

Abstract
Immunotherapy‐based strategies for gastrointestinal carcinomas (GIC) have been exploited so far, but these approaches have to face strong mechanisms of immune escape induced by tumours. We previously demonstrated that sub‐therapeutic doses of an adenovirus expressing IL‐12 genes (AdIL‐12) mediated a potent antitumour effect against subcutaneous (s.c.) colorectal carcinomas (CRC) in mice pre‐treated with low doses of cyclophosphamide (Cy). In our study we used this combination to assess its impact on the immunosuppressive microenvironment. In s.c. CRC model we demonstrated that non‐responder mice failed to decrease Tregs in tumour, spleen and peripheral blood. Reconstitution of Tregs into tumour‐bearing mice treated with combined therapy abolished the antitumoural effect. In addition, Cy + AdIL‐12 modified Tregs functionality by inhibiting the in vitro secretion of IL‐10 and TGF‐β and their ability to inhibit dendritic cells activation. Combined treatment decreased the number of myeloid‐derived suppressor cells (MDSCs) in comparison to non‐treated mice and, interestingly, administration of Tregs restored splenic MDSCs population. Furthermore, combined therapy potently generated specific cytotoxic IFN‐γ‐secreting CD4+ T cells able to eradicate established CRC tumours after adoptive transfer. Finally, we evaluated the combination on disseminated CRC and pancreatic carcinoma (PC). Cy + AdIL‐12 were able to eradicate liver metastatic CRC (47%) and PC tumour nodules (40%) and to prolong animal survival. The results of this study support the hypothesis that Cy + AdIL‐12 might be a valid immunotherapeutic strategy for advanced GIC.
Keywords: Gastrointestinal carcinoma, Immunosuppression, Cyclophosphamide, IL-12, Tregs
1. Introduction
Potent mechanisms of immunosuppression are active during tumour growth limiting the effectiveness of immunotherapy strategies (Zou, 2005). Mechanisms used by malignant tumours to elude immune recognition include tumour‐induced impairment of antigen presentation, activation of negative co‐stimulatory signals and secretion of immunosuppressive factors (Almand et al., 2001; Bell et al., 1999; Ghiringhelli et al., 2005; Nestle et al., 1997). In addition, cancer cells may induce the expansion and/or recruitment of regulatory cell populations that may promote and sustain this immunosuppressive network; these populations include regulatory T cells (Tregs), myeloid‐derived suppressor cells (MDSCs) and distinct subsets of immature and mature regulatory dendritic cells (DCs) (Croci et al., 2007).
Colorectal carcinoma (CRC) is the second most common cause of cancer mortality in western countries and the 9.7% of the most commonly diagnosed cancers worldwide (Ferlay et al., 2010). Twenty five percent of the patients have metastatic disease at diagnosis, and their predicted 5‐year survival among non‐surgical patients is less than 10% (Lorenz et al., 2000). Pancreatic cancer is the fourth leading cause of cancer‐related death and surgery is the only curative option; however, more than 80% of the patients showed local or disseminated recurrence (Lorenz et al., 2000). Therefore, there is an urgent need for new therapeutic strategies for advanced GIC. During the last two decades multiple immunotherapy‐based strategies for GIC have been developed with promising results not only at preclinical stage but also in the clinic (Elkord et al., 2008; Shapira et al., 2010). The loss/down regulation of MHC class I antigens, the lack of co‐stimulatory molecules, defective death receptor signalling, apoptosis of activated T cells, immunosuppressive cytokines, and activation of suppressor T cells have been described as responsible for the failure of anticancer treatment in mice and humans (Mazzolini et al., 2007).
Interleukin 12 (IL‐12), a cytokine with multiple biological effects, is one of the most potent antitumour cytokines (Mazzolini et al., 2003; Trinchieri, 2003). However, high doses of IL‐12 are needed to achieve significant antitumour effects that results in severe toxicity (Leonard et al., 1997). In recent years, gene therapy has emerged as a therapeutic tool to allow desired levels of IL‐12 or other cytokines into tumoural/peritumoural milieu avoiding undesired side effects (Colombo and Forni, 1994). The efficacy of IL‐12 gene transfer for advanced GIC in animal models has been shown consistently by different research groups including ours (Barajas et al., 2001; Caruso et al., 1996; Mazzolini et al., 1999; Putzer et al., 2001). Intratumoural administration of an adenovirus expressing IL‐12 genes (AdIL‐12) in patients with advanced GIC in a phase I trial was a feasible and well‐tolerated procedure that exerted only mild antitumoural activity (Sangro et al., 2004). Immunosuppressive activity induced by tumoural and peritumoural cells seem to be critical and might be responsible for the lack of clinical success in immune‐ and non immune‐based therapies (Rabinovich et al., 2007; Zwirner et al., 2010). Seeking for immunotherapeutic synergies to fight against cancer, it was possible to increase the efficacy of IL‐12, by combining this cytokine with other procedures, such as chemotherapy, immunostimulatory monoclonal antibodies, dendritic cells, and other immunotherapy approaches (Gomez et al., 2001; Melero et al., 2001). Cyclophosphamide (Cy) is a widely known chemotherapeutic agent that displays either immunosuppressive or immunopotentiating effects, depending on the dosage and the duration of administration of this drug (Brodsky et al., 1998; Colvin, 1999). Several mechanisms have been proposed to explain Cy immunomodulatory activity including depletion of tumour‐induced regulatory T cells, production of soluble growth factors, Th2/Th1 shift in cytokine production, and others (Ghiringhelli et al., 2004; Matar et al., 2002; Proietti et al., 1998). However, the precise mechanism is not yet clarified.
Recently, we have shown a novel synergistic combination of Cy and AdIL‐12 for the eradication of CRC in mice. Currently, we explored the mechanisms of action between Cy and AdIL‐12 combination and observed that the potent anticancer effect obtained in advanced GIC is based on their capability to block Tregs secretion of IL‐10 and TGF‐β cytokines, as well as on the loss of their inhibitory activity exerted on DCs. In addition, we observe a decrease in the number of MDSCs after Tregs inhibition. Finally, combined treatment showed synergistic activity not only at the induction of the immune response but also at the effector phase by generating potent IFN‐γ‐secreting CD4+ T cells that were able to eradicate established CRC tumour nodules.
2. Materials and methods
2.1. Animals and cell lines
Six‐ to eight‐week‐old male BALB/c mice and C57BL/6 mice were purchased from CNEA (National Atomic Energy Commission, Ezeiza, Argentina). Animals were maintained at our Animal Resource Facilities (School of Biomedical Sciences, Austral University) in accordance with the experimental ethical committee and the NIH guidelines on the ethical use of animals. The CT26 (colorectal carcinoma), BNL (hepatocellular carcinoma) and Panc02 (pancreatic carcinoma) tumour cell lines were used (kindly provided by Prof. Prieto, University of Navarra, Spain). Cells were maintained in DMEM or RPMI 1640 supplemented with 10% heat‐inactivated FCS, 2 mmol/L l‐glutamine, 100 U/mL streptomycin, and 100 mg/mL penicillin and incubated at 37 °C in a 5% CO2 humidified atmosphere.
2.2. Drugs
Cyclophosphamide (Filaxis, Argentina) was dissolved in sterile water at a concentration of 20 mg/mL and injected i.p. at the indicated dose.
2.3. Adenoviral vectors
Construction of a recombinant adenovirus encoding for IL‐12 (AdIL‐12) was previously described (Mazzolini et al., 1999).
2.4. In vivo experiments
2.4.1. Subcutaneous CRC model
CT26 cells were injected at a dose of 5 × 105 cells s.c. into the right flank of BALB/c mice (day 0). Tumours were allowed to reach approximately 85 mm3 in size before treatment was started. Animals were distributed in different groups and then treated with saline i.p.; Cy (50 mg/kg i.p., day 8); AdIL‐12 (109 TCID50 intratumourally (i.t.), day 9); Cy (50 mg/kg i.p.) + AdIL‐12 (109 TCID50 i.t.). Adenovirus were diluted in saline (final volume 50 μL) and i.t. injected in a single site; no leakage of material was observed after inoculations.
2.4.2. Adoptive transfer of CD4+CD25+ T cells (Tregs)
CT26 tumour‐bearing mice (approximately 85 mm3 in size) were i.p. treated with saline or Cy (50 mg/kg i.p, day 8) + AdIL‐12 (109 TCID50 i.t, day 9). Forty‐eight and 96 h later, mice were injected i.v with 1 × 106 CD4+CD25+ T lymphocytes isolated by magnetic cell sorting from spleen of non‐treated tumour‐bearing mice excised at day 14 of tumour growth evolution.
2.4.3. Adoptive transfer of CD4+CD25− T cells
2.4.3.1. Preventive model
BALB/c mice were simultaneously inoculated with 5 × 105 CT26 cells s.c. into the right flank, and with 2.5 × 106 CD4+CD25− cells i.v., isolated by magnetic cell sorting and in vitro re‐stimulated, from spleen of saline, Cy, AdIL‐12 or Cy + AdIL‐12‐treated mice (see Section 2.13 below).
