

Abstract
Differentiation‐inducing therapy has been proposed to be a novel potential approach to treat malignant gliomas. Glial fibrillary acidic protein (GFAP) is a well‐known specific astrocyte biomarker and acts as a tumor suppressor gene (TSG) in glioma pathogenesis. Previously we reported that a traditional biotoxin cholera toxin could induce malignant glioma cell differentiation characterized by morphologic changes and dramatic GFAP expression. However, the molecular mechanisms underlying GFAP induction are still largely unknown. Here we demonstrate that an oncogenic pathway interleukin‐6/janus kinase‐2/signal transducer and activator of transcription 3 (IL‐6/JAK2/STAT3) cascade mediates the cholera toxin‐induced GFAP expression. Cholera toxin dramatically stimulated GFAP expression at the transcriptional level in C6 glioma cells. Meanwhile, phosphorylation of STAT3 and JAK2 was highly induced in a time‐dependent manner after cholera toxin incubation, whereas no changes of STAT3 and JAK2 were observed. Furthermore, the IL‐6 gene was quickly induced by cholera toxin and subsequent IL‐6 protein secretion was stimulated. Importantly, exogenous recombinant rat IL‐6 can also induce phosphorylation of STAT3 concomitant with GFAP expression while JAK2 specific inhibitor AG490 could effectively block both cholera toxin‐ and IL‐6‐induced GFAP expression. Given that the methylation of the STAT3 binding element can suppress GFAP expression, we detected the methylation status of the critical recognition sequence of STAT3 in the promoter of GFAP gene (−1518 ∼ −1510) and found that it was unmethylated in C6 glioma cells. In addition, neither DNA methyltransferase1 (DNMT1) inhibitor 5‐Aza‐2′‐deoxycytidine (5‐AZa‐CdR) nor silencing DNMT1 can stimulate GFAP expression, indicating that the loss of GFAP expression in C6 cells is not caused by its promoter hypermethylation. Taken together, our findings suggest that activation of a pro‐survival IL‐6/JAK2/STAT3 cascade contributes to cholera toxin‐induced GFAP expression, which implies that a survival‐promoting signal may also play a differentiation‐supporting role in malignant gliomas.
Keywords: Interleukin-6, Janus kinase-2, Signal transducer and activator of transcription 3, Glioma, Glial fibrillary acidic protein, Cholera toxin
Highlights
Cholera toxin activates oncogenic IL‐6/JAK2/STAT3 pathway in C6 glioma cells.
Activation of the pro‐survival IL‐6/JAK2/STAT3 pathway results in GFAP induction.
The promoter of GFAP gene is demethylated in C6 glioma cells.
A survival‐promoting signal may also play a differentiation‐supporting role in malignant gliomas.
Abbreviations
- IL-6
Interleukin-6
- JAK2
Janus kinase-2
- STAT3
Signal transducer and activator of transcription 3
- GFAP
Glial fibrillary acidic protein
- ELISA
enzyme-linked immunosorbent assay
- 5-AZA
5-Aza-2′-deoxycytidine
- DNMT1
DNA methyltransferase1
- siDNMT1
siRNA-mediated knockdown of DNMT1
1. Introduction
Malignant gliomas derived from astrocytes or astroglial precursors are the most common primary tumors of the adult central nervous system (CNS). Current therapies such as irradiation and chemotherapy are often ineffective and fail to improve its prognosis (Bao et al., 2006; Curran et al., 1993). Differentiation‐inducing therapy, which modifies cancer cell differentiation and is the most successful for the treatment of acute myelocytic leukemia (Huang et al., 1988), has been proposed to be a novel potential approach to treat malignant tumors, including gliomas (Leszczyniecka et al., 2001; Li et al., 2007). Despite these advances, a detailed knowledge of the molecular mechanisms that regulate the malignant glioma cell differentiation is still missing.
Glial fibrillary acidic protein (GFAP), an astrocyte‐specific intermediate filament thought to provide structural support to normal astrocytes, is a well‐known biomarker of astrocyte differentiation (Roymans et al., 2001). Early reports demonstrated that transfection of astrocytoma cells with GFAP complementary DNA could effectively inhibit proliferation, tumorigenicity and induce morphologic alteration. Conversely, transfection of antisense GFAP complementary DNA could strongly enhance growth, invasion, and adhesion of astrocytoma cells (Rutka et al., 1994; Rutka and Smith, 1993). Furthermore, a critical observation from histopathological studies shows that there is progressive loss of GFAP expression with increasing astrocytic anaplasia (Duffy et al., 1982). These findings suggest that GFAP may act as a tumor suppressor gene (TSG) in astrocytoma pathogenesis. We previously reported that cholera toxin, the traditional biotoxin and well‐known inducer of accumulation of cellular cAMP, was capable of inducing astrocytic differentiation on malignant gliomas via the PKA/CREB pathway (Li et al., 2007). This astrocytic differentiation was characterized by typical morphological changes as well as dramatic increased expression of GFAP. However, little progress has been made about the critical mechanism underlying the up‐regulation of GFAP induced by cholera toxin.
