaved caspase 3 and PARP fragments did not always increase in proportion with the disappearance of their uncleaved pro‐forms, suggesting that these may be degraded rapidly under these conditions in GIST882 cells.
Figure 3.
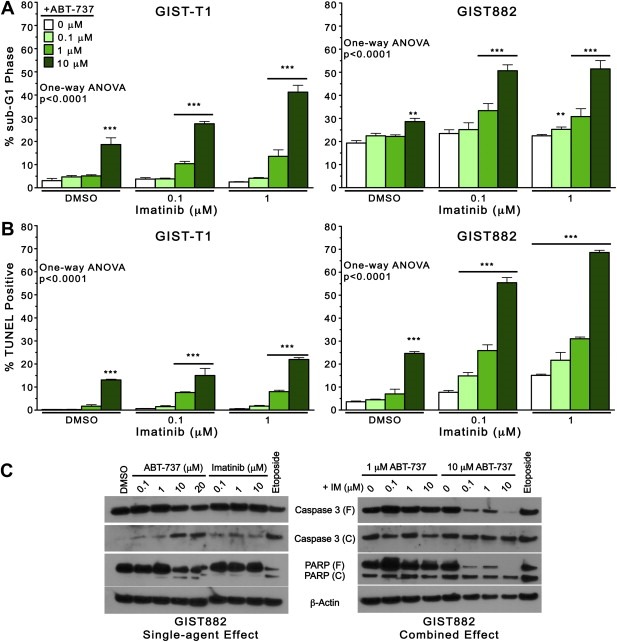
ABT‐737 and imatinib induce apoptosis synergistically in imatinib‐sensitive cells. GIST‐T1 and GIST882 cells were treated with imatinib (0, 0.1, 1 μM) and ABT‐737 (0, 0.1, 1, 10 μM) for 48 h at 37 °C and apoptosis was determined by cell cycle analysis (PI‐labeling of sub‐G1 phase cells) and TUNEL assay, using flow cytometry. Apoptosis was quantified in GIST‐T1 and GIST882 cells by (A) sub‐G1 DNA content, and (B) FITC‐positivity. Columns, mean of duplicate experiments; error bars, SD. Results were analyzed by one‐way ANOVA, and three asterisks (***) represent p < 0.0001 versus DMSO control by Bonferroni's post‐test. (C) Representative western blots of GIST882 cells treated with ABT‐737 and imatinib as single agents (Figure 3C, left) and in combination Figure 3C, right). Cells were treated for 72 h with vehicle (DMSO) or with increasing concentrations of imatinib and/or ABT‐737, and caspase 3 and PARP cleavage were assessed by immunoblotting. Treatment with Etoposide (10 μM) was used as a positive control for caspase activation. β‐actin was used to demonstrate equal protein loading. Abbreviations: (F), full length; (C), cleaved.
3.4. The morphologic features of apoptosis are induced by ABT‐737 in GIST cells
Morphologic confirmation of the characteristic features of apoptosis, including nuclear condensation and fragmentation, cell blebbing, and loss of plasma membrane integrity, is the gold standard for determination of apoptosis (Squier and Cohen, 2001). After 72 h of treatment with ABT‐737 and/or imatinib, apoptotic cell death was evaluated by nuclear morphologic assessment of ethidium bromide/acridine orange (EB/AO) dual stained cells. Representative micrographs of GIST882 cells in Figure 4 demonstrate minimal apoptosis in DMSO‐treated or imatinib‐treated cells, whereas 10 μM ABT‐737, or 10 μM ABT‐737 + 1 μM IM cause superior apoptosis induction, evidenced by chromatin fragmentation, as well as nuclear condensation. Quantitative assessment of normal versus apoptotic GIST882 cells after treatment with 1 μM imatinib and ABT‐737 (0.1, 1, 10, 20 μM) for 72 h confirmed that ABT‐737 enhanced imatinib‐induced apoptosis (Figure 4, right). Importantly, the proportion of apoptotic GIST882 cells by nuclear morphology surpassed 90% with 20 μM ABT‐737. Similar results are available for GIST‐T1 (Supplementary figure 3).
Figure 4.