2.4.3.2. Therapeutic model
CT26 tumour‐bearing mice (approximately 65 mm3 in size) were adoptively transferred with 2.5 × 106 CD4+CD25− cells i.v., isolated by magnetic cell sorting, from saline, Cy, AdIL‐12 or Cy + AdIL‐12‐treated mice.
2.4.4. Pancreatic tumour model
C57BL/6 mice were inoculated in the right flank with 5 × 105 Panc02 cells s.c. When tumours reached 85 mm3 in size, animals were treated with saline, Cy (50 mg/kg i.p., day 8), AdIL‐12 (109 TCID50 i.t., day 9) or Cy + AdIL‐12. Tumour growth was assessed twice a week by calliper.
2.4.5. Liver metastatic CRC model
BALB/c mice received an intra‐hepatic inoculation of 5 × 105 CT26 cells (day 0). Then, mice were distributed in experimental groups and treated with saline; Cy (50 mg/kg i.p., day 8); AdIL‐12 (109 TCID50 i.t., day 9) or Cy + AdIL‐12. At day 20, animals were sacrificed and the volume of metastatic nodules were measured with calliper.
2.5. Sample preparation for Tregs and MDSCs cytometry analysis
BALB/c mice were injected with 5 × 105 CT26 cells s.c. into the right flank (day 0) and tumours were allowed to reach 85 mm3 before the beginning of the treatment. For Tregs analysis, animals were distributed and treated with saline or Cy + AdIL‐12. At day 15 and 22 (7 and 14 days post‐treatment), peripheral blood was collected by cardiac puncture and anticoagulated with EDTA. Single splenic and tumoural cell suspensions were obtained by mechanical disruption and then treated with RBC lysis buffer (0.15 mol/L NH4Cl, 1 mmol/L KHCO3, 0.1 mmol/L Na2‐EDTA) and washed with PBS 1% bovine serum albumin, before flow cytometry analysis. For MDSCs analysis, tumour‐bearing animals were distributed into different groups and treated with saline, Cy, AdIL‐12 or Cy + AdIL‐12. The mice were sacrificed on day 15, spleens were excised and single cell suspensions were prepared for flow cytometry analysis as described below. MDSCs were phenotypically characterized as CD11b+ Gr1+.
2.6. Generation of bone marrow‐derived DCs
DCs were generated from BALB/c mice as described elsewhere (Alaniz et al., 2009). Flow cytometric analysis for CD11c and the capacity to produce IL‐12 after LPS activation was performed to certify the presence and functional status of generated DCs at the end of procedure.
2.7. Cell purification by magnetic cell sorting
Spleen cell suspensions were obtained by mechanical disruption of spleen from CT26‐bearing mice treated with saline, Cy, AdIL‐12 or Cy + AdIL‐12. CD4+CD25+ and CD4+CD25− cells were purified by magnetic cell sorting using a mouse T regulatory cell isolation kit and LD plus MS columns according to the manufacturers' instructions (Miltenyi Biotec, Auburn, CA, USA). CD4+CD25+ cells (Tregs) obtained were >95% in purity by flow cytometric analysis of intracellular Foxp3.
2.8. Expansion of CD4+CD25+ T cells and co‐culture with DCs
Purified CD4+CD25+ T cells were cultured for 48h with rmIL‐2 (10 UI/ml) (Peprotech, Rocky Hill, NJ, USA). Supernatants were collected and used for allogeneic splenocytes proliferation assay and cytokines quantification. Tregs and DCs (ratio = 1:1) were co‐cultured for 24 h and then LPS (Sigma Chemicals, St Louis, MO, USA) was added (1 μg/ml) for additional 24 h.
2.9. Cell labelling
Tregs were labelled with the fluorescent dye Fast DiO (Molecular Probes, Eugene, USA) according to the manufacturer instructions. Briefly, cells were incubated with 3 mM Fast DiO cell solution for 5 min at 37 °C in 5% CO2‐humidified atmosphere and for 15 min at 4 °C and then washed with PBS. After labelling Fast DiO‐stained Tregs were diluted in saline and i.v. injected into mice. Five days later, peripheral blood was collected and mice were sacrificed. Spleen, liver, tumour and regional lymph nodes were excised from each animal and single cell suspensions were obtained by digestion with collagenase I (Calbichem®, Merck KGaA, Darmstadt, Germany). The cell suspension was treated with RBC lysis buffer, washed with PBS 1% bovine serum albumin, fixed in 1% paraformaldehyde and then analysed by flow cytometry (FACSAria, BD).
2.10. Flow cytometry
Peripheral blood mononuclear cells, tumour cells or splenocytes were stained with the following conjugated antibodies: phycoeritrin‐anti‐Foxp3 (eBioscience), PECy5‐anti‐CD4 (BD Biosciences), allophycocyanin‐anti‐CD11b (BD Biosciences), PE‐anti‐Gr1 (BD Bioscience) and their respective isotypes. DCs were stained with APC‐anti‐CD11c (kindly provided by Dr. Gabriel Rabinovich, IByME, Argentina), PE‐anti‐MHCII (BD biosciences), FITC‐ anti‐CD40 (BD biosciences) and their respective isotypes. Cells were fixed with 1% paraformaldehyde and subjected to flow cytometry (FACSAria, BD). Data were analysed using WinMDI software.
2.11. Cytokine quantificationby ELISA
Transforming growth factor‐β (TGF‐β), IL‐10 and IL‐12 concentrations in culture supernatants were measured by specific ELISA kits (OptEIA, BD Biosciences Pharmingen for IL‐10 and IL‐12 and R&D Systems for TGF‐β). Assays were carried out according to the instructions provided by the manufacturers.
2.12. Allogeneic splenocytes proliferation assay
Bone marrow‐derived DCs (1 × 105) from BALB/c mice were treated for 20 min with 50 μg/ml mitomycin C (Sigma Chemical,St. Louis, MO, USA), washed and used as stimulators. Splenocytes (1 × 106) from naïve C57BL/6 mice were plated in U‐bottom 96‐well plates at a ratio of 1:10 (DCs: splenocytes). Assays were carried out in the presence of Tregs supernatants derived from saline, Cy, AdIL‐12 or Cy + AdIL‐12‐treated mice. Cell proliferation was evaluated after 4 days in culture, as described elsewhere (Malvicini et al., 2009).
2.13. In vitro re‐stimulation of CD4+CD25− T cells
BALB/c mice were injected with CT26 cells and treated with Cy, AdL‐12 or Cy + AdIL‐12 as described above. Splenocytes were isolated 14 days after treatment and CD4+CD25− T cells were obtained by immunomagnetic selection. Then, purified CD4+CD25− T cells were pooled, and 4 × 106cells/ml were co‐cultured with mitomycin C‐treated CT26 cells (4 × 105/ml) and mitomycin C‐treated splenocytes (4 × 105/ml) in a 24‐well plate (1 ml/well) with 10 UI/ml rmIL‐2. Seven days later, viable cells were harvested and washed, adjusted to 4 × 106/mL, and co‐cultured again with mitomycin C‐treated CT26 cells and splenocytes. On day 14, CD4+CD25− T cells were used for in vivo adoptive T cells transfer experiments.
2.14. Statistical analysis
Mann Whitney, Kruskal–Wallis tests and Kaplan–Meier, log rank (InStat, GraphPad Software) were used to statistically examine the differences between groups. P < 0.05 was considered statistically significant.
3. Results
3.1. Tregs depletion is required to generate an effective antitumoural response after treatment with Cy +AdIL‐12
We previously demonstrated that the sequential administration of Cy followed by adenoviral gene transfer of IL‐12 induced a synergistic antitumoural effect on s.c. CRC model (CT26) (Malvicini et al., 2009). Two well defined groups were found in Cy + AdIL‐12‐treated mice. The two categories were identified as responders (R) and non‐responders (NR) mice, depending on whether they showed a >50% in tumour size reduction or not. Post‐treatment tumour sizes in NR mice at days 7 and 14 post‐treatment were significantly higher (566 ± 143 and 614 ± 92 mm3, respectively) than in R mice (58 ± 19 and 126 ± 58 mm3, respectively) (p < 0.01) (Figure 1A). To determine whether immunosuppressive T cell‐mediated mechanisms could be involved in the observed differential antitumoural response obtained after treatment with Cy + AdIL‐12, we first analysed the prevalence of Tregs in peripheral blood and spleens using triple‐staining flow cytometry. While the levels of CD4+ T cells were similar for either untreated mice or treated with the combination (Supplementary figure #1), the percentage of Tregs was significantly higher in NR than in R mice (p < 0.05), in both peripheral blood and spleens (Figure 1B and C). In addition, we observed an increase in the percentage of tumour‐infiltrating CD4+ cells in R mice compared to NR and saline groups (p < 0.05; Figure 1D, left panel). The tumour‐infiltrating CD4+CD25+Foxp3+/CD4+ proportion were evaluated in the same experiment. The percentage of Tregs in R was significantly lower than in untreated mice (P < 0.05, Figure 1D, right panel). On day 14, the percentage of intratumoural Tregs in R was lower than in NR and saline‐treated mice (p < 0.05), in agreement with the results obtained in peripheral blood and spleen. These results showed an association between increased circulating and tumour‐infiltrating Tregs with tumour resistance to combined Cy + AdIL‐12 therapy. These results indicate that depletion of Tregs is critical to achieve a therapeutic benefit in CT26 CRC‐bearing mice.
