

Abstract
Centrosome abnormalities occur commonly in cancer, and contribute to chromosomal instability and tumorigenesis. New evidence on a phylogenetically conserved mechanism termed ‘centrosomal clustering’ provides exciting insights into how cells with supernumerary centrosomes adapt to avoid lethal multipolar divisions. Here, we highlight the emerging molecular basis of centrosome clustering, and its impact on asymmetric divisions of stem cells, chromosomal (in)stability and malignant transformation. Finally, pharmacological inhibition of centrosome clustering promises to selectively target tumor cells.
Keywords: Centrosome clustering, Chromosomal instability, Multipolarity, Mitosis, DNA damage, Stem cells
Highlights
Centrosome abnormalities occur commonly in cancer.
Centrosome clustering enables cells with extra centrosomes to divide successfully.
We discuss the molecular basis of centrosome clustering and its biological roles.
Clustering impacts on stem cell division, chromosomal stability and tumorigenesis.
Inhibition of centrosome clustering promises to selectively target tumor cells.
Centrosomes consist of a pair of centrioles embedded in pericentriolar material and act as main microtubule organizing centers (Bettencourt‐Dias and Glover, 2007; Bornens, 2002). Centrosomes duplicate precisely once per cell cycle, analogous to genomes, and function as spindle poles to guide bipolar spindle formation that is essential for accurate chromosome segregation during mitosis (Doxsey, 2001; Krämer et al., 2002).
Abnormalities of diverse tumor suppressors and oncogenes can cause centrosome amplification (Fukasawa, 2007), which occurs through centrosome overduplication during interphase, de novo synthesis of centrosomes or cytokinesis failure (Nigg, 2002). Centrosome amplification is frequent in cancer, and is linked to tumorigenesis and aneuploidy (Nigg, 2002; Lingle et al., 1998; Pihan et al., 1998; Neben et al., 2003; Krämer et al., 2003; Koutsami et al., 2006). The extent of centrosomal aberrations correlates with chromosomal instability (CIN) and malignant behavior in tumor cell lines, mouse tumor models, and human tumors (Nigg, 2002; Lingle et al., 1998; Neben et al., 2003; Krämer et al., 2003; Koutsami et al., 2006; Levine et al., 1991; Pihan et al., 2001).
CIN is a feature commonly observed in human cancers and was first identified almost a century ago (von Hansemann, 1890). One major cause of CIN is mitotic checkpoint overactivation as recently reviewed by Benezra and collegues (Schvartzman et al., 2010). Only recently, using elaborate mouse models, it became clear that CIN does not only correlate with but probably plays a causative part in a substantial proportion of malignancies (Weaver et al., 2007; Sotillo et al., 2007; Baker et al., 2009). Here, we focus on recent discoveries related to another phenomenon intricately linked to CIN, centrosomal clustering, its emerging mechanistic basis, significance for cancer cell survival, tumor progression, role in asymmetric divisions of stem cells, and as a potential tumor‐selective therapeutic target.
1. Implications of centrosome clustering for chromosomal instability and cancer
In mitosis, supernumerary centrosomes can form multipolar spindles, which occur in many tumor types and have long been accused of contributing to CIN and tumorigenesis (Krämer et al., 2002; Nigg, 2002; Boveri, 1929). However, recent findings show that multipolar divisions and the resulting CIN undermine cell viability, frequently leading to cell death (Weaver et al., 2007; Ganem et al., 2009; Brinkley, 2001; Kops et al., 2004). To avoid cell death, many cancer cells induce supernumerary centrosome clustering into two spindle poles thereby enabling bipolar division (Figure 1). Centrosomal clustering was first observed in N115 mouse neuroblastoma cells some 30 years ago (Ring et al., 1982). This observation was rediscovered at the turn of the millennium owing to the observation that the frequent occurrence of supernumerary centrosomes in human breast cancer samples was associated with surprisingly rare abnormal mitoses (Lingle and Salisbury, 1999). Whether or not centrosomal clustering was coupled with reduced CIN was not examined at the time. Indeed, the concept that has crystallized since these pioneering studies is that centrosomal clustering enables cells to successfully divide despite the presence of supernumerary centrosomes (Nigg, 2002; Brinkley, 2001).
Figure 1.
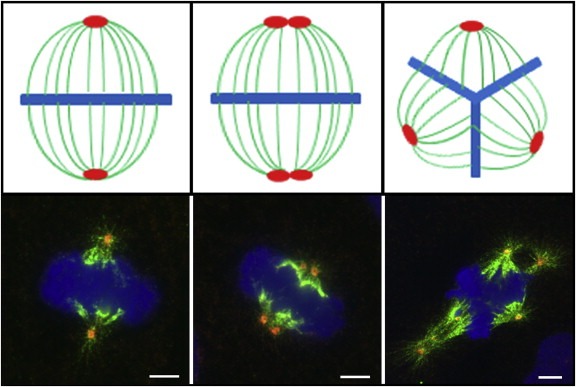
Clustering of supernumerary centrosomes into a bipolar mitotic spindle array. Cartoons (upper panel) illustrating a normal bipolar spindle with two centrosomes (left), a bipolar spindle with clustered supernumerary centrosomes (middle) and a multipolar spindle (right), and corresponding representative immunofluorescence images from the human prostate cancer cell line DU‐145 (lower panel) are shown. Cells were stained for centrosomes (γ‐tubulin, red), microtubules (α‐tubulin, green) and DNA (DAPI, blue). Images have been made by B. M., using an Axiovert 200 M microscope (Zeiss, Jena, Germany).