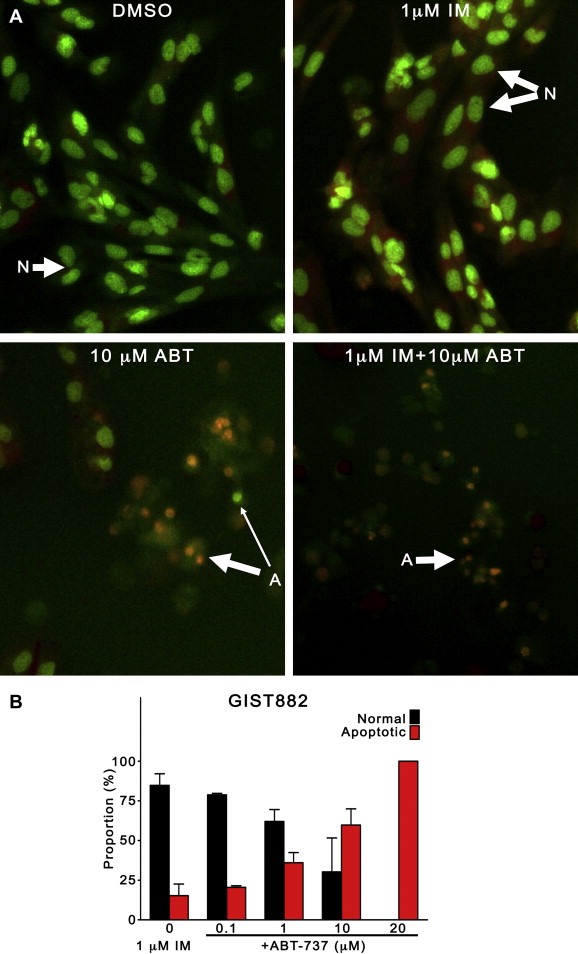
The morphologic features of apoptosis are induced by ABT‐737 in GIST cells. GIST882 cells were treated with imatinib (1 μM) alone, or in combination with ABT‐737 (0.1, 1, 10, 20 μM) for 72 h and apoptotic cell death was evaluated by assessment of nuclear morphology after ethidium bromide/acridine orange (EB/AO) staining. (A) Representative micrographs of GIST882 cells treated with vehicle (DMSO), 1 μM imatinib, 10 μM ABT‐737, or both, demonstrating nuclear fragmentation and condensation in ABT‐737‐treated cells. Original magnification, ×200. Abbreviations: (N), normal nuclei; (A), apoptotic nuclei; Thick arrow, late apoptosis; Thin Arrow, early apoptosis. (B) Quantitative assessment of normal and apoptotic GIST882 cells treated with 1 μM imatinib, alone or with ABT‐737 (0, 0.1, 1, 10, 20 μM).
3.5. Combined treatment with ABT‐737 and imatinib induces apoptosis synergistically to overcome imatinib‐resistance in GIST48IM cells
Having observed that ABT‐737 effectively enhanced apoptosis in GIST cells susceptible to KIT inhibition, we next determined whether combined treatment overcame the imatinib‐resistance phenotype exhibited by GIST48IM cells. We first examined the effect of imatinib and ABT‐737 as single agents (Figure 5A), by cell viability assays at 24, 48 and 72 h. We observed only moderate inhibition (45%) with a high concentration of imatinib (10 μM), and the IC50 of imatinib at 72 h was not reached (Figure 5A, left). In contrast, single‐agent ABT‐737 caused significant growth inhibition in GIST48IM cells, with an IC50 < 1 μM at 72 h (Figure 5A, right). We next evaluated the effect of combined ABT‐737 and imatinib on the viability of GIST48IM cells (Figure 5B), and found that combined treatment exhibited superior inhibition compared with either agent alone (One‐way ANOVA p < 0.0001). However, the degree of synergy observed between imatinib and ABT‐737 in GIST48IM was not as pronounced as in GIST‐T1 or GIST882 cells. Indeed, while isobologram analysis confirmed that the interactions were mostly synergistic in GIST48IM, we also observed three antagonistic, and two nearly‐additive combinations in this cell line (Figure 5C and Supplementary table). This may be explained by observing that, at doses above 10 μM ABT‐737, adding imatinib does not appear to significantly enhance growth inhibition. To determine whether reductions of GIST48IM cell viability were due to apoptosis, we examined the cells morphologically after treatment with ABT‐737 and imatinib for 72 h. Representative micrographs of EB/AO‐stained GIST48IM cells demonstrate that this cell line exhibits greater apoptosis at baseline (DMSO‐treated) than either GIST‐T1 or GIST882 cells (Figure 5D). Moreover, 10 μM ABT‐737, with or without 1 μM imatinib, but not 1 μM imatinib, induced the appearance of characteristic features of apoptosis. Quantification of normal and apoptotic cells treated with 1 μM imatinib and increasing concentrations of ABT‐737 (0, 0.1, 1, 10, 20 μM) confirmed that the proportion of apoptotic cells increased proportionally with ABT‐737 dose, to a maximum close to 100% with 20 μM ABT‐737 (Figure 5E). Using immunoblotting, we also examined the expression of Bcl‐2, Bcl‐xL and Mcl‐1, as well as the cleavage of caspase 3 and PARP, after treatment with DMSO, 1 μM imatinib, 10 μM ABT‐737, or a combination (Figure 5F). We found that Bcl‐2, Bcl‐xL and Mcl‐1 proteins were unchanged by these conditions, whereas caspase 3 and PARP were cleaved with ABT‐737 and 1 μM imatinib + 10 μM ABT‐737, but not by imatinib alone.