The oncogenic role of JAK2/STAT3 has been elucidated in a number of human malignancies, including glioma (Adachi et al., 2006; Bowman et al., 2000; Bromberg et al., 1999). It is known that STAT3 activation can be stimulated by interleukin‐6 (IL‐6) family of cytokines mobilizing gp130 receptor subunits, followed by recruitment of cytosolic JAKs. The latter phosphorylate STAT3, which then translocates into the nucleus and alters gene expression by binding to STAT3‐responsive element (Miller and Gauthier, 2007). Both c‐myc and pim have been identified as target genes of STAT3 and together can promote cell survival and G1 to S cell‐cycle‐transition. In addition, STAT3 is also required for gp130‐mediated maintenance of the pluripotential state of proliferating embryonic stem cells and for the gp130‐induced macrophage differentiation of M1 cells (Hirano et al., 2000). A growing number of tumor derived cell lines as well as samples from human cancers are reported to contain constitutively activated STAT proteins, very frequently STAT3 (Garcia and Jove, 1998). The constitutively activated STAT3 which qualified as a protooncogene is capable of driving transcription and induces cell transformation (Bromberg et al., 1999). In contrast, blockage of JAK2/STAT3 pathway by inhibitors or siRNAs can reduce cell survival and induce apoptosis (Kudo et al., 2009; Kupferman et al., 2009; Yang et al., 2010). Together, these findings provide compelling evidence that aberrant activation of JAK2/STAT3 associated with oncogenesis is not merely adventitious but instead is likely to play a central role in the process of malignant transformation.
DNA methylation at CpG dinucleotides is a major epigenetic modification of mammalian genomes which can regulate several aspects of gene expression, such as genomic imprinting, aging, and tumorigenesis (Bird and Wolffe, 1999; Razin, 1998; Robertson and Jones, 2000). In mammals, DNA methylation is catalyzed primarily by three DNA methyltransferases, DNMT1, DNMT3a, and DNMT3b, and occurs largely at the cytosine residues contained within the dinucleotide sequence cytosine‐phosphate diesterguanine (CpG) (Bestor et al., 1988; Okano et al., 1999). Selective depletion of DNMT1 with DNMT inhibitors or siRNAs has been shown to induce demethylation of promoters and re‐expression of the silenced TSGs (Costello et al., 1996; Foltz et al., 2009). Increasing evidences indicate that GFAP expression during astrocyte differentiation in the developing brain is governed by both extrinsic environmental cues and cell intrinsic programs, where the former involves JAK2/STAT3 pathway and the latter involves the methylation status of the promoter of the gene for astrocyte‐specific protein GFAP in this case (Deneen et al., 2006; Namihira et al., 2009; Takizawa et al., 2001a). Both the activation of JAK2/STAT3 cascade and the demethylation of the binding element of STAT3 in the promoter of GFAP are required for GFAP expression.
In this study, we demonstrate that the traditional biotoxin cholera toxin is capable of activating IL‐6/JAK2/STAT3 cascade, which thereby induce GFAP expression in C6 glioma cells. Our findings suggest that cholera toxin can stimulate a pro‐survival signal to induce GFAP expression, which may pave a way to better understand the molecular mechanisms underlying the cholera toxin‐induced differentiation of malignant glioma cells.
2. Materials and methods
2.1. Cell cultures and reagents
C6 and T98G glioma cells were obtained from the American Type Culture Collection (Manassas, USA). Cells maintained in DMEM (Invitrogen, USA) supplemented with 10% FBS and a humidified atmosphere of 5% CO2 at 37 °C. Cell differentiation was induced with cholera toxin (Sigma, USA) in DMEM containing 1% FBS. Control was treated with an equivalent volume of DMEM containing 1% FBS. Chemical reagents were obtained from the following companies: AG490 (Sigma, USA), reconstitute rat IL‐6(R&D Systems, USA) and 5‐AZa‐CdR (Calbiochem, USA).
2.2. Western blot analysis
Western blot was performed as described (Akagi et al., 2002). The following antibodies were used: antibodies against STAT3, Phospho‐STAT3 (tyr705), JAK2, Phospho‐JAK2 (tyr1007/1008), GFAP (1:1000; Cell Signaling Technology, USA) and β‐actin (1:2000; New England Biolabs, USA), DNMT1 (1:1000; Santa Cruz Biotechnology, USA).
2.3. siRNA‐mediated knockdown of DNMT1 expression
siDNMT1 and negative control (NC) oligonucleotides were purchased from Ribobio, China. C6 cells grown to 30–50% confluence were transfected by using HiPerFect Transfection Reagent (QIAGEN, Germany) according to the manufacturer's protocol. Inhibition of protein expression was assessed by immunoblot analysis.
2.4. Reverse transcription–polymerase chain reaction
Total RNA of treated cultures was extracted using a TRIzol kit (Invitrogen). RNA (1 μg) was used for reverse transcription with a commercial kit (Invitrogen). PCR was performed using an initial step of denaturation (5 min at 94 °C), 30 cycles of amplification (94 °C for 30 s, 53 °C for 1 min and 72 °C for 1 min) and an extension (72 °C for 10 min). PCR products were analyzed on 2% agarose gels. The primers used were as follows. GFAP: forward, 5′‐TGT AGG AGT GGG TAG GGC ‐3′; and reverse, 5′‐CTG AGC AAC CAG GAA TAG AC ‐3′; IL‐6: forward, 5′‐CAG TTG C CT TCT TGG GAC TG‐3′; and reverse, 5′‐ACA GTG CAT CAT CGC TGT TC ‐3′. β‐actin: forward, 5′‐AGG CTC TTT TCC AGC CTT CCT ‐3′; and reverse, 5′‐GTC TTT ACG GAT GTC AAC GTC ACA ‐3′.