The initial description of centrosome clustering noted that cells with supernumerary centrosomes pass through a transient multipolar spindle intermediate before centrosome clustering and bipolar anaphase occurs (Ring et al., 1982). Excitingly, recent data from several laboratories demonstrate that, while passing through the transient multipolar state, merotelic kinetochore‐microtubule attachment errors, defined by the persistent attachment of microtubules from both spindle poles to a single kinetochore, accumulate, and consequently increase the frequency of lagging chromosomes during bipolar anaphase after centrosomal clustering (Ganem et al., 2009; Silkworth et al., 2009). Importantly, this finding implies that cells with amplified centrosomes do not necessarily need to divide in a multipolar fashion to allow low‐level chromosomal missegregation that can fuel tumor progression. Such interpretation also supports the emerging bimodal relationship between aneuploidy and tumorigenesis (Weaver et al., 2007): whereas moderate CIN induced tumorigenesis in mice, high‐level CIN instead suppressed tumor formation in vivo. Suppression of tumor cell growth in this context seems to be brought about by apoptosis induction due to loss of chromosomes encoding genes required for maintenance of cell viability (Kops et al., 2004). Similarly, in patients with breast, ovarian, gastric and non‐small cell lung cancer, extreme CIN is associated with improved prognosis relative to tumors with intermediate CIN levels (Birkbak et al., 2011).
As already mentioned above several studies have recently provided substantial evidence for a causative role of CIN in malignant transformation. Similarly, the key question of whether supernumerary centrosomes are simply a passenger phenotype or can induce malignancy has now been addressed by constructing flies that overexpress SAK (also known as polo‐like kinase 4 (PLK4)), a kinase important for centriole replication (Basto et al., 2008). Flies overexpressing SAK contain extra centrosomes in about 60% of their somatic cells. Although many of the fly cells with supernumerary centrosomes initially form multipolar spindles, they ultimately cluster into bipolar arrays, resulting in only slightly increased CIN levels. Nevertheless, larval brain cells of these animals can generate metastatic tumors when transplanted into the abdomens of wild‐type hosts (Basto et al., 2008).
Similar to flies, mice overexpressing the centrosomal protein ninein‐like (NINL) show centrosome amplification as detected in mouse embryonic fibroblasts from the transgenic animals and develop tumors of breast, ovary, and testicles at 10–15 months of age (Shao et al., 2010). Whether or not extra centrosomes are clustered into bipolar mitoses in this system was not examined. Although NINL overexpression will certainly have additional effects other than centrosome amplification these data nevertheless hint for a role of supernumerary centrosomes in tumorigenesis in mammals as well.
The ability to cluster supernumerary centrosomes into a bipolar mitotic spindle array is not a specific trait of tumor cells. For example, during physiological hepatocyte polyploidization, primary binuclear hepatocytes – which naturally contain four centrosomes in G2 phase – efficiently cluster pairs of centrosomes at opposite spindle poles, leading to the generation of mononuclear 4n progeny (Guidotti et al., 2003). Recent data confirm that polyploid mouse hepatocytes in most cases reorganize their spindles into a bipolar mitotic array from an intermediate multipolar state, a process associated with lagging chromosomes in 25–50% of tetraploid hepatocytes undergoing bipolar anaphase and resulting in a high rate of aneuploidy (Duncan et al., 2010). Interestingly, however, a small percentage of tetraploid mouse hepatocytes underwent successful tripolar divisions, producing viable offspring. Also, likely as a consequence of centrosome clustering, during liver regeneration in mice, which is associated with excessive polyploidization, about 20% of hepatocytes missegregate one or more chromosomes at each mitosis (Putkey et al., 2002). Furthermore, several studies showed that both non‐transformed Drosophila melanogaster (D. melanogaster) neuroblasts and diverse types of human cells that have been manipulated to contain supernumerary centrosomes by either PLK4 overexpression or treatment with cytochalasin D to inhibit cytokinesis, can cluster multiple centrosomes into a bipolar spindle array both in vitro and in vivo (Ganem et al., 2009; Basto et al., 2008; Quintyne et al., 2005; Kwon et al., 2008; Yang et al., 2008).
Collectively, it seems that not only cancer cells but also non‐transformed cell types can cluster supernumerary centrosomes into bipolar mitotic spindles. Initial evidence implicates supernumerary centrosomes and centrosomal clustering in tumorigenesis in flies. However, data generated in mouse hepatocytes show that neither centrosome amplification nor centrosome clustering or multipolar cell division with subsequent aneuploidy necessarily leads to malignant transformation in mammals. From those data it can also be concluded that multipolar divisions are not universally lethal. However, as these experiments have been performed using polyploid hepatocytes, surviving multipolar divisions might well be a peculiarity of polyploid cells which better tolerate the loss of multiple chromosomes.
It appears that more insights into the molecular and cellular basis of centrosomal clustering are needed. This topic, along with data on deregulation of the genes and proteins mechanistically involved in centrosomal clustering in diverse malignanices, are discussed in the next section.