Figure 5.
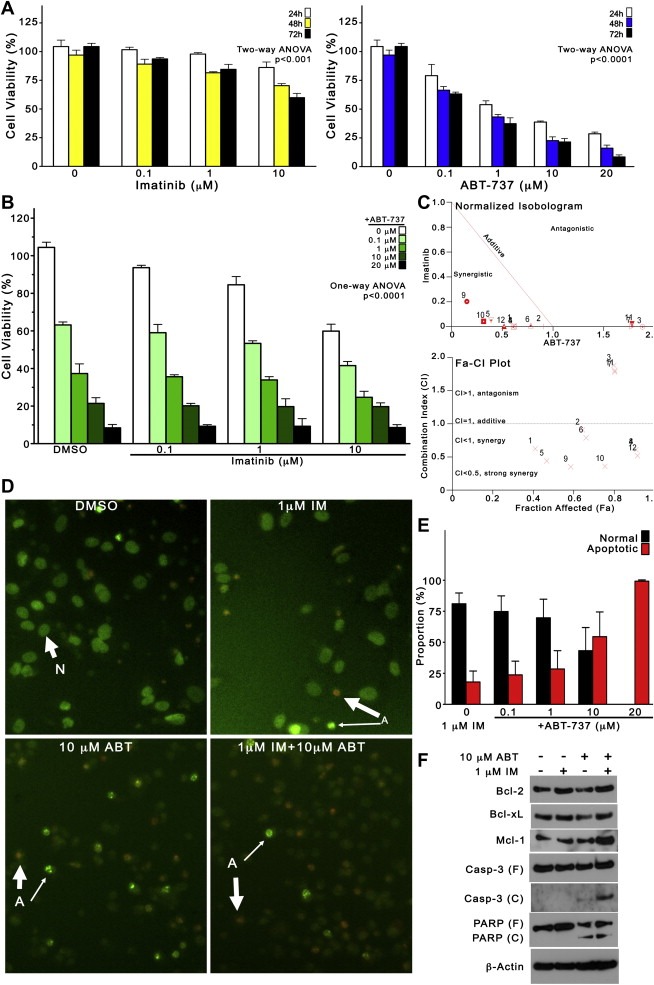
Combined treatment with ABT‐737 and imatinib induces apoptosis synergistically to overcome imatinib‐resistance in GIST48IM cells. (A) The antiproliferative effect of single‐agent imatinib (left) and single‐agent ABT‐737 (right) in imatinib‐resistant GIST48IM cells was examined after 24, 48 and 72 h of treatment, using the MTS cell viability assay. Columns, mean of duplicate experiments; error bars, SD. Results were analyzed by two‐way ANOVA. (B) The effect of combined ABT‐737 (0, 0.1, 1, 10, 20 μM) and imatinib (0, 0.1, 1, 10 μM) on the viability of GIST48IM cells at 72 h. Columns, mean of duplicate experiments; error bars, SD. Results were analyzed by one‐way ANOVA. (C) Normalized isobologram (top) and Fa‐CI plot (bottom) of GIST48IM cells, graphically depicting synergistic, additive, and antagonistic interactions between imatinib and ABT‐737 in this cell line. (D) To determine whether reductions of GIST48IM cell viability were due to apoptosis, nuclear morphology was assessed by EB/AO staining after treatment with ABT‐737 and imatinib for 72 h. Representative micrographs of ethidium bromide/acridine orange‐stained GIST48IM cells. Original magnification, ×200. Abbreviations: (N), normal nuclei; Thick arrow, late apoptosis; (A), apoptotic nuclei; Thin Arrow, early apoptosis. (E) Quantification of normal and apoptotic cells treated with 1 μM imatinib alone, or combined with ABT‐737 (0.1, 1, 10, 20 μM). (F) Immunoblot analysis of the expression of Bcl‐2, Bcl‐xL and Mcl‐1, as well as the cleavage of caspase 3 and PARP, after treatment with DMSO, 1 μM imatinib, 10 μM ABT‐737, or a combination for 72 h. Actin was used to demonstrate equal loading. Abbreviations: (F), full length; (C), cleaved.