2.5. Enzyme‐linked immunosorbent assay
Culture medium from C6 cells treated with cholera toxin (10 ng/ml) was harvested at the indicated points in time. Rat IL‐6 protein levels in the culture medium were detected by enzyme‐linked immunosorbent assay using Quantikine M (R&D Systems) according to the manufacturer's protocol.
2.6. DNA methylation analyses
Bisulfite treatment of genomic DNA was performed as previously described (Honoki et al., 2004; Strelkov and Davie, 2002). Briefly, genomic DNA was extracted with a DNeasy Blood & Tissue Kit (QIAGEN). The extracted DNA was modified by bisulfite using EpiTect Bisulfite Kits (QIAGEN) according to the protocol. For bisulfite sequencing, PCR was performed with the following primer sets; GFAP‐BS‐F: 5′‐ GGA GAG GTA AGT AGT TAG GTT TGG TTT ‐3′, GFAP‐BS‐R: 5′‐ TAC AAT ACA AAC TCC CAA CTC AAT AAA ‐3′ (annealing temperature: 50 °C, 40 cycles). The primer was designed by using MethPrimer (www.urogene.org/methprimer/index1.html) (Li and Dahiya, 2002). PCR products were recycled with QIAquick Gel Extraction Kit (QIAGEN) and subcloned with a TOPO TA cloning kit (Invitrogen). For each sample, 10 clones were sequenced.
2.7. Statistical analysis
Data are presented as mean ± SD of three separated experiments if not noticed. A difference with a P value 0.05 by ANOVA was considered statistical significant.
3. Results
3.1. Cholera toxin induces differentiation of C6 glioma cells
Microscopic observation of C6 glioma cells treated for 48 h with 10 ng/ml cholera toxin revealed major alterations in morphology. We first observed that the cell numbers were dramatically reduced after cholera toxin incubation compared with control group. Interestingly, unlike the mainly polygonal morphology of the control, the shape of cholera toxin‐treated cells was similar to that of mature astrocytes, with smaller round cell bodies and much longer, fine, tapering processes (Figure 1A). Importantly, GFAP protein, an established biomarker of mature astrocytes was strongly up‐regulated even after 24 h treatment of cholera toxin (Figure 1B). Moreover, the mRNA of GFAP was also highly up‐regulated in a time‐dependent manner induced by cholera toxin (Figure 1C). Together, these results are consistent with what we found previously (Li et al., 2007) and indicate that cholera toxin can stimulate GFAP expression at the transcriptional level.
Figure 1.

Cholera toxin induces differentiation of C6 malignant glioma cells. (A), morphologic transformation (Original magnification: ×200). C6 cells were incubated with 10 ng/ml cholera toxin for 48 h. (B), Time‐dependent effect of GFAP protein expression. (C), Time‐dependent effect of GFAP mRNA level. C6 cells were incubated with 10 ng/ml cholera toxin for the indicated times.
3.2. Cholera toxin activates JAK2/STAT3 pathway in C6 glioma cells
As the transcriptional stimulation of GFAP mRNA can result from the activation of its transcriptional factors, we tested the phosphorylation of STAT3 on the tyr705 residue (P‐STAT3), a confirmed transcriptional factor of GFAP (Takizawa et al., 2001a). As shown in Figure 2A, treatment of 10 ng/ml cholera toxin elevated the P‐STAT3 expression within 12 h of stimulation and induction of P‐STAT3 increased dramatically in 24 h and continuously until 48 h. Nevertheless, no change in level of STAT3 was observed during the stimulation period, suggesting that the up‐regulation of P‐STAT3 might be attributed to the activation of upstream factors. Consistently, the phosphorylation of JAK2 on the tyr1007/1008 residue (P‐JAK2) which can phosphorylated STAT3 on the tyr705 residue directly was up‐regulated in a time‐dependent manner induced by cholera toxin concomitant with unaltered JAK2 expression (Figure 2B). These results suggest that cholera toxin is capable of activating JAK2/STAT3 pathway in C6 glioma cells.
Figure 2.
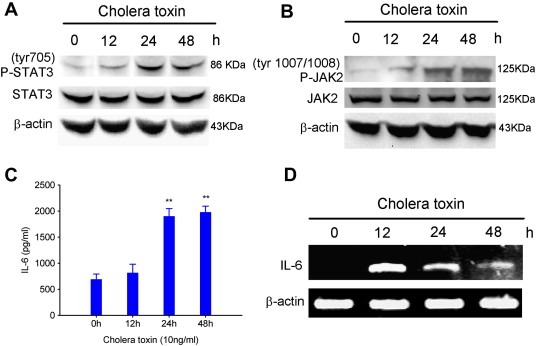
Cholera toxin activates IL‐6/JAK2/STAT3 pathway in C6 glioma cells. Time‐dependent effects of phosphorylation of STAT3(tyr705). (A), phosphorylation of JAK2(tyr1007/1008). (B), IL‐6 protein expression. (C) and mRNA level. (D). C6 cells were incubated with 10 ng/ml cholera toxin for the indicated times, ∗∗, P < 0.01.