Figure 1.
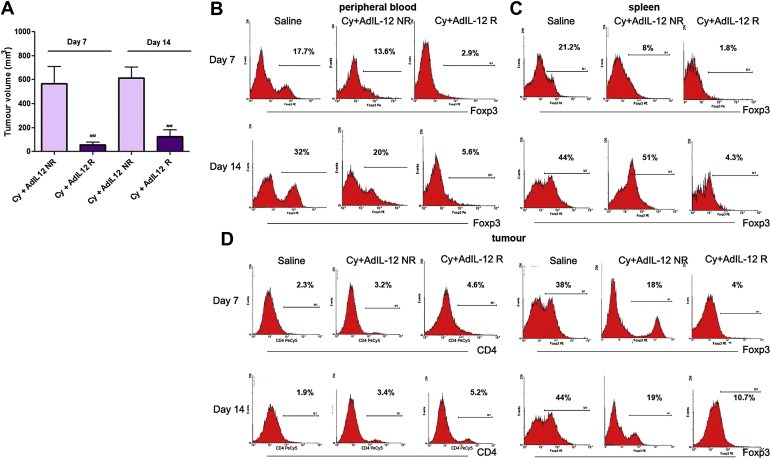
Assessment of Tregs in mice R (responder) and NR (non‐responders) to combined treatment Cy + AdIL‐12 in s.c CT26 tumour model. (A) Tumour volume in R and NR mice. BALB/c mice (n = 35) were s.c. inoculated with 5 × 105 CT26 cells into the right flank (day 0) and tumours were allowed to reach 85 mm3 before the treatment began. Animals were treated with Cy (50 mg/kg i.p., day 8) + AdIL‐12 (109 TCID50 i.t., day 9) (n = 12) or with saline (n = 6). Tumour growth was assessed twice a week by calliper. Tumour volumes at days 7 and 14 after treatment are expressed as mean (bars, SEM). The experiment was performed four times. Mann Whitney test, Cy + AdIL‐12 NR vs. Cy + AdIL‐12 R: (##p < 0.01). Percentage of CD4+CD25+Foxp3+/CD4+ cells in peripheral blood (B) or spleen (C) of R and NR mice. Mann Whitney test, Peripheral blood, day 7 and day 14: Cy + AdIL‐12 R vs. saline (∗∗p < 0.01) and vs. Cy + AdIL‐12 NR (#p < 0.05). Spleen, day 7: Cy + AdIL‐12 R vs. saline and Cy + AdIL‐12 NR (#p < 0.05); Cy + AdIL‐12 NR vs. saline (∗p < 0.05); day 14: Cy + AdIL‐12 R vs. saline and Cy + AdIL‐12 NR (∗and #p < 0.05). (D) Percentage of tumour‐infiltrating CD4+ Tcells (left) and CD4+CD25+Foxp3+/CD4+ cells (right). Mann–Whitney test, CD4+ Tcells, day 7: Cy + AdIL‐12 R vs. saline, (∗p < 0.05); day 14: Cy + AdIL‐12 R vs. saline and Cy + AdIL‐12 NR (#p < 0.05); Cy + AdIL‐12 NR vs. saline (∗p < 0.05). CD4+CD25+Foxp3+/CD4+ cells, day 7: Cy + AdIL‐12 R vs. saline, (∗p < 0.05); day 14: Cy + AdIL‐12 R vs. saline and Cy + AdIL‐12 NR (#p < 0.05). The results shown represent four independent experiments.
To confirm the inhibitory effect of Tregs on the antitumoural efficacy of combined therapy, we adoptively transferred CD4+CD25+Foxp3+ T cells into CT26 tumour‐bearing mice that were treated with the combination. Thus, 1 × 106 CD4+CD25+Foxp3+ T cells derived from untreated tumour‐bearing mice were administered i.v. on days 11 and 13 after tumour inoculation (days 2 and 4 after the combined treatment with Cy + AdIL‐12). As a result, adoptive transfer of Tregs significantly abolished the antitumoural effect achieved with the combined treatment (mean tumour volume ± SE [mm3] at day 28, Cy + AdIL‐12: 144 ± 81 vs. Cy + AdIL‐12+Tregs: 3191 ± 394, p < 0.01; Figure 2A). Moreover, animal survival was strongly reduced after adoptive transfer of Tregs, showing a similar value in saline group (Figure 2B). The trafficking behaviour of administered Tregs was also analysed. Interestingly, FastDio‐stained Tregs injected i.v. revealed that Tregs were distributed in different organs, with higher levels in peripheral blood, lymph nodes and tumours (Figure 2C). Altogether, these results strongly suggest that, at least in this tumour model, CD4+CD25+Foxp3+ T cells should be inhibited in order to achieve a potent antitumoural immune response.
Figure 2.
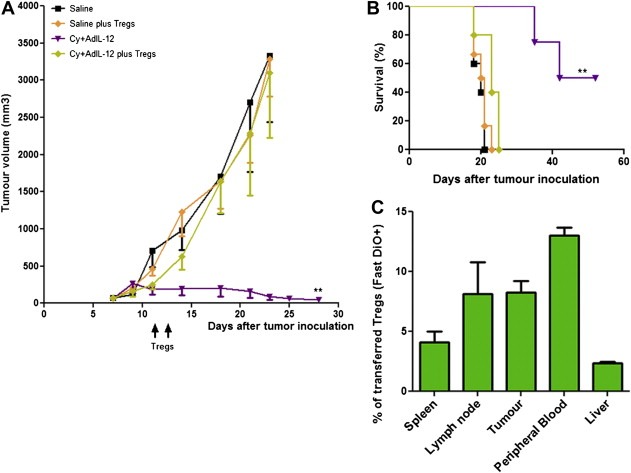
Re‐infusion of Tregs in CT26‐bearing mice treated with the combination Cy + AdIL‐12. (A) Tumour growth: BALB/c mice were s.c. inoculated with 5 × 105 CT26 cells into the right flank (day 0) and tumours were allowed to reach 85 mm3 before the treatment began. Animals (n = 6/group) were treated with saline or Cy (50 mg/kg i.p., day 8) + AdIL‐12 (109 TCID50 i.t., day 9). In addition,saline or Cy + AdIL‐12‐treated mice were inoculated i.v. on days 11 and 13 with 1 × 106 CD4+CD25+ T cells (Tregs) isolated from untreated tumour‐bearing mice. Tumour volume was assessed twice a week by calliper. The experiment was performed four times. Data are expressed as mean (bars, SEM). Kruskal–Wallis test, day 28: Cy + AdIL‐12 vs. saline and Cy + AdIL‐12+Tregs (∗p < 0.05). B) Survival: Kaplan–Meier, log rank test, (∗∗p < 0.01). C) In vivo distribution pattern of re‐infused Tregs: Cy + AdIL‐12‐treated mice were inoculated with 1 × 106 fastDiO‐dyed CD4+CD25+ T cells (i.v; days 11 and 13) and biodistribution was analysed by flow cytometry. The results shown represent three independent experiments.
3.2. Combined therapy with Cy + AdIL‐12 reverts the inhibitory effects of Tregs on DCs activation
We investigated the effects of Cy + AdIL‐12 on the immunosuppressive functions of Tregs derived from CT26 tumour‐bearing mice. Thus, we decided to assess the effects of combined therapy on the ability of Tregs to modulate not only the number but also the maturation and/or functional status of DCs. To this end, naïve DCs were co‐cultured with Tregs isolated from different experimental groups. When DCs were cultured with saline‐derived Tregs there was a significant reduction in the production of IL‐12 as well as in the expression of MHC‐II and the CD40 co‐stimulatory molecules induced by LPS (p < 0.05, Figure 3A and B). In contrary, when DCs were cultured in the presence of Cy +AdIL‐12‐treated mice‐derived Tregs, they showed a similar pattern of activation, IL‐12 production and MHC‐II‐CD40 expression phenotype than the observed in control DCs after LPS stimulation.
Figure 3.