4. Discussion
Despite its overwhelming success as the standard of care in GIST, evidence abounds that imatinib is unable to kill GIST cells efficiently. Evasion of apoptosis through acquired imatinib‐resistant mutations, and the ability to enter cytostatic states, allow GIST subclones to survive imatinib monotherapy. Currently, there are limited therapeutic options for patients with imatinib‐refractory GIST. Sunitinib malate, which targets KIT, PDGFR‐α, and vascular endothelial growth factor receptor (VEGFR), is the only FDA‐approved therapy for imatinib‐resistant GIST, but delays progression by only 20 weeks compared with placebo (Demetri et al., 2006). Other second‐generation TKIs, including nilotinib and sorafenib, are often used off‐label or in clinical trials, as treatment options for imatinib‐resistance and/or sunitinib‐resistance. However, it is well‐known that individual patients can harbor diverse TKI‐resistant subclones within single lesions, and among different metastatic lesions, and it is therefore unlikely that second‐ and third‐line therapies based on KIT inhibition will achieve cure. Rational combination regimens may be a more effective approach to augment imatinib therapy, overcome resistance, and achieve durable clinical remissions.
We and others have previously found that imatinib‐induced apoptosis occurs in GIST cells and human tumor tissue (McAuliffe et al., 2009; Trent et al., 2006). In imatinib‐sensitive GIST cells, apoptosis occurs partly through the BIM upregulation and its subsequent antagonism of pro‐survival Bcl‐2 proteins, but also through a variety of other intracellular stresses, including H2AX‐mediated transcriptional arrest and ER stress, which also activate the intrinsic pathway of apoptosis (Liu et al., 2007; Nakatani et al., 2006). However, apoptosis is not the only effect of imatinib treatment, even in sensitive models. For instance, Liu and colleagues have demonstrated that a substantial proportion of GIST882 cells does not undergo apoptosis after imatinib, but enters a quiescent state (Liu et al., 2008). Others have shown that imatinib induces autophagy as a survival pathway (Gupta et al., 2010). As the antitumor effects of imatinib in GIST appear to be mediated by both cytostatic and cytotoxic effects, we explored Bcl‐2 inhibition as a therapeutic approach to enhance GIST eradication. Activation of the intrinsic pathway of apoptosis through Bcl‐2 inhibition has been shown to enhance TKI‐induced apoptosis and overcome resistance in other hematologic and solid tumor models, but this approach has not been evaluated in GIST. We hypothesized that the Bcl‐2 inhibitor ABT‐737 would effectively enhance imatinib‐induced cytotoxicity by targeting the apoptotic pathway downstream and independently of KIT inhibition. The primary objectives of this study were (1) to determine whether ABT‐737 enhanced imatinib‐induced apoptosis in imatinib‐sensitive GIST cell lines, (2) to determine whether the effective in vitro concentration of ABT‐737 was physiologically attainable for GIST patients in a clinical trial, and (3) to examine whether inhibition of Bcl‐2 could overcome imatinib‐resistance in GIST cells.
Herein, we provide preclinical evidence that ABT‐737 combines synergistically with imatinib to inhibit proliferation and induce apoptosis of GIST cells, irrespective of their underlying sensitivity or resistance to imatinib. The synergistic interaction between imatinib and ABT‐737 may be explained by the distinct but complementary mechanisms of activation of the intrinsic pathway of apoptosis, which may achieve greater antagonism of Bcl‐2 proteins than either agent alone. In our study, ABT‐737 enhanced imatinib‐induced cytotoxicity in GIST‐T1 and GIST882 cells in parallel with their initial sensitivity to imatinib. In contrast, ABT‐737 as a single agent was highly active against the imatinib‐resistant GIST48IM cells, independent of imatinib. Thus, it is possible that the imatinib‐resistant phenotype resulting from secondary KIT exon 17 mutation in GIST48IM may render these cells sensitive to the pro‐apoptotic effects of ABT‐737. Alternatively, ABT‐737 cytotoxicity may depend on the expression profile of pro‐survival Bcl‐2 proteins, and be independent of KIT signaling. Although we did not examine directly the extent of functional inhibition of Bcl‐2 proteins in our cell lines, the published literature on ABT‐737 has consistently demonstrated that its pro‐apoptotic effects are directly proportional to the specific inhibition of Bcl‐2 and Bcl‐xL and inversely proportional to expression of Mcl‐1 (Tse et al., 2008). Moreover, compound A‐793844, an enantiomer of ABT‐737 with 100‐fold lower affinity for Bcl‐2 and Bcl‐xL, exhibited no cytotoxicity in GIST cells in this study, suggesting that apoptosis was a direct result of Bcl‐2/Bcl‐xL inhibition.