3.3. Cholera toxin stimulates IL‐6 expression at the transcriptional level in C6 glioma cells
The JAK2/STAT3 pathway can be activated by IL‐6 family cytokines, including IL‐6, IL‐27, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF) and cardiotrophin‐1 (CT‐1) (Koch et al., 2007; Miller and Gauthier, 2007). Previously we reported that cholera toxin‐induced GFAP induction through cAMP/PKA/CREB pathway (Li et al., 2007). Moreover, it was found that the transcription of IL‐6 gene by cAMP requires CRE sequence in its promoter region (Nagashima et al., 2003). Thus we propose that cholera toxin may induce IL‐6 expression and thereby choose IL‐6 as our candidate gene. In fact, we observed that the protein level of secreted IL‐6 in the culture medium of C6 glioma cells was significantly increased after 24 h cholera toxin incubation (Figure 2C), consistent with GFAP induction. Furthermore, RT‐PCR showed that the mRNA level of IL‐6 was also dramatically increased in a time‐dependent manner and reached maximum value by 12 h after cholera toxin treatment (Figure 2D). These results indicate that cholera toxin can induce IL‐6 expression at the transcriptional level.
3.4. IL‐6/JAK2/STAT3 pathway is involved in cholera toxin‐induced up‐regulation of GFAP
To further test if activation of IL‐6/JAK2/STAT3 pathway contributes to the up‐regulation of GFAP induced by cholera toxin, C6 cells were treated with recombinant rat IL‐6 protein. As Figure 3A shown, analogous to cholera toxin, the exogenous IL‐6 resulted in dramatic STAT3 phosphorylation after 48 h incubation, meanwhile, GFAP was also strongly induced. Furthermore, JAK2 specific inhibitor AG490 could significantly block the increase of GFAP induced by cholera toxin (Figure 3B) and IL‐6 (Figure 3C). These results suggest that activation of IL‐6/JAK2/STAT3 pathway plays an important role in cholera toxin‐induced GFAP expression.
Figure 3.
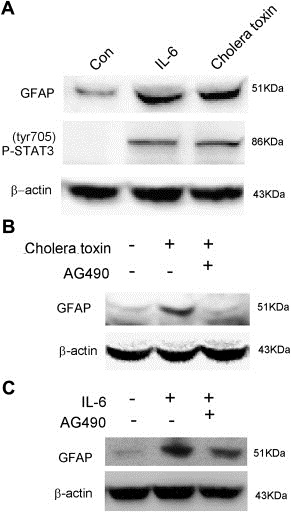
IL‐6/JAK2/STAT3 pathway is involved in cholera toxin‐induced up‐regulation of GFAP. (A), C6 cells were incubated with recombinant rat IL‐6 (100 ng/ml) or 10 ng/ml cholera toxin for 48h. (B), C6 cells were pretreated with 12.5 μM AG490 for 2 h and then treated with 10 ng/ml cholera toxin for an additional 48 h. (C), C6 cells were pretreated with 12.5 μM AG490 for 2 h and then treated with 100 ng/ml recombinant rat IL‐6 for further 48 h.
3.5. The methylation status of GFAP promoter is demethylated in C6 glioma cells
Given that the methylation of the STAT3 binding element can negatively regulate GFAP expression during astrocyte differentiation (Takizawa et al., 2001a), we investigated the methylation status of the particular STAT3 recognition sequence (TTCCGAGAA, −1518 to −1510) of GFAP gene promoter in C6 glioma cells (Figure 4A). Bisulfite sequencing analysis showed that the CpG dinucleotide in the STAT3 binding side in the GFAP promoter was unmethylated in C6 glioma cells treated with cholera toxin for 48 h while that in control cells which express low level of GFAP was barely methylated as well (Figure 4B). This finding proposes a hypothesis that the loss of GFAP expression in C6 glioma cells is not caused by the methylation of the binding site of STAT3. To further support this hypothesis, we next examined a possibility whether inhibit methyltransferase could stimulate GFAP expression. As shown in Figure 4C, both methyltransferase inhibitor (5‐AZa‐CdR) and small interfering RNA (siDNMT1) failed to induce GFAP expression. Taken together, these data suggest that the binding site of STAT3 in the promoter of GFAP in C6 glioma cells is unmethylated and the loss of GFAP expression in C6 cells is not caused by its promoter hypermethylation.
Figure 4.

Cholera toxin‐induced GFAP expression is independent on its promoter demethylation in C6 glioma cells. (A), A schematic of the particular binding side of STAT3 in the promoter of GFAP gene. (B), C6 glioma cells were treated with or without cholera toxin for 48 h, then the methylation status was measured by bisulfite sequencing analysis. (C), C6 glioma cells were treated with 10 μM 5‐AZA‐CdR or transfected with 50 nM siDNMT1 oligonucleotides for 48 h for western blotting to evaluate DNMT1 and GFAP expression.