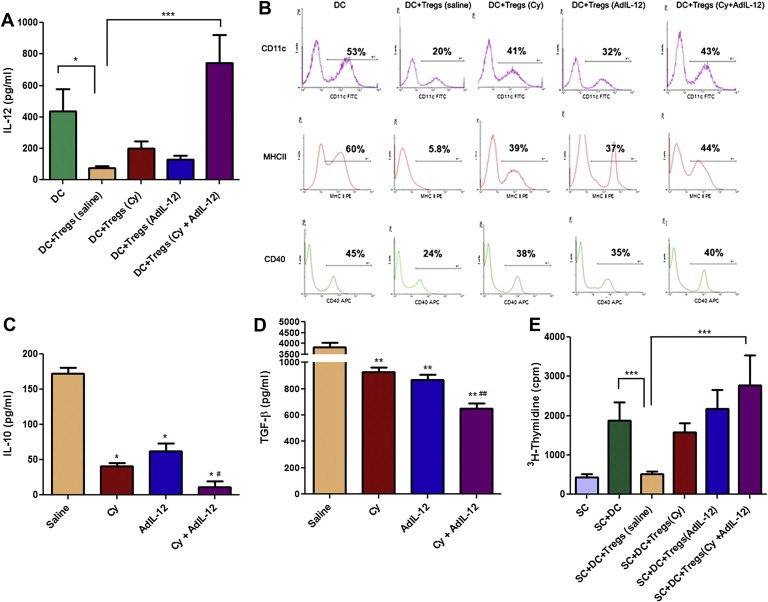
Effect of combined therapy Cy + AdIL‐12 on the interaction DC/Tregs. A) Quantification of IL‐12: Data are expressed as mean (bars, SEM). Kruskal–Wallis and Dunn's multiple comparisons test: DC vs. DC+Tregs (saline), ∗p < 0.05; DC+Tregs (saline) vs. DC+Tregs (Cy + AdIL‐12), ∗∗∗p < 0.001. B) Phenotype of DCs (CD11c+MHC‐II+CD40+). Mann Whitney test: DC vs. DC+Tregs (saline), ∗p < 0.05; DC+Tregs (saline) vs. DC+Tregs (Cy + AdIL‐12), ∗p < 0,05. The results shown represent three independent experiments. IL‐10 (C)and TGF‐β (D) secretion by Tregs: Mann Whitney test, saline vs. Cy, AdIL‐12, and Cy + AdIL‐12, ∗p < 0.05; Cy + AdIL‐12 vs. Cy and AdIL‐12; #p < 0.05. (E) Effect of Tregs supernatants (SN) on allogeneic spleen cell (SC) proliferation under stimulation with mitomycin C‐treated DC. (3H‐Thymidine). Mann Whitney test, SC+DC vs. SC+DC+Tregs SN (saline), ∗∗∗p < 0.001; SC+DC+Tregs SN (saline) vs. SC+DC+Tregs SN (Cy + AdIL‐12), ∗∗∗p < 0.001.
3.3. Combined therapy with Cy + AdIL‐12 reverts Tregs‐immunosuppressive phenotype by reducing IL‐10 and TGF‐β production
We investigate in vitro the secretion of IL‐10 and TGF‐β by Tregs derived from spleens of CT26 tumour‐bearing mice, untreated or treated with Cy, AdIL‐12 or with a combination of both. We found that Tregs from untreated mice (saline group) produced high levels of IL‐10 and TGF‐β (mean ± SEM [mm3]: 172 ± 8, and 3820 ± 213 pg/ml, respectively; (Figure 3C and D). In contrast, we observed that Tregs of mice treated with a single agent (Cy or AdIL‐12) produced significantly lower amounts of both cytokines (IL‐10: 41 ± 5 or 62 ± 11 pg/ml, respectively [p < 0,05 vs. saline]; TGF‐β: 926 ± 35 or 866 ± 42 pg/ml, respectively [p < 0,01 vs. saline]). Importantly, Cy + AdIL‐12 induced further inhibition of IL‐10 and TGF‐β production by Tregs, in comparison to saline and single treatments (11 ± 8 pg/ml; p < 0,05 and 647 ± 43 pg/ml; p < 0,01, respectively). Consistently with these results, supernatants of Tregs from untreated tumour‐bearing mice inhibited DCs stimulated allogeneic splenocytes proliferation (Figure 3E). However, when supernatants of Tregs derived from CT26‐bearing animals treated with Cy + AdIL‐12 were assayed, a strong proliferation activity was detected. Tregs cell viability from all groups was confirmed by trypan blue exclusion test (not shown); no evidence of apoptosis was observed by TUNEL assay in all groups (data no shown). Altogether, these results suggest that treatment of CT26‐bearing mice with Cy + AdIL‐12 reverts the inhibitory activity of Tregs on DCs, probably by reduction of IL‐10 and/or TGF‐β secretion.
3.4. Depletion of MDSCs induced by the combination of Cy with AdIL‐12 is reverted after re‐infusion of Tregs
It has been demonstrated that MDSCs contribute to generate an immunosuppressive microenvironment leading to tumour progression (Huang et al., 2006). We decided to investigate whether Cy + AdIL‐12 could affect the number of splenic MDSCs. The prevalence of CD11b+Gr1+ cells (Bronte et al., 1998) was determined in spleens derived from CT26 tumour‐bearing mice treated with Cy, AdIL‐12 or the combination by flow cytometry. We observed that Cy or AdIL‐12 as a single treatment induced a slight decrease in the amount of MDSCs in comparison to the saline group. However, Cy + AdIL‐12 induced a 3‐fold decrease in the percentage of splenic MDSCs (p < 0.01; Figure 4A). Interestingly, these reduced levels of MDSCs induced after Cy + AdIL‐12 treatment returned to baseline when Tregs from untreated tumour‐bearing mice were adoptively transferred (Figure 4B). These results suggest that the combined treatment affect the proportion of MDSCs. Also, the restitution of MDCSs levels after adoptive transfer of Tregs in Cy + AdIL‐12‐treated mice suggests a cross‐regulation between both cell types in our tumour model.
Figure 4.
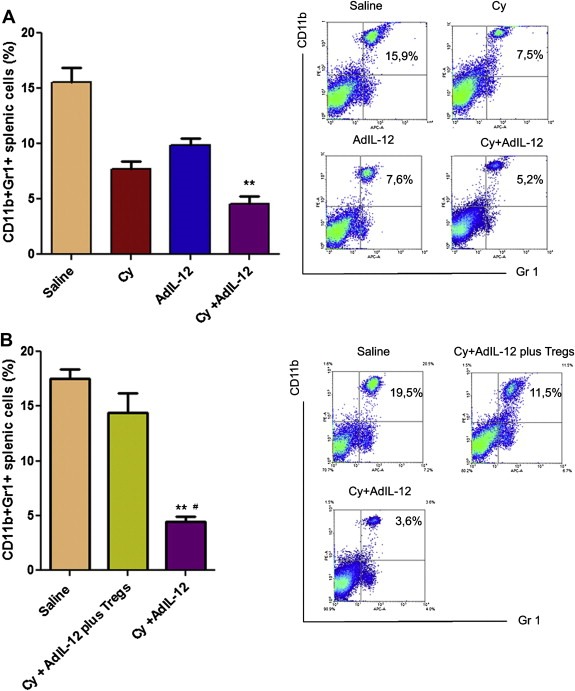
(A) Effect of combined treatment Cy + AdIL‐12 on splenic MDSCs (CD11b+Gr1+): Mann Whitney test, saline vs. Cy + Ad‐12 (∗∗p < 0.01). Data are representative of three independent experiments (n = 5/group per experiment) B) Effect of in vivo administration of Tregs on splenic MDSCs in mice treated with the combination Cy + AdIL‐12. Mann Whitney test, Cy + AdIL‐12 vs. Saline, ∗∗p < 0,01 and Cy + AdIL‐12 plus Tregs, #p < 0.05. Data are representative of two independent experiments (n = 6/group per experiment).
3.5. Adoptive transfer of in vitro expanded specific IFN‐γ secreting CD4+ T lymphocytes derived from Cy + AdIL‐12‐treated mice has potent antitumoural effects
We have previously found that sequential treatment of mice bearing CT26 with Cy followed by AdIL‐12 significantly increased IFN‐γ secretion by CD4+ T lymphocytes (Malvicini et al., 2009). Here, we decided to further assess whether in vitro expanded CD4+CD25− T cells (from here on CD4+) might have a direct therapeutic activity in vivo after i.v. injection in two models. In a therapeutic model (Figure 5A), CD4+ T cells from mice treated with Cy or AdIL‐12 as single agents were devoid of any significant effect on tumour growth and survival. On the contrary, CD4+ T cells from mice treated with Cy + AdIL‐12 showed a significant reduction of tumour volume (89% vs. saline group at day 23, p < 0,05; 81% and 78% vs. Cy and AdIL‐12 groups, respectively; at day 28, p < 0,05) and an increase in animal survival (p < 0.01). Similar result was obtained when CD4+ T cells from combined treatment were adoptively transferred in a preventive tumour model (Figure 5B). On the other hand, we investigated the in vitro cytotoxic activity of CD4+ T cells used for in vivo experiments and saw that cells derived from mice receiving the combined treatment displayed a significantly higher lytic activity against CT26 cells (p < 0.001 vs. saline; p < 0.01 vs. Cy or AdIL‐12) and produced higher levels of IFN‐γ in comparison with control group (p < 0.001 vs. saline; p < 0.01 vs. Cy or AdIL‐12) (Figure 5C). The specificity of cytotoxic effect was confirmed against BNL cells.
Figure 5.