Given the limited availability of imatinib‐resistant GIST cell lines, this study assessed only one imatinib‐resistant cell line (GIST48IM). As such, these results may not be generalizable to all forms of imatinib‐resistance. However, GIST48IM cells are highly representative of the major resistance mechanism observed clinically, as these cells were established from a patient with GIST whose tumor initially harbored an exon 11 mutation, and which progressed during imatinib therapy with an exon 17 imatinib‐resistant, secondary mutation. Additionally we included the imatinib‐resistant undifferentiated sarcoma cell line A204 as a control in cell proliferation assays, and found that this cell line endured >20 μM ABT‐737 with moderate cytotoxicity (not shown). As such, the results obtained in GIST48IM cells suggest that ABT‐737 may be an important therapy for imatinib‐resistant GIST patients. Further, whereas the current study provides evidence that Bcl‐2 inhibition is an effective addition to imatinib therapy in GIST cells, future work will extend the work to in vivo models of GIST, including xenotransplanted mice.
One of the aims of our study was to determine whether the dose of ABT‐737 required to kill GIST cells in vitro was clinically feasible. There is limited pharmacologic data available from human trials of ABT‐263, the orally bioavailable analog of ABT‐737. Nevertheless, peak plasma concentrations (C max) from 3 to 14 μM have been achieved in mice and dogs receiving 10–100 mg/kg/day, in the absence of toxicity (Tse et al., 2008). The synergism we observed in GIST cells was most apparent with low‐dose combinations (0.1 and 1 μM ABT‐737 and 0.1 μM and 1 μM imatinib), suggesting that the dose of ABT‐737 required for single‐agent inhibition can be reduced in combination with imatinib. Importantly, while most chemotherapy regimens currently employed for soft‐tissue sarcomas were developed empirically, the combination of ABT‐737 and imatinib was developed via a rational approach that considered complementary mechanisms of action as the therapeutic goal. In this way, we may maximize the antitumor effects of both drugs, while minimizing their potential cross‐resistance. Additionally, the safety profiles of both drugs in humans have been previously established to be tolerable, and there appears to be little overlap in normal organ toxicity.
In summary, our findings have demonstrated that simultaneous inhibition of oncogenic KIT signaling and direct engagement of apoptosis may be an effective combinatorial approach to in GIST. ABT‐737 was shown to synergistically enhance imatinib‐induced cytotoxicity via apoptosis, in imatinib‐sensitive and ‐resistant GIST cell lines. Our data indicate that the cytotoxicity of imatinib in susceptible GIST cells can be augmented by the addition of a pro‐apoptotic agent, thereby suggesting that resistant cells may be prevented from emerging a priori. Further, the efficacy of ABT‐737 against imatinib‐refractory GIST cells suggests that this may be a suitable strategy to overcome established imatinib‐resistance. Importantly, the synergistic effects of ABT‐737 and imatinib suggest that rational drug combinations with independent, but complementary, mechanisms warrant further clinical investigation. Further studies involving drug combinations of rational design are needed to eventually translate into new therapies for patients with imatinib‐resistant, metastatic GIST.
Supporting information
The following are the supplementary data related to this article:
Supplementary table. Overall results from isobologram (synergy) analyses of imatinib/ABT‐737 combinations in GIST‐T1, GIST882, and GIST48IM cells, performed for growth inhibition and apoptosis. The combinations are numbered sequentially (# 1–12 for growth inhibition; # 1–6 for apoptosis) and these correspond to the numbers in the normalized isobolograms and Fa‐CI plots (Figures 2 and 5, and Supplementary figures 1 and 2). Abbreviations: (CI), combination index; (Fa), fraction affected (%growth inhibition or % TUNEL‐positive); Legend: CI < 0.5, strong synergy; CI < 1 synergy; CI = 1 additive (italics); and CI > 1, antagonism (underlined).