4. Discussion
The suppression of GFAP expression in neural precursor cells (NPCs) during embryonic stage is due to two key events, one is the inactivation of JAK2/STAT3 signal, and the other is the methylation of the recognition sequence of STAT3 in the promoter of GFAP. Both the cell‐external cues and cell intrinsic programs govern the astrocyte cell fate during brain development (Takizawa et al., 2001a). Here we demonstrate that during cholera toxin‐induced astrocytic differentiation of C6 malignant glioma cells, the accumulation of GFAP is strongly triggered by the activation of a pro‐survival IL‐6/JAK2/STAT3 pathway; however, the suppression of GFAP expression in C6 cells is not caused by its promoter hypermethylation.
Current therapies for cancer are highly toxic and often nonspecific. Differentiation therapy, using agents that modify cancer cell differentiation, has shown promise in the spectrum of agents used against tumors (Leszczyniecka et al., 2001). Notably, all‐trans‐retinoic acid has been used as an agent to induce cell differentiation in clinical treatment of acute promyelocytic leukemia (APL) (Huang et al., 1988), demonstrating the remarkable efficacy of differentiation therapy in treatment of cancers. Such excellent effects, however, were not reproduced in solid tumors. Differentiation agents for malignant gliomas remain a real challenge. In the present study, we demonstrate that secretion of IL‐6 triggered by cholera toxin plays an important role in GFAP induction. Interleukin‐6 (IL‐6)‐type cytokines are major regulators of inflammation and thereby contribute to the neuropathology and pathophysiology associated with inflammation of the central nervous system (CNS) (Koch et al., 2007). Furthermore, it is well established that GFAP is not only a reliable marker for gliogenesis but also works as a tumor suppressor gene in malignant glioma pathogenesis (Duffy et al., 1982; Rutka et al., 1994; Rutka and Smith, 1993).Therefore, our findings support those previous reports demonstrating that proinflammatory cytokines might work as potential differentiation‐supporting agents against malignant gliomas (Koch et al., 2007; Slegers and Joniau, 1996; Takanaga et al., 2004).
GFAP is a member of the family of 8‐ to 10‐nm intermediate filaments and is specific for the cytoskeleton of the astrocyte (McKeown‐Longo et al., 1984). It appears to stabilize the astrocyte's cytoskeleton and helps to maintain astrocyte cell shape through interactions between GFAP filaments, the nuclear membrane, and the plasma membrane (Duffy et al., 1982; Goldman and Chiu, 1984). It has been shown that overexpression of GFAP can effectively induce morphologic alteration of malignant astrocytoma (Rutka and Smith, 1993). In the present study, we found that cholera toxin can induce morphologic alteration of C6 glioma cells characterized by long and thin processes extending concomitant with GFAP stimulation; however, although the exogenous IL‐6 could dramatically trigger GFAP expression, no morphologic changes could be observed (Supplementary 1), indicating that the cholera toxin‐induced morphologic changes of C6 cells are independent of GFAP induction and GFAP may not be the morphologic fate determinant during the cholera toxin‐induced differentiation of malignant glioma cells.
It is clear that STAT3 recognition sequences have been identified within a regulatory region of the GFAP gene promoter (Bonni et al., 1997; Nakashima et al., 1999). Importantly, a critical site between −1518 and −1510 within these potential STAT3 binding sites is evolutionarily conserved between mouse, rat, and human GFAP genes and is essential for GFAP expression by gp130‐stimulating cytokines (Bonni et al., 1997; Nakashima et al., 1999). Further analysis reveals that a point mutation in this STAT3 recognition sequence or overexpression of a dominant‐negative form of STAT3 in neuroepithelial cells blocks the GFAP expression induced by gp130‐stimulating cytokines (Bonni et al., 1997; Nakashima et al., 1999; Ochiai et al., 2001; Rajan and McKay, 1998; Takizawa et al., 2001b; Yanagisawa et al., 2000).Therefore, GFAP expression is largely dependent on the activation of STAT3 and the presence of its critical recognition element in the promoter. Our results here demonstrate that cholera toxin is capable of activating JAK2/STAT3 pathway, thereby triggering GFAP expression in C6 glioma cells. Interestingly, in differentiation‐resistant T98G glioma cells, cholera toxin failed to stimulate GFAP expression concurrent with no changes of phosphorylation of STAT3 and JAK2 (Supplementary 2), suggesting that the activation of JAK2/STAT3 signal is a critical event that is responsible for the cholera toxin‐triggered GFAP expression. Furthermore, although the pro‐survival cascade IL‐6/JAK2/STAT3 was strongly activated in C6 glioma cells by cholera toxin, the astrocytic differentiation effects instead of enhanced malignant properties were observed. One possible explanation might be that the oncogenic roles of IL‐6/JAK2/STAT3 were conquered by other tumor suppressive signals stimulated by cholera toxin, such as GSK‐3β (Li et al., 2010).
With regard to cell intrinsic programs, DNA methylation is an epigenetic modification that plays an important role in cancer initiation and progression by suppression of genes essential for the control of normal cell growth and differentiation (Costello and Plass, 2001; Robertson, 2001). Previous reports have demonstrated that methylation of the CpG site in a particular STAT3 recognition sequence between −1518 and −1510 in the GFAP promoter can abolish the accessibility of STAT3 and transcriptional activation, thereby inhibiting GFAP expression (Takizawa et al., 2001a). Interestingly, our present work revealed that the particular binding site of STAT3 in the GFAP promoter in C6 malignant glioma cells, however, was unmethylated per se. In addition, both DNMT inhibitor 5‐AZa‐CdR and siDNMT1 failed to trigger GFAP expression. These data indicate that activation of IL‐6/JAK2/STAT3 pathway can directly induce GFAP expression on account of the demethylation status of GFAP promoter in C6 glioma cells, which is different from that in NPC during embryonic stage.