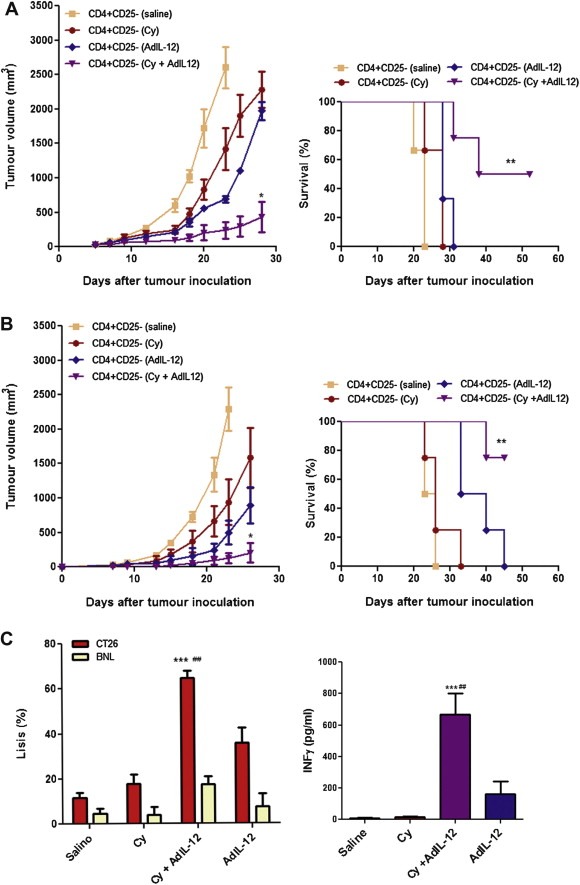
Antitumour effect of in vitro expanded CD4+ T‐lymphocytes from CT26‐bearing mice treated with Cy, AdIL‐12 or Cy + AdIL‐12. (A) Therapeutic model: BALB/c mice were s.c. inoculated with 5 × 105 CT26 cells into the right flank (day 0) and when tumours reached 65 mm3 in size (day 6) the mice were treated with vitro expanded CD4+ cells (2,5 × 106 i.v) from different experimental groups (n = 4/group) (left panel). Data are expressed as mean (bars, SEM). Mann Whitney test, days 23 and 28: CD4+CD25− (Cy + AdIL‐12) vs. CD4+CD25− (saline, Cy and AdIL‐12), ∗p < 0.05. Survival (right panel). Kaplan–Meier, log rank test, ∗∗p < 0.01. Data are representative of three independent experiments. B) Preventive model: BALB/c mice (n = 4/group) were simultaneously inoculated with 5 × 105 CT26 cells s.c. into the right flank and adoptive transfer of 2,5 × 106 CD4+ cells at the same time (day 0) (left panel). Data are expressed as mean (bars, SEM). Mann Whitney test, days 23 and 26:CD4+CD25− (Cy + AdIL‐12) vs. CD4+CD25− (saline, Cy and AdIL‐12), ∗p < 0.05. Survival (right panel). Kaplan–Meier, log rank test, ∗∗p < 0.01. Data are representative of three independent experiments. C) Specific CTL activity (leftt panel): CD4+ T cells from different experimental groups (n = 4/group) were isolated and stimulated in vitro with mitomycin C‐treated splenocytes and CT26 cells for 5 days. Specific CTL activity was evaluated against CT26 and BNL cells. The percentage of specific cytotoxicity was quantified whit the LDH Cytotoxicity Detection Kit and calculated according to the following formula: ([abs 492 nm experimental ‐ abs 492 nm background])/[abs 492 nm maximum ‐ abs492 nm background]) × 100. Mann Whitney test, Cy + AdIL‐12 vs. saline, ∗∗∗p < 0.001; Cy + AdIL‐12 vs. Cy and AdIL‐12, ###p < 0.01. Data represent the mean of triplicate cultures. Quantification of IFNγ (right panel): The amount of IFN‐γ secreted by CD4+T cells was evaluated after its isolation and in vitro stimulation as described above. Data are expressed as mean (bars, SEM). Kruskal–Wallis and Dunn's multiple comparisons test: Cy + AdIL‐12 vs. saline, ∗∗∗p < 0.001; Cy + AdIL‐12 vs. Cy and AdIL‐12, ##p < 0.01.
3.6. Cy + AdIL‐12 is highly effective in two stringent gastrointestinal cancer models
In order to examine whether combined treatment exert antitumoural effects in aggressive tumour models we investigated its therapeutic efficacy in a liver metastatic CRC model using CT26 cells and also against an established pancreatic cancer model using Panc02 cells. For this purpose, we injected CT26 cells into the liver of mice (day 0), treated with Cy (day 10) and 24 h later with AdIL‐12. Figure 6A (left panel) shows that no significant antimetastatic effect was observed when only Cy and AdIL‐12 were administered, whereas the combined therapy showed a significant reduction in metastases growth (98% vs. saline group, at day 20, p < 0.001). Therapeutic efficacy of the combination was superior to each individual agent (mean metastases volume±SE [mm3] at day 20: Cy + AdIL‐12: 15,6 ± 11,7; Cy: 191 ± 87,4; and AdIL‐12: 306 ± 167; p < 0.01) and complete metastases regressions were observed in 7 out of 15 animals (47% vs. 0% in saline and single agents groups). In addition, survival of mice receiving combined therapy was significantly higher than the controls (p < 0.001; Figure 6A; right panel). Remarkably, Cy followed by AdIL‐12 showed a potent antitumoural effect on Panc02 tumour nodules. Animals treated only with Cy or AdIL‐12 showed a significant reduction in tumour volume with respect to saline group (70% and 74% at day 54, respectively). However, no complete tumour regression was observed in mice treated with a single therapy. In contrast, the combined therapy resulted in a marked reduction in tumour volume (95% vs. saline group, at day 54; p < 0,05) and complete tumour regressions in 40% of animals (4/10) (Figure 6B, left panel). Importantly, the combined treatment produced a significant reduction in tumour growth in comparison to the effects of each single agent (mean tumour volume ± SE [mm3] at day 54; Cy + AdIL‐12: 209 ± 127; Cy: 1330 ± 358; AdIL‐12: 1177 ± 173; p < 0.05). Survival of mice receiving combined therapy was also significantly increased in comparison to mice receiving individual therapy or saline (p < 0.001, Figure 6B; right panel). Analysis of the in vivo interaction between both treatments was performed by the fractional product method (FTV) (Yokoyama et al., 2000). Figure 6C summarises the relative tumour volume of different groups at 4 different time points. On day 29 after treatment, in the combination group there was a 1.3‐fold improvement in the antitumour efficacy when compared to the expected additive effect. On days 33 and 36, Cy + AdIL‐12 showed a 1.4‐fold increase in the inhibition of tumour growth over an additive effect (expected fractional tumour volume). Moreover, on day 40 the increase was of 1.7‐fold over the additive effect. These results allow us to conclude that Cy + AdIL‐12 has a synergistic effect on pancreatic carcinoma growth inhibition. Similar doses of control adenovirus (Adβ‐Gal), alone or in combination with Cy, did not produce any significant change of tumour growth in both experimental models (data no shown). In both tumour models, the combined Cy plus AdIL‐12 strategy was well tolerated with no signs of toxicity (data not shown). Altogether, these results show that the antitumour effects of the combined treatment can be achieved in very aggressive tumour models including pancreatic cancer.
Figure 6.
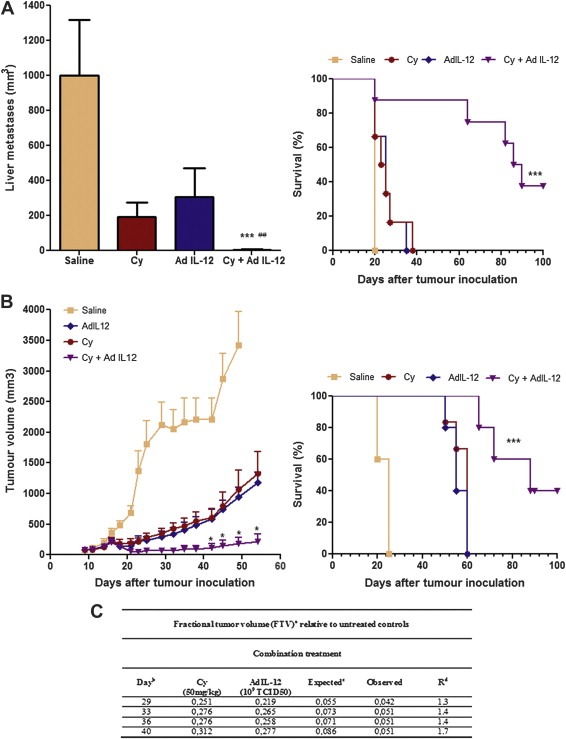
Effect of combined treatment Cy + AdIL‐12 on aggressive GIC models. A) Experimental liver metastases of CRC (CT26). Left panel, Liver metastases volume: BALB/c mice received an intra‐hepatic injection of 5 × 105 CT26 cells (day 0). Mice were distributed in different experimental groups and treated with saline (n = 6); Cy (50 mg/kg i.p; day 8, n = 6); AdIL‐12 (109 TCID50 i.t; day 9, n = 6); or Cy + AdIL‐12 (n = 8). Metastases volume was measured at day 20. Data are representative of three independent experiments. Mann Whitney test: Cy + AdIL‐12 vs. saline (∗∗∗p < 0.001); Cy + AdIL‐12 vs. AdIL‐12 or Cy (##p < 0.01). Right panel, Survival: Kaplan–Meier, log rank test, (∗∗∗p < 0.001). B) Pancreatic tumour model (Panc02). Left Panel, Tumour growth: C57BL/6 mice were inoculated s.c. in the right flank with 5 × 105 Panc02 cells. Treatments began when tumours reached 85 mm3 in size. Animals were treated with saline (n = 6), Cy (50 mg/kg i.p; day 8, n = 5), AdIL‐12 (109 TCID50 i.t; day 9, n = 5), or Cy + AdIL‐12 (n = 6). Tumour growth was assessed twice a week by calliper. Data are expressed as mean (bars, SEM). Data are representative of three independent experiments. Mann Whitney test: Cy + AdIL‐12 vs. Cy or AdIL‐12 (∗p < 0.05). Right panel, Survival: Kaplan–Meier, log rank test (∗∗∗p < 0.001). C) Analysis of the in vivo interaction between Cy and AdIL‐12 by the fractional product method (FTV) in Panc02 model. a FTV (experimental mean tumour volume)/(control mean tumour volume); b Day after treatment onset; c (Cy mean FTV) × (AdIL‐12 mean FTV); d R = Expected FTV/Observed FTV. A ratio >1 indicates a synergistic effect, and a ratio <1 indicates a less than additive effect.