Supplementary figure 1 Isobologram analysis of synergy with respect to growth inhibition for imatinib/ABT‐737 combinations in GIST‐T1 cells. Normalized isobologram (top), and Fraction affected‐Combination Index (Fa‐CI) plot (bottom). All but one imatinib/ABT‐737 combination was synergistic in this analysis, and all combinations in GIST‐T1 achieved greater than 80% growth inhibition.
{kind=link}
Supplementary figure 2 Isobologram analyses of synergy with respect to apoptosis (% TUNEL‐positive) for imatinib/ABT‐737 combinations in GIST‐T1 (A) and GIST882 (B) cells. Normalized isobolograms (top), and Fraction affected‐Combination Index (Fa‐CI) plots (bottom). All combinations were strongly synergistic with regard to apoptosis in both GIST‐T1 and GIST882 cells.
{kind=link}
Supplementary figure 3 GIST‐T1 cells were treated with imatinib (1 μM) and ABT‐737 (0, 0.1, 1, 10, 20 μM) for 72 h and apoptotic cell death was evaluated by assessment of nuclear morphology after ethidium bromide/acridine orange (EB/AO) staining. (A) Representative micrographs of GIST‐T1 cells treated with vehicle (DMSO), imatinib (1 μM) and/or ABT‐737 (10 μM), demonstrating increased chromatin fragmentation, nuclear and cytoplasmic condensation in ABT‐737‐treated cells. Original magnification, ×200. Abbreviations: (N), normal nuclei; (A), apoptotic nuclei; Thick arrow, late apoptosis; Thin Arrow, early apoptosis. (B) Quantitative assessment of normal and apoptotic cells treated with 1 μM imatinib and increasing concentrations of ABT‐737 (0, 0.1, 1, 10, 20 μM).
{kind=link}
Acknowledgements
This work was supported by the Center for Clinical and Translational Sciences, which is funded by National Institutes of Health Clinical and Translational Award TL1 RR024147 from the National Center for Research Resources, and by NIH/NCI grant 1K23CA109060‐02. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Supplementary material 1.
Supplementary data related to this article can be found online at doi:10.1016/j.molonc.2010.10.003.
Reynoso David, Nolden Laura K., Yang Dan, Dumont Sarah N., Conley Anthony P., Dumont Amaury G.P., Zhou Kim, Duensing Anette, Trent Jonathan C., (2011), Synergistic induction of apoptosis by the Bcl‐2 inhibitor ABT‐737 and imatinib mesylate in gastrointestinal stromal tumor cells, Molecular Oncology, 5, doi: 10.1016/j.molonc.2010.10.003.
References
- Agaram, N.P. , Wong, G.C. , 2008. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 47, (10) 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, S. , Yu, L.K. , 2006. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 66, (18) 9153–9161. [DOI] [PubMed] [Google Scholar]
- Benjamin, R.S. , Debiec-Rychter, M. , 2009. Gastrointestinal stromal tumors II: medical oncology and tumor response assessment. Semin. Oncol. 36, (4) 302–311. [DOI] [PubMed] [Google Scholar]
- Blanke, C.D. , Demetri, G.D. , 2008. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 26, (4) 620–625. [DOI] [PubMed] [Google Scholar]
- Blanke, C.D. , Rankin, C. , 2008. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J. Clin. Oncol. 26, (4) 626–632. [DOI] [PubMed] [Google Scholar]
- Blay, J.Y. , Le Cesne, A. , 2007. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J. Clin. Oncol. 25, (9) 1107–1113. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , Talalay, P. , 1981. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur. J. Biochem. 115, (1) 207–216. [DOI] [PubMed] [Google Scholar]
- Chou, T.C. , 2008. Preclinical versus clinical drug combination studies. Leuk. Lymphoma. 49, (11) 2059–2080. [DOI] [PubMed] [Google Scholar]
- Cragg, M.S. , Jansen, E.S. , 2008. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J. Clin. Invest. 118, (11) 3651–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMatteo, R.P. , Lewis, J.J. , 2000. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann. Surg. 231, (1) 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri, G.D. , van Oosterom, A.T. , 2006. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 368, (9544) 1329–1338. [DOI] [PubMed] [Google Scholar]