2. Mechanisms of centrosome clustering
Mechanistically, multiple cellular systems are involved in the clustering of supernumerary centrosomes in normal and tumor cells (Figure 2). Three recent studies show that extra centrosomes activate a MAD2‐dependent delay of anaphase onset in different cell types, which is required for centrosomal clustering and suppression of multipolar mitosis (Basto et al., 2008; Kwon et al., 2008; Yang et al., 2008). MAD2 is a central component of the spindle assembly checkpoint (SAC) that blocks dissolution of sister chromatids at metaphase until microtubule attachment at kinetochores is complete and spindle tension is established (Weaver et al., 2007). From those results it can be assumed that, although the SAC does not recognize abnormal spindles per se (Sluder et al., 1997), multipolarity is accompanied by improper kinetochore attachment or insufficient tension and thereby activates the SAC and leads to metaphase arrest. In line with this interpretation an RNA interference (RNAi) screen in D. melanogaster S2 cells suggested that knockdown of components of the actin cytoskeleton and actin‐dependent cortical force generators including the formin FORM3/INF2, the myosin MYO10, the microtubule plus‐end‐tracking protein CLIP190 as well as several cell‐matrix adhesion molecules (Turtle, Echinoid, CAD96CA, CG33171, FIT1) induces spindle multipolarity through interference with the interphase cell adhesion pattern (Kwon et al., 2008). When cells round up during mitosis, retraction fibers (actin‐rich structures linked to the sites of former adhesion during interphase) remain attached to the extracellular substrate and promote interaction of astral spindle microtubules with the cell cortex (Thery et al., 2005). Disturbance of the connection between cell‐matrix adhesion proteins and actin cytoskeleton on the one hand and spindle microtubule components on the other hand might therefore cause reduced spindle tension thereby inhibiting centrosome clustering. Indeed, in elegant experiments it has been shown that O‐ and Y‐shaped fibronectin‐coated micropatterns, allowing for multidirectional distribution of retraction fiber formation, caused increased frequencies of multipolar spindles (Kwon et al., 2008). On the other hand, bipolar arrangements of adhesive contacts induced by H‐shaped micropatterns promoted bipolar mitoses.
Figure 2.
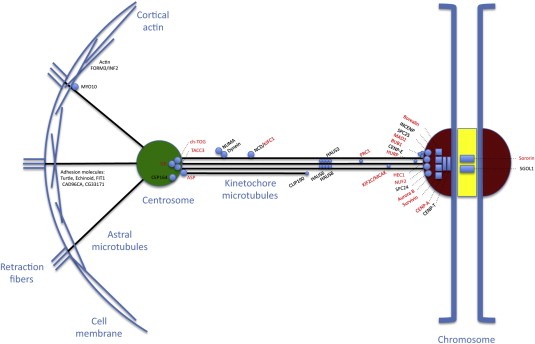
Molecular mechanisms and proteins involved in centrosome clustering. Sketch showing a mitotic cell with some of the proteins that have been implicated in centrosomal clustering. Proteins in red are known to be overexpressed in human cancer cells.
Another factor that prevents spindle multipolarity by clustering of supernumerary centrosomes is spindle tension itself (Figure 3) (Kwon et al., 2008; Leber et al., 2010). Before chromosome segregation, kinetochores of sister chromatids attach to microtubules of opposite spindle poles. This configuration is achieved through a trial‐and‐error process in which correct attachments exert tension across the centromere, which stabilizes kinetochore‐microtubule interactions. Incorrect attachments exert less tension and are destabilized, providing a new opportunity to bi‐orient (Liu et al., 2009). The mitotic kinase aurora B, the enzymatically active component of the chromosomal passenger complex (CPC), localizes to the inner centromere between sister kinetochores, and regulates chromosome‐spindle attachments by phosphorylating kinetochore substrates, including the NDC80 microtubule‐binding complex (Liu et al., 2009; Ruchaud et al., 2007; Wei et al., 2007). The CPC, composed of aurora B and its regulatory subunits INCENP, survivin, and borealin is a key regulator of chromosome segregation and cytokinesis. Since tension across centromeres widens spatial separation of the CPC and thereby aurora B from its kinetochore substrates, substrate phosphorylation is reduced resulting in stabilized microtubule‐kinetochore interactions (Liu et al., 2009).
Figure 3.
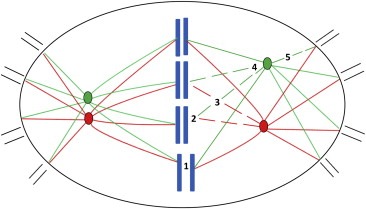
Spindle tension is required for centrosomal clustering. Model of centrosomal clustering (left spindle half) and mechanisms involved in its prevention via reduction of spindle tension (right spindle half). Centrosome clustering is brought about by microtubule tension‐dependent uniform positioning of individual centrosomes resulting in the formation of two spindle poles. Spindle tension can be disrupted by reduction of chromatid cohesion (1), disturbed microtubule‐kinetochore attachment (2), reduced microtubule generation (3), disturbed microtubule bundling and centrosome attachment (4), or interference with the interphase cell adhesion pattern by disruption of components of the actin cytoskeleton (5).
Using genome‐wide RNAi screening in human cancer cells with extra centrosomes both NDC80 complex and CPC components were found to be involved in centrosomal clustering (Leber et al., 2010). In addition, shugoshin‐like 1 (SGOL1), a protein previously known to be involved in sensing spindle tension at budding yeast kinetochores (Indjeian et al., 2005), is necessary for centrosome clustering. Importantly, SGOL1 also contributes to the recruitment of the CPC to centromeres (Boyarchuk et al., 2007; Kawashima et al., 2007; Vanoosthuyse et al., 2007) while itself is loaded onto histone H2A after histone phosphorylation by BUB1 in yeast and human cells (Kawashima et al., 2010; Yamagishi et al., 2010). Fittingly, BUB1 knockdown does cause centrosome declustering as well (Sluder et al., 1997). Another recently identified centromeric recruitment factor for the CPC is haspin (Wang et al., 2010; Kelly et al., 2010). Haspin phosphorylates histone H3, thereby creating a docking site for survivin in both Xenopus and human cells. Interestingly, depletion of haspin leads to the generation of multiple spindle poles and disruption of mitotic spindle structure in U2OS and HeLa cells as a consequence of acentriolar pole formation and centriole disengagement (Dai et al., 2009).