4. Discussion
A number of immunotherapy strategies for advanced GIC are currently under preclinical and clinical evaluation (Mazzolini et al., 2007). However, there is a frustrating inconsistency in the correlation between biological and clinical responses. The reasons for the mentioned data discrepancies are manifold but the presence of strong mechanisms of immune escape induced by tumours appears to be important (Clark et al., 2009; Croci et al., 2007). Thus, it seemed reasonable to explore immunotherapy strategies aimed at inducing reversion of tolerogenic processes in tumour‐bearing hosts, such as those induced by Tregs (Zou, 2006).
We have previously demonstrated the synergistic antitumoural effect of sequential systemic administration of sub‐optimal Cy doses followed by immuno‐gene therapy with AdIL‐12 in mice with CRC (Malvicini et al., 2009). One important finding was that, regardless of the individual response of each mouse, the synergistic antitumour activity was associated with depletion of regulatory T cells.
We analysed the levels of Tregs after treatment with the combination and classified them according to the treatment response in R and NR mice. As a result, we observed that NR mice failed to significantly decrease Tregs significantly in spleen, peripheral blood, and inside tumours. The relevance of Tregs depletion to achieve a therapeutic response in our model was consistent when the efficacy of Cy + AdIL‐12 was completely abolished following reconstitution of Tregs by adoptive transfer. Importantly, the levels of Tregs reached with adoptive therapy in Cy + AdIL‐12‐treated mice (Figure 2C) were similar to the percentages of Tregs found in NR mice (Figure 1B; 1D). Considering that Tregs are up regulated in cancer and that they induce a detrimental effect on the immune system, strategies aimed at reducing Tregs number may increase the efficacy of any immunotherapy (Curiel et al., 2004; Ormandy et al., 2005; Sakaguchi et al., 2001). A myriad of inhibitory mechanisms have been proposed to explain the immunosuppression induced by Tregs, including secretion of cytokine suppressors and the induction of apoptosis/cell cycle arrest on effector T cells (Tang and Bluestone, 2008). It has also been suggested that Tregs could modulate the maturation and/or function of DCs (Larmonier et al., 2007; Onishi et al., 2008). Indeed, intravital microscopy studies have suggested that Tregs contact DCs more frequently than potential effector T cell targets (Bluestone and Tang, 2005). Taking into account the current data regarding Tregs function in cancer and to the way tumour cells drive Tregs induction, we decided to investigate the immunosuppressive effects of Tregs derived from tumour‐bearing mice and the effects of the combined Cy and AdIL‐12 treatment on their regulatory capacity (Beyer and Schultze, 2006; Ghiringhelli et al., 2005).
Previously we demonstrated that in vivo treatment with Cy + AdIL‐12 affected the maturation status of DCs. Currently, we provide new evidence showing that this effect is mediated, at least in part, by Tregs. Co‐cultures of naïve DC with Tregs from untreated animal tumours resulted in a significant inhibition of IL‐12 production as well as in MHC‐II and CD40 expression by DCs. More importantly, the inhibitory activity of Tregs on the maturation/activation status of DCs was completely abolished when Tregs derived from mice that received Cy + AdIL‐12 were used.
The mechanisms used by Tregs to generate immunosuppression remain conflictive and controversial. Direct cell–cell interaction between Tregs and their target cells (effector T cells or DCs) has been established as a prerequisite to exert their immunoregulatory activity (Vignali et al., 2008). On the other hand, the role of soluble factors, such as IL‐10 and TGF‐β has also been investigated extensively (Dieckmann et al., 2001; Takahashi et al., 1998; Thornton and Shevach, 1998). In the present work, we have detected high levels of both cytokines in supernatants of Tregs isolated from spleen of untreated tumours. These supernatants containing high levels of IL‐10 and TGF‐β were able to inhibit allogeneic splenocytes proliferation under stimulation with DCs. It is important to note that Cy + AdIL‐12 significantly reduced the production of IL‐10 and TGF‐β by Tregs and also abolished their inhibitory effect on lymphocyte proliferation. Altogether, our results suggest that treatment of CRC‐bearing mice with Cy + AdIL‐12 not only induces CD4+CD25+Foxp3+ T cell depletion but also modifies its functionality, at least by reduction of IL‐10 and TGF‐β production. Our present results strongly suggest that depletion of Tregs and/or inhibition of Tregs ability to induce immunosuppression are key immunoregulatory mechanisms associated with the synergistic antitumour effects achieved with Cy + AdIL‐12.
Recently, myeloid‐derived suppressor cells (MDSCs) have been recognized as a cell population that can negatively regulate T‐cell functions (Gabrilovich et al., 2001). These cells can act by inhibiting both the innate and adaptive immune responses (Huang et al., 2006). MDSCs are a heterogeneous population of myeloid cells that includes macrophages, granulocytes and other cells that express Ly‐6C/G (recognized by Gr‐1 antibody) and CD11b in mice and suppress immune responses in vivo and in vitro (Gabrilovich and Nagaraj, 2009). It has been observed that elimination of MDSC by antibodies or cytotoxic agents such as gemcitabine in mouse tumour models increased antitumour responses (Ko et al., 2010; Suzuki et al., 2005) In our study, we analysed the phenotype and frequency of CD11b+Gr‐1+ MDSCs in spleen of mice with CT26 tumours treated with Cy, AdIL‐12 or a combination of both. While the administration of Cy or AdIL‐12 as single agents induced a slight decrease in the percentage of MDSCs in comparison with the saline group, the combination of Cy + AdIL‐12 produced a significant reduction of MDSCs in the spleens of tumour‐bearing mice. However, other immunosuppressive CD11b populations, such as F4/80+ and CD11c+ cells, might be involved but were not analysed.
In agreement with our findings in CT26 tumour‐bearing mice, recent data published by Medina‐Echeverz et al. (2010), demonstrated that Cy in combination with IL‐12 depleted not only Tregs but also tumour‐infiltrating monocytic‐MDCs (Mo‐MDSCs) in another colorectal carcinoma model (MC38 cells). Using this approach, they observed that recruitment of inflammatory monocytes/macrophages (IMC) into tumour microenvironment after the elimination of Mo‐MDSC by Cy + IL‐12 has a key role for the observed antitumoural effect (Medina‐Echeverz et al., 2010)
In our tumour model the immune rejection of established CT26 tumours was clearly affected by negative regulatory mechanisms linked to an increase in number and function of Tregs and MDSCs. We demonstrated that these negative influences could be overcome by the combination of Cy with AdIL‐12. More importantly, the combined therapy not only has reverted immunosuppressive mechanisms induced during tumour progression but also generated a strong cytotoxic immune response against tumour cells mediated by specific IFN‐γ secreting CD4+ T lymphocytes.
The majority of immune therapy strategies to treat cancer have directed to the generation of CD8+ T cell responses against tumour‐associated antigens. CD4+T cells have the ability to sustain CTL responses but can also function as effector cells either by production of potent cytokines (such as IFN‐γ) or by exhibiting anticancer cytotoxic activity (Appay et al., 2002; Haigh et al., 2008; Spaapen et al., 2010). In our work, the in vitro expanded specific IFN‐γ secreting CD4+ T lymphocytes generated by Cy + AdIL‐12 have demonstrated to be effective in both a preventive and a therapeutic model using CT26 tumour model. We assume that the induction of high cytolytic activity and the potent Th1‐type response in vivo might be the cause for the profound antitumour effects observed and could be also exploited in the future in combination with other immunostimulatory strategies.