- Dupart, J.J. , Trent, J.C. , 2009. Insulin-like growth factor binding protein-3 has dual effects on gastrointestinal stromal tumor cell viability and sensitivity to the anti-tumor effects of imatinib mesylate in vitro. Mol. Cancer. 8, 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, V.L. , Rock, E.P. , 2007. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin. Cancer Res. 13, (5) 1367–1373. [DOI] [PubMed] [Google Scholar]
- Gordon, P.M. , Fisher, D.E. , 2010. Role for the proapoptotic factor BIM in mediating imatinib-induced apoptosis in a c-KIT-dependent gastrointestinal stromal tumor cell line. J. Biol. Chem. 285, (19) 14109–14114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, A. , Roy, S. , 2010. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc. Natl. Acad. Sci. U S A. 107, (32) 14333–14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, (1) 57–70. [DOI] [PubMed] [Google Scholar]
- Heinrich, M.C. , Corless, C.L. , 2003. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 299, (5607) 708–710. [DOI] [PubMed] [Google Scholar]
- Hirota, S. , Isozaki, K. , 1998. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 279, (5350) 577–580. [DOI] [PubMed] [Google Scholar]
- Jayanthan, A. , Howard, S.C. , 2009. Targeting the Bcl-2 family of proteins in Hodgkin lymphoma: in vitro cytotoxicity, target modulation and drug combination studies of the Bcl-2 homology 3 mimetic ABT-737. Leuk. Lymphoma. 50, (7) 1174–1182. [DOI] [PubMed] [Google Scholar]
- Kuroda, J. , Kimura, S. , 2008. ABT-737 is a useful component of combinatory chemotherapies for chronic myeloid leukaemias with diverse drug-resistance mechanisms. Br. J. Haematol. 140, (2) 181–190. [DOI] [PubMed] [Google Scholar]
- Lasota, J. , Miettinen, M. , 2008. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 53, (3) 245–266. [DOI] [PubMed] [Google Scholar]
- Liegl, B. , Kepten, I. , 2008. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J. Pathol. 216, (1) 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K.H. , Huang, M.J. , 2008. Molecular analysis of secondary kinase mutations in imatinib-resistant gastrointestinal stromal tumors. Med. Oncol. 25, (2) 207–213. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Tseng, M. , 2007. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res. 67, (6) 2685–2692. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Perdreau, S.A. , 2008. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res. 68, (21) 9015–9023. [DOI] [PubMed] [Google Scholar]
- McAuliffe, J.C. , Hunt, K.K. , 2009. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann. Surg. Oncol. 16, (4) 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miselli, F. , Negri, T. , 2008. Is autophagy rather than apoptosis the regression driver in imatinib-treated gastrointestinal stromal tumors?. Transl. Oncol. 1, (4) 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani, H. , Araki, K. , 2006. STI571 (Glivec) induces cell death in the gastrointestinal stromal tumor cell line, GIST-T1, via endoplasmic reticulum stress response. Int. J. Mol. Med. 17, (5) 893–897. [PubMed] [Google Scholar]
- Nishida, T. , Kanda, T. , 2008. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 99, (4) 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf, T. , Elmore, S.W. , 2005. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 435, (7042) 677–681. [DOI] [PubMed] [Google Scholar]
- Paoluzzi, L. , Gonen, M. , 2008. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood. 112, (7) 2906–2916. [DOI] [PubMed] [Google Scholar]
- Reichardt, P. , 2008. Novel approaches to imatinib- and sunitinib-resistant GIST. Curr. Oncol. Rep. 10, (4) 344–349. [DOI] [PubMed] [Google Scholar]
- Reynolds, C.P. , Maurer, B.J. , 2005. Evaluating response to antineoplastic drug combinations in tissue culture models. Methods Mol. Med. 110, 173–183. [DOI] [PubMed] [Google Scholar]
- Ribble, D. , Goldstein, N.B. , 2005. A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol. 5, 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, S. , Ou, W. , 2006. Gastrointestinal stromal tumours overexpress fatty acid synthase. J. Pathol. 209, (3) 369–375. [DOI] [PubMed] [Google Scholar]