CENPA (Hori et al., 2008), the centromere‐specific histone H3 variant, CENPT (Hori et al., 2008), a component of the linker structure connecting the centromere with outer kinetochore components, sororin (also known as cell division cycle associated 5 (CDCA5)) (Schmitz et al., 2007), a protein involved in sister chromatid cohesion, and the augmin complex (Lawo et al., 2009) (which promotes microtubule‐dependent microtubule amplification within the mitotic spindle), are necessary for centrosome clustering as well (Kwon et al., 2008; Leber et al., 2010) (Figure 3). Similar to haspin depletion, knockdown of augmin complex components leads to the formation of acentriolar spindle poles and centrosome fragmentation in addition to centrosomal declustering (Leber et al., 2010; Lawo et al., 2009; Uehara et al., 2009; Einarson et al., 2004; Wu et al., 2008b).Most recently, hepatoma up‐regulated protein (HURP) has been shown to be required for centrosome clustering in cells with supernumerary centrosomes as well (Breuer et al., 2010). This observation is noteworthy as HURP serves as an attachment‐ and tension‐sensitive kinetochore‐microtubule stabilizing factor during mitosis (Koffa et al., 2006; Sillje et al., 2006; Wong and Fang, 2006).
Together, these findings support the notion that loss of centromere tension results in centrosome declustering. Indeed, when pulling forces are measured directly across multipolar spindles in cancer cells with supernumerary centrosomes, depletion of NDC80, CPC and augmin complexes or SGOL1 result in substantially reduced spindle tension, as indicated by shorter inter‐kinetochore distances and BUBR1 labeling of kinetochores in multipolar metaphase cells (Leber et al., 2010). In addition, knockdown of haspin as well as components of the augmin complex has been shown to reduce tension at sister kinetochores (Dai et al., 2009; Uehara et al., 2009). However, these data also suggest that at least some of the proteins involved in the clustering of supernumerary centrosomes might contribute to centriole cohesion and bipolar spindle formation in cells with a regular centrosome content as well.
Several of the proteins of the chromosomal passenger and NDC80 complexes including aurora B, survivin, borealin, NUF2 and highly expressed in cancer 1 (HEC1) as well as sororin and HURP have been found to be overexpressed in a wide variety of cancer types (Carmena and Earnshaw, 2003; Bischoff et al., 1998; Adams et al., 2001; Altieri, 2003; Chang et al., 2006; Hayama et al., 2006; Ferretti et al., 2010; Nguyen et al., 2010; Tsou et al., 2003). Furthermore, overexpression of HEC1, a component of the NDC80 complex as well as aurora B have been implicated in tumor formation in mouse models (Diaz‐Rodriguez et al., 2008; Nguyen et al., 2009). These findings have already led to the development of potent and selective inhibitors of aurora B kinase which are currently in early clinical trials with patients with different kinds of malignancies (Taylor and Peters, 2008). Although only little information is available about individual members of the augmin complex because it has been identified only recently, one study demonstrates mutation of the augmin subunit HAUS3 in breast cancer (Shah et al., 2009). Taken together, these data suggest that proteins involved in centrosome clustering in cancer cells with supernumerary centrosomes are frequently overexpressed in human cancers, suggesting that clustering of extra centrosomes into a bipolar spindle array might indeed be important for cancer cell survival and/or progression.
Several studies report that centrosome clustering also relies on microtubule‐based motors and microtubule‐bundling proteins that organize spindle poles in both normal and tumor cells with supernumerary centrosomes (Quintyne et al., 2005; Kwon et al., 2008; Leber et al., 2010). For example, depletion of the minus‐end directed motor dynein causes declustering of centrosomes and subsequent spindle multipolarity in tumor cells as well as in non‐transformed cells engineered to contain extra centrosomes (Quintyne et al., 2005; Leber et al., 2010). Mechanistically, in mitosis the dynein complex is responsible for targeting nuclear mitotic apparatus protein (NUMA) to spindle poles, where it focuses microtubule minus ends and tethers them to the centrosomes. However, whether delocalization of dynein from spindle microtubules is responsible for the generation of multipolar spindles as initially suggested, remains controversial (Quintyne et al., 2005; Nguyen et al., 2008). Whereas in D. melanogaster S2 cells depletion of dynein does not substantially increase the frequency of multipolar mitoses, another minus‐end directed motor, non‐claret disjunctional (NCD), seems to take over the role of dynein in suppressing multipolarity in fly cells (Kwon et al., 2008). In acentrosomal D. melanogaster oocytes NCD is necessary for efficient bundling of microtubules at spindle poles. Also, mitotic centromere‐associated kinesin KIF2C/MCAK, which functions as microtubule depolymerase and is believed to be a key component of the error correction mechanism at kinetochores, plays a role in centrosomal clustering in flies (Kwon et al., 2008). Interestingly, analogous to SGOL1 for the CPC, SGOL2 serves to recruit KIF2C/MCAK to the inner centromere (Huang et al., 2007).