In conclusion, our data uncovered that cholera toxin can seize an oncogenic cascade IL‐6/JAK2/STAT3 to induce GFAP expression, which implies that a survival‐promoting signal may also play a differentiation‐supporting role in malignant gliomas. We also provided the first evidence that the critical binding side of STAT3 (−1518 ~ −1510) in the promoter of GFAP in C6 malignant glioma cells is unmethylated. These founding may pave a way for better understanding the molecular mechanism underling the cholera toxin‐induced differentiation of malignant gliomas.
Supporting information
The following are the Supplementary data related to this article:
IL‐6 does not induce morphologic alteration of C6 glioma cells. C6 cells were incubated with recombinant rat IL‐6 (100 ng/ml) or 10 ng/ml cholera toxin for 48 h. Original magnification: ×200.
{kind=link}
Cholera toxin does not induce differentiation of T98G glioma cells. A, Morphology (Original magnification: ×200). T98G cells were incubated with 100 ng/ml cholera toxin for 48 h. B, The expression of STAT3, Phospho‐STAT3 (tyr705) and GFAP were detected by western blotting. C, The expression of JAK2 and Phospho‐JAK2 (tyr1007/1008) were detected by western blotting. T98G cells were incubated with 100 ng/ml cholera toxin for the indicated times.
{kind=link}
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 30830111 and 30801408), National Natural Science Foundation of Guangdong Province (No. 8451008901000297), Chinese Postdoctoral Science (No. 20100480824) and Guangzhou Scientific and Technological Programs (No. 2008Z1‐E561).
Supplementary material 1.
Supplementary data related to this article can be found online at doi:10.1016/j.molonc.2011.03.003.
Shu Minfeng, Zhou Yuxi, Zhu Wenbo, Wu Sihan, Zheng Xiaoke and Yan Guangmei, (2011), Activation of a pro‐survival pathway IL‐6/JAK2/STAT3 contributes to glial fibrillary acidic protein induction during the cholera toxin‐induced differentiation of C6 malignant glioma cells, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.03.003.
References
- Adachi, Y. , Aoki, C. , Yoshio-Hoshino, N. , Takayama, K. , Curiel, D.T. , Nishimoto, N. , 2006. Interleukin-6 induces both cell growth and VEGF production in malignant mesotheliomas. International Journal of Cancer. 119, 1303–1311. [DOI] [PubMed] [Google Scholar]
- Akagi, T. , Murata, K. , Shishido, T. , Hanafusa, H. , 2002. v-Crk activates the phosphoinositide 3-kinase/AKT pathway by utilizing focal adhesion kinase and H-Ras. Molecular and Cellular Biology. 22, 7015–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, S. , Wu, Q. , McLendon, R.E. , Hao, Y. , Shi, Q. , Hjelmeland, A.B. , Dewhirst, M.W. , Bigner, D.D. , Rich, J.N. , 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 444, 756–760. [DOI] [PubMed] [Google Scholar]
- Bestor, T. , Laudano, A. , Mattaliano, R. , Ingram, V. , 1988. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. Journal of Molecular Biology. 203, 971–983. [DOI] [PubMed] [Google Scholar]
- Bird, A.P. , Wolffe, A.P. , 1999. Methylation-induced repression–belts, braces, and chromatin. Cell. 99, 451–454. [DOI] [PubMed] [Google Scholar]
- Bonni, A. , Sun, Y. , Nadal-Vicens, M. , Bhatt, A. , Frank, D.A. , Rozovsky, I. , Stahl, N. , Yancopoulos, G.D. , Greenberg, M.E. , 1997. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 278, 477–483. New York, N.Y [DOI] [PubMed] [Google Scholar]
- Bowman, T. , Garcia, R. , Turkson, J. , Jove, R. , 2000. STATs in oncogenesis. Oncogene. 19, 2474–2488. [DOI] [PubMed] [Google Scholar]
- Bromberg, J.F. , Wrzeszczynska, M.H. , Devgan, G. , Zhao, Y. , Pestell, R.G. , Albanese, C. , Darnell, J.E. , 1999. Stat3 as an oncogene. Cell. 98, 295–303. [DOI] [PubMed] [Google Scholar]
- Costello, J.F. , Berger, M.S. , Huang, H.S. , Cavenee, W.K. , 1996. Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Research. 56, 2405–2410. [PubMed] [Google Scholar]
- Costello, J.F. , Plass, C. , 2001. Methylation matters. Journal of Medical Genetics. 38, 285–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, W.J. , Scott, C.B. , Horton, J. , Nelson, J.S. , Weinstein, A.S. , Fischbach, A.J. , Chang, C.H. , Rotman, M. , Asbell, S.O. , Krisch, R.E. , 1993. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. Journal of the National Cancer Institute. 85, 704–710. [DOI] [PubMed] [Google Scholar]
- Deneen, B. , Ho, R. , Lukaszewicz, A. , Hochstim, C.J. , Gronostajski, R.M. , Anderson, D.J. , 2006. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 52, 953–968. [DOI] [PubMed] [Google Scholar]