- Sambol, E.B. , Ambrosini, G. , 2006. Flavopiridol targets c-KIT transcription and induces apoptosis in gastrointestinal stromal tumor cells. Cancer Res. 66, (11) 5858–5866. [DOI] [PubMed] [Google Scholar]
- Shoemaker, A.R. , Oleksijew, A. , 2006. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 66, (17) 8731–8739. [DOI] [PubMed] [Google Scholar]
- Squier, M.K. , Cohen, J.J. , 2001. Standard quantitative assays for apoptosis. Mol. Biotechnol. 19, (3) 305–312. [DOI] [PubMed] [Google Scholar]
- Steinert, D.M. , Oyarzo, M. , 2006. Expression of Bcl-2 in gastrointestinal stromal tumors: correlation with progression-free survival in 81 patients treated with imatinib mesylate. Cancer. 106, (7) 1617–1623. [DOI] [PubMed] [Google Scholar]
- Taguchi, T. , Sonobe, H. , 2002. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab. Invest. 82, (5) 663–665. [DOI] [PubMed] [Google Scholar]
- Trent, J.C. , Beach, J. , 2003. A two-arm phase II study of temozolomide in patients with advanced gastrointestinal stromal tumors and other soft tissue sarcomas. Cancer. 98, (12) 2693–2699. [DOI] [PubMed] [Google Scholar]
- Trent, J.C. , Ramdas, L. , 2006. Early effects of imatinib mesylate on the expression of insulin-like growth factor binding protein-3 and positron emission tomography in patients with gastrointestinal stromal tumor. Cancer. 107, (8) 1898–1908. [DOI] [PubMed] [Google Scholar]
- Tse, C. , Shoemaker, A.R. , 2008. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, (9) 3421–3428. [DOI] [PubMed] [Google Scholar]
- Tuveson, D.A. , Willis, N.A. , 2001. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 20, (36) 5054–5058. [DOI] [PubMed] [Google Scholar]
- Wardelmann, E. , Merkelbach-Bruse, S. , 2006. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin. Cancer Res. 12, (6) 1743–1749. [DOI] [PubMed] [Google Scholar]
- Yang, D. , Ylipaa, A. , 2010. An integrated study of aberrant gene copy number and gene expression in GIST and LMS. Technol. Cancer Res. Treat. 9, (2) 171–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary table. Overall results from isobologram (synergy) analyses of imatinib/ABT‐737 combinations in GIST‐T1, GIST882, and GIST48IM cells, performed for growth inhibition and apoptosis. The combinations are numbered sequentially (# 1–12 for growth inhibition; # 1–6 for apoptosis) and these correspond to the numbers in the normalized isobolograms and Fa‐CI plots (Figures 2 and 5, and Supplementary figures 1 and 2). Abbreviations: (CI), combination index; (Fa), fraction affected (%growth inhibition or % TUNEL‐positive); Legend: CI < 0.5, strong synergy; CI < 1 synergy; CI = 1 additive (italics); and CI > 1, antagonism (underlined).
Supplementary figure 1 Isobologram analysis of synergy with respect to growth inhibition for imatinib/ABT‐737 combinations in GIST‐T1 cells. Normalized isobologram (top), and Fraction affected‐Combination Index (Fa‐CI) plot (bottom). All but one imatinib/ABT‐737 combination was synergistic in this analysis, and all combinations in GIST‐T1 achieved greater than 80% growth inhibition.
Supplementary figure 2 Isobologram analyses of synergy with respect to apoptosis (% TUNEL‐positive) for imatinib/ABT‐737 combinations in GIST‐T1 (A) and GIST882 (B) cells. Normalized isobolograms (top), and Fraction affected‐Combination Index (Fa‐CI) plots (bottom). All combinations were strongly synergistic with regard to apoptosis in both GIST‐T1 and GIST882 cells.
Supplementary figure 3 GIST‐T1 cells were treated with imatinib (1 μM) and ABT‐737 (0, 0.1, 1, 10, 20 μM) for 72 h and apoptotic cell death was evaluated by assessment of nuclear morphology after ethidium bromide/acridine orange (EB/AO) staining. (A) Representative micrographs of GIST‐T1 cells treated with vehicle (DMSO), imatinib (1 μM) and/or ABT‐737 (10 μM), demonstrating increased chromatin fragmentation, nuclear and cytoplasmic condensation in ABT‐737‐treated cells. Original magnification, ×200. Abbreviations: (N), normal nuclei; (A), apoptotic nuclei; Thick arrow, late apoptosis; Thin Arrow, early apoptosis. (B) Quantitative assessment of normal and apoptotic cells treated with 1 μM imatinib and increasing concentrations of ABT‐737 (0, 0.1, 1, 10, 20 μM).