Most recently integrin‐linked kinase (ILK) has been shown to mediate centrosome clustering via transforming acidic coiled‐coil 3 (TACC3) and colonic and hepatic tumor overexpressed gene (ch‐TOG), two centrosomal proteins involved in stabilization of microtubule minus ends at spindle poles (Fielding et al., 2011). In addition, depletion of protein regulator of cytokinesis 1 (PRC1), a microtubule‐bundling protein with most prominent activity during central spindle formation that is also important for the establishment of kinetochore tension, leads to spindle multipolarity (Leber et al., 2010). These data are further evidence for the suggestion that the mechanisms responsible for holding supernumerary centrosomes together might be similar to the forces that bundle microtubules into a bipolar spindle array in cells with two centrosomes or even without centrosomes.
In further support of this concept, depletion of CEP164, a centrosomal protein implicated in anchoring microtubules at the centrosome, leads to spindle pole disintegration when tension is applied, ultimately resulting in increased acentrosomal spindle poles in human cells with and without extra centrosomes (Leber et al., 2010). Consistently, knockdown of several proteins involved in centrosomal clustering (e. g. ch‐TOG, CEP164, SGOL1, augmin complex components) causes the formation of multiple centrosomal spindle poles but also leads to varying proportions of acentrosomal poles. This suggests that inhibition of centrosomal clustering and interference with microtubule bundling into two spindle poles occur simultaneously, due to overlapping mechanisms (Leber et al., 2010).
Similar to CPC and NDC80 complex components, the majority of microtubule‐based motors and microtubule‐bundling proteins involved in centrosomal clustering including TACC3, ch‐TOG, ILK, PRC1, KIF2C/MCAK and the human NCD homolog KIFC1 (also known as HSET) are frequently overexpressed in different types of human cancer (Peset and Vernos, 2008; Charrasse et al., 1995; Ishikawa et al., 2008; Nakamura et al., 2007; Hannigan et al., 2005; Shimo et al., 2007; Carter et al., 2006; De et al., 2009; Grinberg‐Rashi et al., 2009). In addition, overexpression of the kinesin KIFC1/HSET has been shown to mediate resistance against docetaxel in breast cancer cells (De et al., 2009). Since taxanes induce spindle multipolarity at low concentrations (Chen and Horwitz, 2002), high‐level expression of KIFC1/HSET might counteract this effect and prevent taxane‐treated cells from multipolarity‐induced cell death by enabling bipolar spindle formation through centrosomal clustering. Also, expression levels of PRC1 were found to strongly correlate with aneuploidy levels, which themselves were associated with poor clinical outcome in several cancer types (Carter et al., 2006), findings that might as well be explained by prevention of multipolarity‐induced cell death and CIN induction through centrosomal clustering.
3. Asymmetric stem cell division, centrosomes and cancer
Besides inducing tolerable levels of CIN, supernumerary centrosomes disrupt asymmetric stem cell division leading to expansion of the stem cell pool and tumor formation, at least in flies (Basto et al., 2008). Whereas several elegant studies have unequivocally demonstrated the link between supernumerary centrosomes and CIN in vitro, no data is available to prove that induction of CIN is the mechanism by which extra centrosomes may cause tumors in mammals. Therefore, disruption of asymmetric stem cell division by extra centrosomes should be considered as a plausible alternative mechanism of transformation in the mammalian system as well.
When stem cells divide, their daughters either self‐renew stem cell identity or initiate differentiation. The balanced choice between these alternate fates is critical to maintain stem cell numbers and to rein in their potentially dangerous capacity for long‐term proliferation. Symmetric division allows stem cell expansion during embryogenesis and replacement of stem cells after injury but might also harbor the risk for tumorigenesis. Recent studies have highlighted the importance of the stem cell niche as a source of local extrinsic signals that specify stem cell self‐renewal. In the context of such a niche, developmentally regulated orientation of the mitotic spindle directs whether the outcome of a stem cell division is asymmetric or symmetric.
Studies of both mouse radial glia progenitors and D. melanogaster male germ stem cells showed that when the spindle is oriented perpendicular to the interface with the niche, upon cleavage, one daughter can maintain contact with the niche while the other is displaced away and is free to initiate differentiation (Figure 4A). By contrast, spindle orientation parallel to the niche interface allows both daughters to inherit attachments to, and receive local self‐renewal signals from the niche (Figure 4B). Strikingly, differential labeling of mother centrosomes in both flies and mice revealed that it is always the mother centrosome that remains next to the niche in the new stem cell while the daughter centrosome enters the differentiating daughter cell (Yamashita et al., 2007; Wang et al., 2009). Why centrosome age seemingly does not impact on daughter cell fate during symmetric stem cell divisions remains to be elucidated.
Figure 4.
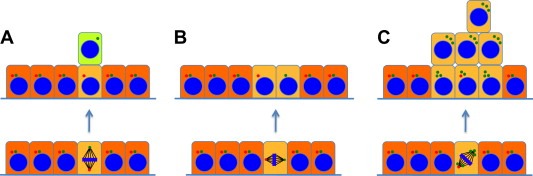
Centrosome behavior during stem cell division. (A) During asymmetric division, which generates one stem cell and one differentiated daughter, the mitotic spindle is oriented perpendicular to the plane of the epithelium. In D. melanogaster male germ stem cells and mouse radial glia progenitor cells, the mother centrosome (red) is always located close to the niche, thereby remaining within the developing new stem cell whereas the daughter centrosome (green) migrates toward the opposite side of the cell, leading to its segregation into the emerging differentiated daughter cell. (B) Symmetric division in the plane of the epithelium generates two stem cells. (C) Supernumerary centrosomes interfere with spindle orientation during asymmetric stem cell division, resulting in disturbed asymmetric division, aberrant expansion of the stem cell pool (hyperproliferation) and ultimately cancer.