The use of low‐dose Cy combined with sub‐therapeutic AdIL‐12 induces significant regressions in two different and aggressive tumour models: the CT26 liver metastatic model and the pancreatic cancer model. In this study we have shown by the fractional product method that the combined treatment has also a synergistic effect for pancreatic carcinoma. Thus, Cy + AdIL‐12 based treatment is a promising immunotherapy among the limited therapeutic options suggesting the possibility of achieving high efficacy with IL‐12 without toxicity in more malignant tumour models. This strategy could be improved in patients with disseminated disease by the use of tumour‐targeted expression of IL‐12 (Kerkar et al., 2010). In conclusion, Cy + AdIL‐12 demonstrated efficacy to block Tregs and MDSCs‐mediated immunosuppressive tumour environment and, simultaneously, to amplify the effector phase of the immune response by the induction of a potent antitumour CTL activity. Considering our previous clinical experience of separate usage of IL‐12 gene transfer and Cy in patients with cancer, it will be of interest to evaluate this combination as a potential therapeutic strategy in advanced GI carcinomas clinically.
Supporting information
Supplementary data
{kind=link}
Acknowledgements
We would like to thank, Soledad Arregui and Guillermo Gastón for their technical assistance. This work was supported in part by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) grant PICT‐2005/34788 (PM, OGS and GM); PICTO‐CRUP 2005/31179 (GM); AECID 2008 D/022066/08 (GM); PIP‐CONICET 2009‐2011 (PM). MM, JB and CA are fellows from ANPCyT. FP is a fellow from CONICET.
Supplementary data 1.
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molonc.2011.03.007.
Malvicini Mariana, Ingolotti Mariana, Piccioni Flavia, Garcia Mariana, Bayo Juan, Atorrasagasti Catalina, Alaniz Laura, Aquino Jorge B., Espinoza Jaime A., Gidekel Manuel, Scharovsky O. Graciela, Matar Pablo and Mazzolinia Guillermo, (2011), Reversal of gastrointestinal carcinoma‐induced immunosuppression and induction of antitumoural immunity by a combination of cyclophosphamide and gene transfer of IL‐12, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.03.007.
Contributor Information
Pablo Matar, Email: matarpablo@hotmail.com.
Guillermo Mazzolini, Email: gmazzoli@cas.austral.edu.ar.
References
- Alaniz, L. , Rizzo, M. , Malvicini, M. , Jaunarena, J. , Avella, D. , Atorrasagasti, C. , Aquino, J.B. , Garcia, M. , Matar, P. , Silva, M. , Mazzolini, G. , 2009. Low molecular weight hyaluronan inhibits colorectal carcinoma growth by decreasing tumor cell proliferation and stimulating immune response. Cancer Lett.. 278, 9–16. [DOI] [PubMed] [Google Scholar]
- Almand, B. , Clark, J.I. , Nikitina, E. , van Beynen, J. , English, N.R. , Knight, S.C. , Carbone, D.P. , Gabrilovich, D.I. , 2001. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J. Immunol.. 166, 678–689. [DOI] [PubMed] [Google Scholar]
- Appay, V. , Zaunders, J.J. , Papagno, L. , Sutton, J. , Jaramillo, A. , Waters, A. , Easterbrook, P. , Grey, P. , Smith, D. , McMichael, A.J. , Cooper, D.A. , Rowland-Jones, S.L. , Kelleher, A.D. , 2002. Characterization of CD4(+) CTLs ex vivo. J. Immunol.. 168, 5954–5958. [DOI] [PubMed] [Google Scholar]
- Barajas, M. , Mazzolini, G. , Genove, G. , Bilbao, R. , Narvaiza, I. , Schmitz, V. , Sangro, B. , Melero, I. , Qian, C. , Prieto, J. , 2001. Gene therapy of orthotopic hepatocellular carcinoma in rats using adenovirus coding for interleukin 12. Hepatology. 33, 52–61. [DOI] [PubMed] [Google Scholar]
- Bell, D. , Chomarat, P. , Broyles, D. , Netto, G. , Harb, G.M. , Lebecque, S. , Valladeau, J. , Davoust, J. , Palucka, K.A. , Banchereau, J. , 1999. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J. Exp. Med.. 190, 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer, M. , Schultze, J.L. , 2006. Regulatory T cells in cancer. Blood. 108, 804–811. [DOI] [PubMed] [Google Scholar]
- Bluestone, J.A. , Tang, Q. , 2005. How do CD4+CD25+ regulatory T cells control autoimmunity?. Curr. Opin. Immunol.. 17, 638–642. [DOI] [PubMed] [Google Scholar]
- Brodsky, R.A. , Petri, M. , Smith, B.D. , Seifter, E.J. , Spivak, J.L. , Styler, M. , Dang, C.V. , Brodsky, I. , Jones, R.J. , 1998. Immunoablative high-dose cyclophosphamide without stem-cell rescue for refractory, severe autoimmune disease. Ann. Intern. Med.. 129, 1031–1035. [DOI] [PubMed] [Google Scholar]
- Bronte, V. , Wang, M. , Overwijk, W.W. , Surman, D.R. , Pericle, F. , Rosenberg, S.A. , Restifo, N.P. , 1998. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J. Immunol.. 161, 5313–5320. [PMC free article] [PubMed] [Google Scholar]
- Caruso, M. , Pham-Nguyen, K. , Kwong, Y.L. , Xu, B. , Kosai, K.I. , Finegold, M. , Woo, S.L. , Chen, S.H. , 1996. Adenovirus-mediated interleukin-12 gene therapy for metastatic colon carcinoma. Proc. Natl. Acad. Sci. U S A. 93, 11302–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, C.E. , Beatty, G.L. , Vonderheide, R.H. , 2009. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett.. 279, 1–7. [DOI] [PubMed] [Google Scholar]
- Colombo, M.P. , Forni, G. , 1994. Cytokine gene transfer in tumor inhibition and tumor therapy: where are we now?. Immunol. Today. 15, 48–51. [DOI] [PubMed] [Google Scholar]
- Colvin, O.M. , 1999. An overview of cyclophosphamide development and clinical applications. Curr. Pharm. Des. 5, 555–560. [PubMed] [Google Scholar]
- Croci, D.O. , Zacarias Fluck, M.F. , Rico, M.J. , Matar, P. , Rabinovich, G.A. , Scharovsky, O.G. , 2007. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol. Immunother. 56, 1687–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel, T.J. , Coukos, G. , Zou, L. , Alvarez, X. , Cheng, P. , Mottram, P. , Evdemon-Hogan, M. , Conejo-Garcia, J.R. , Zhang, L. , Burow, M. , Zhu, Y. , Wei, S. , Kryczek, I. , Daniel, B. , Gordon, A. , Myers, L. , Lackner, A. , Disis, M.L. , Knutson, K.L. , Chen, L. , Zou, W. , 2004. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med.. 10, 942–949. [DOI] [PubMed] [Google Scholar]
- Dieckmann, D. , Plottner, H. , Berchtold, S. , Berger, T. , Schuler, G. , 2001. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J. Exp. Med.. 193, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkord, E. , Hawkins, R.E. , Stern, P.L. , 2008. Immunotherapy for gastrointestinal cancer: current status and strategies for improving efficacy. Expert Opin. Biol. Ther.. 8, 385–395. [DOI] [PubMed] [Google Scholar]
- Ferlay, J. , Shin, H.R. , Bray, F. , Forman, D. , Mathers, C. , Parkin, D.M. , 2010. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer. [DOI] [PubMed] [Google Scholar]
- Gabrilovich, D.I. , Nagaraj, S. , 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol.. 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich, D.I. , Velders, M.P. , Sotomayor, E.M. , Kast, W.M. , 2001. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J. Immunol.. 166, 5398–5406. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli, F. , Larmonier, N. , Schmitt, E. , Parcellier, A. , Cathelin, D. , Garrido, C. , Chauffert, B. , Solary, E. , Bonnotte, B. , Martin, F. , 2004. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol.. 34, 336–344. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli, F. , Puig, P.E. , Roux, S. , Parcellier, A. , Schmitt, E. , Solary, E. , Kroemer, G. , Martin, F. , Chauffert, B. , Zitvogel, L. , 2005. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med.. 202, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, G.G. , Hutchison, R.B. , Kruse, C.A. , 2001. Chemo-immunotherapy and chemo-adoptive immunotherapy of cancer. Cancer Treat. Rev.. 27, 375–402. [DOI] [PubMed] [Google Scholar]
- Haigh, T.A. , Lin, X. , Jia, H. , Hui, E.P. , Chan, A.T. , Rickinson, A.B. , Taylor, G.S. , 2008. EBV latent membrane proteins (LMPs) 1 and 2 as immunotherapeutic targets: LMP-specific CD4+ cytotoxic T cell recognition of EBV-transformed B cell lines. J. Immunol.. 180, 1643–1654. [DOI] [PubMed] [Google Scholar]