- Duffy, P.E. , Huang, Y.Y. , Rapport, M.M. , 1982. The relationship of glial fibrillary acidic protein to the shape, motility, and differentiation of human astrocytoma cells. Experimental Cell Research. 139, 145–157. [DOI] [PubMed] [Google Scholar]
- Foltz, G. , Yoon, J.G. , Lee, H. , Ryken, T.C. , Sibenaller, Z. , Ehrich, M. , Hood, L. , Madan, A. , 2009. DNA methyltransferase-mediated transcriptional silencing in malignant glioma: a combined whole-genome microarray and promoter array analysis. Oncogene. 28, 2667–2677. [DOI] [PubMed] [Google Scholar]
- Garcia, R. , Jove, R. , 1998. Activation of STAT transcription factors in oncogenic tyrosine kinase signaling. Journal of Biomedical Science. 5, 79–85. [DOI] [PubMed] [Google Scholar]
- Goldman, J.E. , Chiu, F.C. , 1984. Growth kinetics, cell shape, and the cytoskeleton of primary astrocyte cultures. Journal of Neurochemistry. 42, 175–184. [DOI] [PubMed] [Google Scholar]
- Hirano, T. , Ishihara, K. , Hibi, M. , 2000. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 19, 2548–2556. [DOI] [PubMed] [Google Scholar]
- Honoki, K. , Tsujiuchi, T. , Mori, T. , Yoshitani, K. , Tsutsumi, M. , Takakura, Y. , Mii, Y. , 2004. Expression of the p16INK4a gene and methylation pattern of CpG sites in the promoter region in rat tumor cell lines. Molecular Carcinogenesis. 39, 10–14. [DOI] [PubMed] [Google Scholar]
- Huang, M.E. , Ye, Y.C. , Chen, S.R. , Chai, J.R. , Lu, J.X. , Zhoa, L. , Gu, L.J. , Wang, Z.Y. , 1988. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 72, 567–572. [PubMed] [Google Scholar]
- Koch, M. , May, U. , Kuhns, S. , Drechsler, H. , Adam, N. , Hattermann, K. , Wirtz, S. , Rose-John, S. , Scheller, J. , 2007. Interleukin 27 induces differentiation of neural C6-precursor cells into astrocytes. Biochemical and Biophysical Research Communications. 364, 483–487. [DOI] [PubMed] [Google Scholar]
- Kudo, M. , Jono, H. , Shinriki, S. , Yano, S. , Nakamura, H. , Makino, K. , Hide, T. , Muta, D. , Ueda, M. , Ota, K. , Ando, Y. , Kuratsu, J. , 2009. Antitumor effect of humanized anti-interleukin-6 receptor antibody (tocilizumab) on glioma cell proliferation. Laboratory investigation. Journal of Neurosurgery. 111, 219–225. [DOI] [PubMed] [Google Scholar]
- Kupferman, M.E. , Jayakumar, A. , Zhou, G. , Xie, T. , Dakak-Yazici, Y. , Zhao, M. , Ju, J. , Mandal, M. , Jasser, S. , Madden, T. , Myers, J.N. , Priebe, W. , 2009. Therapeutic suppression of constitutive and inducible JAK\STAT activation in head and neck squamous cell carcinoma. Journal of Experimental Therapeutics & Oncology. 8, 117–127. [PubMed] [Google Scholar]
- Leszczyniecka, M. , Roberts, T. , Dent, P. , Grant, S. , Fisher, P.B. , 2001. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacology & Therapeutics. 90, 105–156. [DOI] [PubMed] [Google Scholar]
- Li, L.C. , Dahiya, R. , 2002. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 18, 1427–1431. (Oxford, England) [DOI] [PubMed] [Google Scholar]
- Li, Y. , Lu, H. , Huang, Y. , Xiao, R. , Cai, X. , He, S. , Yan, G. , 2010. Glycogen synthase kinases-3beta controls differentiation of malignant glioma cells. International Journal of Cancer. 127, 1271–1282. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Yin, W. , Wang, X. , Zhu, W. , Huang, Y. , Yan, G. , 2007. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proceedings of the National Academy of Sciences of the United States of America. 104, 13438–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown-Longo, P.J. , Hanning, R. , Mosher, D.F. , 1984. Binding and degradation of platelet thrombospondin by cultured fibroblasts. The Journal of Cell Biology. 98, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, F.D. , Gauthier, A.S. , 2007. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 54, 357–369. [DOI] [PubMed] [Google Scholar]
- Nagashima, A.C. , Giacomini, D. , Castro, C.P. , Pereda, M.P. , Renner, U. , Stalla, G.K. , Arzt, E. , 2003. Transcriptional regulation of interleukin-6 in pituitary folliculo-stellate TtT/GF cells. Molecular and Cellular Endocrinology. 201, 47–56. [DOI] [PubMed] [Google Scholar]
- Nakashima, K. , Yanagisawa, M. , Arakawa, H. , Kimura, N. , Hisatsune, T. , Kawabata, M. , Miyazono, K. , Taga, T. , 1999. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 284, 479–482. New York, N.Y [DOI] [PubMed] [Google Scholar]
- Namihira, M. , Kohyama, J. , Semi, K. , Sanosaka, T. , Deneen, B. , Taga, T. , Nakashima, K. , 2009. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Developmental Cell. 16, 245–255. [DOI] [PubMed] [Google Scholar]