Recently, Basto and coworkers (Basto et al., 2008) demonstrated that extra centrosomes can indeed initiate tumorigenesis in D. melanogaster overexpressing SAK. Most cells with supernumerary centrosomes initially formed multipolar spindles, but these spindles ultimately became bipolar owing to centrosomal clustering. Surprisingly, the frequency of aneuploidy was only slightly increased. Instead, spindle orientation and thereby asymmetric division of larval neural stem cells was compromised by the extra centrosomes, leading to hyperproliferation of neuroblasts and malignant transformation. A likely explanation for these findings is that amplified centrosomes interfere with asymmetric stem cell division, resulting in hyperproliferation with subsequent induction of CIN that leads to malignant transformation (Basto et al., 2008) (Figure 4C). Consistent with such a scenario is the same sequence of events in D. melanogaster larval neuroblasts containing mutations in genes that directly control asymmetric cell division (Caussinus and Gonzalez, 2005).
The reason why asymmetric division fails in cells with supernumerary centrosomes remains unclear. One possibility is that the asymmetric features of mother versus daughter centrosomes which for example determine microtubule nucleation potentials are disturbed by clustering of multiple centrosomes. Alternatively, astral microtubule organization, which is important for the interaction between cell cortex and spindle poles and therefore inherently linked to asymmetric division might be corrupted by centrosome clustering. Third, supernumerary centrosomes seem to prevent asymmetric localization of polarity determinants like MUD (the D. melanogaster homolog of the human spindle pole protein NUMA) (Caussinus and Gonzalez, 2005), what in turn might induce spindle positioning defects and disturbed asymmetric division.
To separately assess the contribution of centrosome defects versus CIN in tumorigenesis, Castellanos and coworkers studied the tumorigenic potential of multiple D. melanogaster larval brain tissue mutants defective in various aspects of centrosome biogenesis (Castellanos et al., 2008). Mutations affecting proteins required for centriole replication, pericentriolar matrix recruitment and centrosome function resulted in frequent tumor formation despite only a small fraction of cells having abnormal karyotypes. Consistently, mutations known to induce CIN, including defects in DNA replication and spindle assembly checkpoint chromatin condensation and cytokinesis did not give rise to tumors. These results again suggest that in tissues where self‐renewing asymmetric divisions are frequent, centrosome‐related disturbed stem cell division rather than induction of CIN might initiate malignant transformation.
Do these thought‐provoking results imply that centrosome aberrations do indeed cause cancer, however not via CIN as initially thought, but rather by perturbing stem cell division? Given the possible cell‐type‐ and organism‐specific effects, and the presence of moderate CIN along with the perturbed stem cell divisions, this conclusion seems premature. Furthermore, we urgently need insights into centrosome function in mammalian cancer stem cells. Answering the question of whether supernumerary centrosomes contribute to mammalian tumorigenesis by disruption of asymmetric division of cancer stem cells, induction of CIN, or both will be most rewarding.
4. Inhibition of centrosomal clustering as a novel anti‐cancer treatment strategy
Supernumerary centrosomes almost exclusively occur in a wide variety of neoplastic disorders but only rarely in non‐transformed cells. Therefore, inhibition of centrosomal clustering with consequential induction of multipolar spindles and subsequent cell death would specifically target tumor cells with no effect on normal cells with a regular centrosome content (Nigg, 2002; Brinkley, 2001).
Recently, griseofulvin has been identified as to inhibit centrosomal clustering (Rebacz et al., 2007). Griseofulvin has been used for many years for the treatment of dermatophyte infections (Loo, 2006). Mechanistically, it inhibits mitosis in sensitive fungi (Gull and Trinci, 1973) and mammalian cells (Grisham et al., 1973) but whether mitotic arrest is a consequence of microtubule depolymerization or some other action on microtubules in both fungi and human cells is still unclear (Grisham et al., 1973; Weber et al., 1976). Despite extensive studies, the mechanism by which the drug inhibits mitosis in human cells remains unclear. Although griseofulvin has been reported to bind to mammalian brain tubulin and to inhibit microtubule polymerization in vitro, it does so only at concentrations significantly higher than those needed for spindle multipolarity induction in cancer cells with extra centrosomes (Panda et al., 2005). Also, whether griseofulvin binds to tubulin directly or to microtubule associated proteins remains conflicting (Panda et al., 2005; Wehland et al., 1977; Roobol et al., 1977). Already more than 30 years ago it was reported that griseofulvin treatment induces spindle multipolarity with each mitotic center containing two centrioles in HeLa cells (Grisham et al., 1973). While at lower concentrations the drug leads to multipolar spindles with centrosomes at each pole in cells with extra centrosomes, at higher concentrations on top spindle multipolarity with acentrosomal spindle pole formation is induced, consistent with the above concept that clustering extra centrosomes in cancer cells might be similar to focusing microtubules into a bipolar spindle array in normal cells. For detailed mechanistic understanding it will be important to clearly determine the sequence of events: Does the drug at low concentrations indeed cause declustering of supernumerary centrosomes with subsequent multipolar spindle formation or does spindle multipolarity occur first with successive distribution of centrosomes to each pole?
Additional evidence for an effect of griseofulvin on centrosomal clustering comes from the finding that the drug, in contrast to other microtubule interacting compounds, induces hepatomas in mice and rats (Epstein et al., 1967). In these animals the majority of hepatocytes are polyploid and therefore contain supernumerary centrosomes which are usually efficiently clustered into bipolar spindle arrays (Guidotti et al., 2003; Duncan et al., 2010).