- Huang, B. , Pan, P.Y. , Li, Q. , Sato, A.I. , Levy, D.E. , Bromberg, J. , Divino, C.M. , Chen, S.H. , 2006. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res.. 66, 1123–1131. [DOI] [PubMed] [Google Scholar]
- Kerkar, S.P. , Muranski, P. , Kaiser, A. , Boni, A. , Sanchez-Perez, L. , Yu, Z. , Palmer, D.C. , Reger, R.N. , Borman, Z.A. , Zhang, L. , Morgan, R.A. , Gattinoni, L. , Rosenberg, S.A. , Trinchieri, G. , Restifo, N.P. , 2010. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res.. 70, 6725–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, J.S. , Rayman, P. , Ireland, J. , Swaidani, S. , Li, G. , Bunting, K.D. , Rini, B. , Finke, J.H. , Cohen, P.A. , 2010. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res.. 70, 3526–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larmonier, N. , Marron, M. , Zeng, Y. , Cantrell, J. , Romanoski, A. , Sepassi, M. , Thompson, S. , Chen, X. , Andreansky, S. , Katsanis, E. , 2007. Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol. Immunother.. 56, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard, J.P. , Sherman, M.L. , Fisher, G.L. , Buchanan, L.J. , Larsen, G. , Atkins, M.B. , Sosman, J.A. , Dutcher, J.P. , Vogelzang, N.J. , Ryan, J.L. , 1997. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 90, 2541–2548. [PubMed] [Google Scholar]
- Lorenz, M. , Staib-Sebler, E. , Hochmuth, K. , Heinrich, S. , Gog, C. , Vetter, G. , Encke, A. , Muller, H.H. , 2000. Surgical resection of liver metastases of colorectal carcinoma: short and long-term results. Semin. Oncol.. 27, 112–119. [PubMed] [Google Scholar]
- Malvicini, M. , Rizzo, M. , Alaniz, L. , Pinero, F. , Garcia, M. , Atorrasagasti, C. , Aquino, J.B. , Rozados, V. , Scharovsky, O.G. , Matar, P. , Mazzolini, G. , 2009. A novel synergistic combination of cyclophosphamide and gene transfer of interleukin-12 eradicates colorectal carcinoma in mice. Clin. Cancer Res.. 15, 7256–7265. [DOI] [PubMed] [Google Scholar]
- Matar, P. , Rozados, V.R. , Gervasoni, S.I. , Scharovsky, G.O. , 2002. Th2/Th1 switch induced by a single low dose of cyclophosphamide in a rat metastatic lymphoma model. Cancer Immunol. Immunother.. 50, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzolini, G. , Murillo, O. , Atorrasagasti, C. , Dubrot, J. , Tirapu, I. , Rizzo, M. , Arina, A. , Alfaro, C. , Azpilicueta, A. , Berasain, C. , Perez-Gracia, J.L. , Gonzalez, A. , Melero, I. , 2007. Immunotherapy and immunoescape in colorectal cancer. World J. Gastroenterol.. 13, 5822–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzolini, G. , Prieto, J. , Melero, I. , 2003. Gene therapy of cancer with interleukin-12. Curr. Pharm. Des.. 9, 1981–1991. [DOI] [PubMed] [Google Scholar]
- Mazzolini, G. , Qian, C. , Xie, X. , Sun, Y. , Lasarte, J.J. , Drozdzik, M. , Prieto, J. , 1999. Regression of colon cancer and induction of antitumor immunity by intratumoral injection of adenovirus expressing interleukin-12. Cancer Gene Ther.. 6, 514–522. [DOI] [PubMed] [Google Scholar]
- Medina-Echeverz, J. , Fioravanti, J. , Zabala, M. , Ardaiz, N. , Prieto, J. , Berraondo, P. , 2010. Successful colon cancer eradication after chemoimmunotherapy is associated with profound phenotypic change of intratumoral myeloid cells. J. Immunol.. 186, 807–815. [DOI] [PubMed] [Google Scholar]
- Melero, I. , Mazzolini, G. , Narvaiza, I. , Qian, C. , Chen, L. , Prieto, J. , 2001. IL-12 gene therapy for cancer: in synergy with other immunotherapies. Trends Immunol.. 22, 113–115. [DOI] [PubMed] [Google Scholar]
- Nestle, F.O. , Burg, G. , Fah, J. , Wrone-Smith, T. , Nickoloff, B.J. , 1997. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen-presenting cells. Am. J. Pathol.. 150, 641–651. [PMC free article] [PubMed] [Google Scholar]
- Onishi, Y. , Fehervari, Z. , Yamaguchi, T. , Sakaguchi, S. , 2008. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. U S A. 105, 10113–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormandy, L.A. , Hillemann, T. , Wedemeyer, H. , Manns, M.P. , Greten, T.F. , Korangy, F. , 2005. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res.. 65, 2457–2464. [DOI] [PubMed] [Google Scholar]
- Proietti, E. , Greco, G. , Garrone, B. , Baccarini, S. , Mauri, C. , Venditti, M. , Carlei, D. , Belardelli, F. , 1998. Importance of cyclophosphamide-induced bystander effect on T cells for a successful tumor eradication in response to adoptive immunotherapy in mice. J. Clin. Invest.. 101, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzer, B.M. , Stiewe, T. , Rodicker, F. , Schildgen, O. , Ruhm, S. , Dirsch, O. , Fiedler, M. , Damen, U. , Tennant, B. , Scherer, C. , Graham, F.L. , Roggendorf, M. , 2001. Large nontransplanted hepatocellular carcinoma in woodchucks: treatment with adenovirus-mediated delivery of interleukin 12/B7.1 genes. J. Natl. Cancer Inst.. 93, 472–479. [DOI] [PubMed] [Google Scholar]
- Rabinovich, G.A. , Gabrilovich, D. , Sotomayor, E.M. , 2007. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol.. 25, 267–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi, S. , Sakaguchi, N. , Shimizu, J. , Yamazaki, S. , Sakihama, T. , Itoh, M. , Kuniyasu, Y. , Nomura, T. , Toda, M. , Takahashi, T. , 2001. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev.. 182, 18–32. [DOI] [PubMed] [Google Scholar]
- Sangro, B. , Mazzolini, G. , Ruiz, J. , Herraiz, M. , Quiroga, J. , Herrero, I. , Benito, A. , Larrache, J. , Pueyo, J. , Subtil, J.C. , Olague, C. , Sola, J. , Sadaba, B. , Lacasa, C. , Melero, I. , Qian, C. , Prieto, J. , 2004. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J. Clin. Oncol.. 22, 1389–1397. [DOI] [PubMed] [Google Scholar]
- Shapira, S. , Lisiansky, V. , Arber, N. , Kraus, S. , 2010. Targeted immunotherapy for colorectal cancer: monoclonal antibodies and immunotoxins. Expert Opin. Investig. Drugs. 19, (Suppl 1) S67–S77. [DOI] [PubMed] [Google Scholar]
- Spaapen, R.M. , Groen, R.W. , van den Oudenalder, K. , Guichelaar, T. , van Elk, M. , Aarts-Riemens, T. , Bloem, A.C. , Storm, G. , Martens, A.C. , Lokhorst, H.M. , Mutis, T. , 2010. Eradication of medullary multiple myeloma by CD4+ cytotoxic human T lymphocytes directed at a single minor histocompatibility antigen. Clin. Cancer Res.. 16, 5481–5488. [DOI] [PubMed] [Google Scholar]
- Suzuki, E. , Kapoor, V. , Jassar, A.S. , Kaiser, L.R. , Albelda, S.M. , 2005. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res.. 11, 6713–6721. [DOI] [PubMed] [Google Scholar]
- Takahashi, T. , Kuniyasu, Y. , Toda, M. , Sakaguchi, N. , Itoh, M. , Iwata, M. , Shimizu, J. , Sakaguchi, S. , 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol.. 10, 1969–1980. [DOI] [PubMed] [Google Scholar]
- Tang, Q. , Bluestone, J.A. , 2008. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol.. 9, 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, A.M. , Shevach, E.M. , 1998. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med.. 188, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri, G. , 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol.. 3, 133–146. [DOI] [PubMed] [Google Scholar]
- Vignali, D.A. , Collison, L.W. , Workman, C.J. , 2008. How regulatory T cells work. Nat. Rev. Immunol.. 8, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, Y. , Dhanabal, M. , Griffioen, A.W. , Sukhatme, V.P. , Ramakrishnan, S. , 2000. Synergy between angiostatin and endostatin: inhibition of ovarian cancer growth. Cancer Res.. 60, 2190–2196. [PubMed] [Google Scholar]
- Zou, W. , 2005. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer. 5, 263–274. [DOI] [PubMed] [Google Scholar]
- Zou, W. , 2006. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol.. 6, 295–307. [DOI] [PubMed] [Google Scholar]
- Zwirner, N.W. , Croci, D.O. , Domaica, C.I. , Rabinovich, G.A. , 2010. Overcoming the hurdles of tumor immunity by targeting regulatory pathways in innate and adaptive immune cells. Curr. Pharm. Des. 16, 255–267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data