- Ochiai, W. , Yanagisawa, M. , Takizawa, T. , Nakashima, K. , Taga, T. , 2001. Astrocyte differentiation of fetal neuroepithelial cells involving cardiotrophin-1-induced activation of STAT3. Cytokine. 14, 264–271. [DOI] [PubMed] [Google Scholar]
- Okano, M. , Bell, D.W. , Haber, D.A. , Li, E. , 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Rajan, P. , McKay, R.D. , 1998. Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 18, 3620–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin, A. , 1998. CpG methylation, chromatin structure and gene silencing-a three-way connection. The EMBO Journal. 17, 4905–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, K.D. , 2001. DNA methylation, methyltransferases, and cancer. Oncogene. 20, 3139–3155. [DOI] [PubMed] [Google Scholar]
- Robertson, K.D. , Jones, P.A. , 2000. DNA methylation: past, present and future directions. Carcinogenesis. 21, 461–467. [DOI] [PubMed] [Google Scholar]
- Roymans, D. , Vissenberg, K. , De Jonghe, C. , Grobben, B. , Claes, P. , Verbelen, J.P. , Van Broeckhoven, C. , Slegers, H. , 2001. Phosphatidylinositol 3-kinase activity is required for the expression of glial fibrillary acidic protein upon cAMP-dependent induction of differentiation in rat C6 glioma. Journal of Neurochemistry. 76, 610–618. [DOI] [PubMed] [Google Scholar]
- Rutka, J.T. , Hubbard, S.L. , Fukuyama, K. , Matsuzawa, K. , Dirks, P.B. , Becker, L.E. , 1994. Effects of antisense glial fibrillary acidic protein complementary DNA on the growth, invasion, and adhesion of human astrocytoma cells. Cancer Research. 54, 3267–3272. [PubMed] [Google Scholar]
- Rutka, J.T. , Smith, S.L. , 1993. Transfection of human astrocytoma cells with glial fibrillary acidic protein complementary DNA: analysis of expression, proliferation, and tumorigenicity. Cancer Research. 53, 3624–3631. [PubMed] [Google Scholar]
- Slegers, H. , Joniau, M. , 1996. Lipopolysaccharide-enhanced expression of interleukin-6 in dibutyryl cyclic AMP-differentiated rat C6 glioma. Journal of Neurochemistry. 66, 466–473. [DOI] [PubMed] [Google Scholar]
- Strelkov, I.S. , Davie, J.R. , 2002. Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogene-transformed mouse fibroblasts. Cancer Research. 62, 75–78. [PubMed] [Google Scholar]
- Takanaga, H. , Yoshitake, T. , Yatabe, E. , Hara, S. , Kunimoto, M. , 2004. Beta-naphthoflavone disturbs astrocytic differentiation of C6 glioma cells by inhibiting autocrine interleukin-6. Journal of Neurochemistry. 90, 750–757. [DOI] [PubMed] [Google Scholar]
- Takizawa, T. , Nakashima, K. , Namihira, M. , Ochiai, W. , Uemura, A. , Yanagisawa, M. , Fujita, N. , Nakao, M. , Taga, T. , 2001. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Developmental Cell. 1, 749–758. [DOI] [PubMed] [Google Scholar]
- Takizawa, T. , Yanagisawa, M. , Ochiai, W. , Yasukawa, K. , Ishiguro, T. , Nakashima, K. , Taga, T. , 2001. Directly linked soluble IL-6 receptor-IL-6 fusion protein induces astrocyte differentiation from neuroepithelial cells via activation of STAT3. Cytokine. 13, 272–279. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, M. , Nakashima, K. , Arakawa, H. , Ikenaka, K. , Yoshida, K. , Kishimoto, T. , Hisatsune, T. , Taga, T. , 2000. Astrocyte differentiation of fetal neuroepithelial cells by interleukin-11 via activation of a common cytokine signal transducer, gp130, and a transcription factor, STAT3. Journal of Neurochemistry. 74, 1498–1504. [DOI] [PubMed] [Google Scholar]
- Yang, F. , Brown, C. , Buettner, R. , Hedvat, M. , Starr, R. , Scuto, A. , Schroeder, A. , Jensen, M. , Jove, R. , 2010. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Molecular Cancer Therapeutics. 9, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the Supplementary data related to this article:
IL‐6 does not induce morphologic alteration of C6 glioma cells. C6 cells were incubated with recombinant rat IL‐6 (100 ng/ml) or 10 ng/ml cholera toxin for 48 h. Original magnification: ×200.
Cholera toxin does not induce differentiation of T98G glioma cells. A, Morphology (Original magnification: ×200). T98G cells were incubated with 100 ng/ml cholera toxin for 48 h. B, The expression of STAT3, Phospho‐STAT3 (tyr705) and GFAP were detected by western blotting. C, The expression of JAK2 and Phospho‐JAK2 (tyr1007/1008) were detected by western blotting. T98G cells were incubated with 100 ng/ml cholera toxin for the indicated times.