Findings similar to those reported for griseofulvin have recently been described for the microtubule‐modulating noscapinoid EM011 (Karna et al., 2011). In contrast to griseofulvin, EM011 seems to induce centrosome amplification prior to declustering, thereby potentially reducing its specificity to cancer cells with supernumerary centrosomes. Further supporting the candidacy of centrosomal clustering for a largely cancer‐selective target, at low drug concentrations sufficient for spindle multipolarity induction in cancer cells, microtubule poisons including nocodazole and taxol induce greater cell death in tumor cells than in non‐transformed cells (Brito and Rieder, 2009).
In addition to drugs, siRNAs to the kinesin KIFC1/HSET, the NDC80 complex subunit HEC1, aurora B, survivin, sororin, SGOL1, the augmin complex subunits HAUS3 and FAM29A (also known as HAUS6) as well as ILK and PRC1 lead to cell death through inhibition of centrosomal clustering in tumor cells with amplified centrosomes but not in normal cells (Kwon et al., 2008; Leber et al., 2010; Fielding et al., 2011). Identified in an RNAi screen performed in D. melanogaster S2 cells, siRNAs to KIFC1/HSET and Myo10 increased the frequency of spindle multipolarity in human cancer cells harboring supernumerary centrosomes as well (Kwon et al., 2008). Especially KIFC1/HSET might constitute an interesting therapeutic target as knockdown of the protein had no effect on cell division in diploid control cells but largely decreased viability of tumor cells with extra centrosomes by inducing multipolar anaphases and subsequent apoptosis (Kwon et al., 2008).
Also, small molecule inhibition of ILK, HEC1 and aurora B suppresses tumor cell growth in tissue culture as well as in animals (Huang et al., 2007; Wu et al., 2008a; Wilkinson et al., 2007; Kalra et al., 2009). Therefore, induction of multipolar spindles seems to induce cell death irrespective of the underlying mechanism that induced them. By contrast, although inhibition of monopolar spindle 1 (MPS1, also known as TTK), a dual‐specificity kinase required for the maintenance of SAC activation, inhibits centrosomal clustering and induces aberrant cell divisions in cells with supernumerary centrosomes, it does not cause selective cytotoxicity in cells with amplified centrosomes compared to cells with a regular centrosome content (Kwiatkowski et al., 2010). Therefore, it might be concluded that whereas SAC inhibition per se equally targets all cells, selective inhibition of centrosomal clustering through specific targeting may provide a therapeutic window to specifically target cells with supernumerary centrosomes.
Importantly, prior to cell death, cells with inhibited HEC1 or aurora B have multipolar spindles, lagging chromosomes and subsequent aneuploidy and polyploidy (Wu et al., 2008a; Wilkinson et al., 2007). Therefore, a possible downside of centrosomal cluster inhibition as a potential treatment approach might be the induction of cell clones with additional chromosomal abnormalities. On the optimistic side, such a risky scenario seems relatively unlikely, as multipolar cell division mostly leads to gross CIN and cell death (Ganem et al., 2009; Kops et al., 2004).
4.1. DNA damage response, supernumerary centrosomes and cancer therapy
Another striking and tantalizing issue that is emerging is the interplay between the centrosome division cycle, centrosomal clustering and DNA damage responses (DDR). Indeed, a host of key DDR factors including ataxia‐telangiectasia mutated (ATM), ataxia‐telangiectasia mutated and Rad3 related (ATR), CHK1 and CHK2 kinases, BRCA1 and other adapter and repair proteins, reside directly on centrosomes and regulate their function, providing checkpoint mechanisms to guard entry into mitosis (Krämer et al., 2004; Zhang et al., 2007; Parvin, 2009). From a broader perspective, both endogenous DNA damage signaling with the ensuing cellular senescence or death in response to oncogene‐induced replication stress (Jackson and Bartek, 2009; Halazonetis et al., 2008), and the lethal consequences of multipolar mitoses in cells with supernumerary centrosomes, provide complementary intrinsic anti‐cancer barriers (Löffler et al., 2007; Dodson et al., 2004; Dodson et al., 2007; Robinson et al., 2007). Those tumor cells that do survive and progress to invasive stages have commonly eliminated or bypassed such fail‐safe mechanisms, for example through p53 mutations and centrosomal clustering, respectively. Importantly, while defects and selective adaptations of the two barriers allow tumor cell outgrowth, these cancer cell‐selective changes concomitantly create cancer‐selective vulnerabilities that can be exploited for the benefit of patients (Kwon et al., 2008; Leber et al., 2010; Rebacz et al., 2007; Jackson and Bartek, 2009; Halazonetis et al., 2008). This notion can be illustrated by innovative treatments targeting DNA damage repair, signaling and checkpoints (Jackson and Bartek, 2009; Halazonetis et al., 2008), and a broadly analogous scenario is emerging for inhibitors of centrosomal clustering (Kwon et al., 2008; Leber et al., 2010; Rebacz et al., 2007).
Further to the intimate interplay between the DDR machinery and centrosomes, supernumerary centrosomes are inducible by various genotoxic insults including ionizing radiation and chemotherapeutic drugs, usually resulting in death of the affected cells by mitotic catastrophe (Löffler et al., 2007; Dodson et al., 2004; Dodson et al., 2007; Robinson et al., 2007). Whether some cases of acquired chemo‐ or radioresistance, a serious obstacle in cancer treatment, might be attributable to centrosome clustering remains to be seen. A possible relevance of these findings for cancer stem cells and asymmetric divisions would certainly be important to investigate. In any case, given that only a proportion of cells within a tumor mass contains extra centrosomes, the efficacy of inhibiting centrosomal clustering might be increased when combined with DNA‐damaging drugs or irradiation. As centrosome amplification after DNA damage occurs during a prolonged G2 phase arrest (Dodson et al., 2004; Loncarek et al., 2010) and many transformed cells have an intact G2/M but disrupted G1/S checkpoint, such a combined strategy would still predominantly target tumor cells and spare healthy tissues.
5. Conclusions and prospects
Conceptually, centrosomal clustering emerges as a powerful adaptive survival strategy that enables cells to successfully divide in the presence of supernumerary centrosomes (Table 1). In flies, clustering unbalances asymmetric divisions within stem cell compartments and thereby contributes to tumor formation, possibly also through induction of low‐level CIN. Whether disturbed stem cell division and/or CIN as a consequence of centrosome clustering also cause neoplastic transformation of mammalian cells has to be further explored. In this regard it has to be kept in mind that at least polyploid mouse hepatocytes divide by employing centrosome clustering and creating aneuploid offspring on a regular basis without malignant transformation.
Table 1.
Studies dealing with centrosome clustering in non‐transformed and cancer cells.
| Year | Title | Reference |
|---|---|---|
| 1982 | Mitosis in a cell with multiple centrioles | 23 |
| 1999 | Altered centrosome structure is associated with abnormal mitoses in human breast tumors | 24 |
| 2001 | Managing the centrosome numbers game: from chaos to stability in cancer cell division | 21 |
| 2002 | Centrosome aberrations: cause or consequence of cancer progression? | 6 |
| 2003 | Liver cell polyploidization: a pivotal role for binuclear hepatocytes | 29 |
| 2005 | Spindle multipolarity is prevented by centrosomal clustering | 32 |
| 2007 | Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype‐based screen | 90 |
| 2008 | Centrosome amplification can initiate tumorigenesis in flies | 27 |
| 2008 | Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes | 33 |
| 2008 | Extra centrosomes and/or chromosomes prolong mitosis in human cells. | 34 |
| 2008 | Delocalization of the microtubule motor dynein from mitotic spindles by the human papillomavirus E7 oncoprotein is not sufficient for induction of multipolar mitoses | 73 |
| 2009 | A mechanism linking extra centrosomes to chromosomal instability | 20 |
| 2009 | Multipolar spindle pole coalescence is a major source of kinetochore mis‐attachment and chromosome missegregation in cancer cells | 25 |
| 2010 | Proteins required for centrosome clustering in cancer cells | 37 |
| 2010 | The ploidy conveyor of mature hepatocytes as a source of genetic variation | 30 |
| 2010 | HURP permits MTOC sorting for robust meiotic spindle bipolarity, similar to extra centrosome clustering in cancer cells | 56 |
| 2010 | Small‐molecule kinase inhibitors provide insight into Mps1 cell cycle function | 104 |
| 2011 | A critical role of integrin‐linked kinase, ch‐TOG and TACC3 in centrosome clustering in cancer cells | 75 |
| 2011 | A novel microtubule‐modulating noscapinoid triggers apoptosis by inducing spindle multipolarity via centrosome amplification and declustering | 99 |
The high degree of selectivity and addiction of tumor cells that harbor extra centrosomes to centrosomal clustering indicates that drugs interfering with this mechanism may join the growing armamentarium of promising therapeutic options in oncology. High‐throughput screens for centrosome clustering inhibitors, as well as more detailed mechanistic understanding of the signaling and effector pathways involved in centrosomal clustering should accelerate the pace of both basic discovery and clinical applications in this field. However, centrosome clustering is no specialty of tumor cells and a certain percentage of cells – especially when polyploid – seems to be able to survive multipolar divisions. Finally, further insights into the functional interplay between centrosome biology and the DDR machinery hold promise for design of smart innovative combinatorial approaches to personalized treatment of cancer.
Finally, it should be considered that alternatively to centrosome clustering supernumerary centrosomes can also be inactivated, allowing only two centrosomes to function as spindle poles during mitosis. Accordingly, in flies overexpressing SAK some of the supernumerary centrosomes failed to organize microtubules, thereby allowing the spindle to be operationally bipolar (Basto et al., 2008). Since the inactivated centrosomes contained significantly less γ‐tubulin than those at the spindle poles, loss of pericentriolar material may, at least in part, account for the inactivation of extra centrosomes. Given the paucity of data about the mechanisms and frequency of selective inactivation of extra centrosomes and completely unexplored means of dealing with extra centrosomes like extrusion and degradation, these might be rewarding fields for further research.
Competing interests statement
The authors declare no competing financial interests.
Acknowledgments
We apologize to those authors whose work is not cited because of space limitations. A.K. is supported by the Deutsche Forschungsgemeinschaft (KR 1981/3‐1), the Deutsche Krebshilfe, the Deutsche José Carreras Leukämie‐Stiftung and the Interdisciplinary Research Program of the National Center for Tumor Diseases Heidelberg. J.B. is supported by the Danish National Research Foundation, the Danish Cancer Society, the Lundbeck Foundation, and the European Commission (projects: Biomedreg, Infla‐Care, DDResponse).
Krämer Alwin, Maier Bettina and Bartek Jiri, (2011), Centrosome clustering and chromosomal (in)stability: A matter of life and death, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.05.003.
References
- Adams, R.R. , 2001. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma. 110, 65–74. [DOI] [PubMed] [Google Scholar]
- Altieri, D.C. , 2003. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 22, 8581–8589. [DOI] [PubMed] [Google Scholar]
- Baker, D.J. , Jin, F. , Jeganathan, K.B. , van Deursen, J.M. , 2009. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 16, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto, R. , 2008. Centrosome amplification can initiate tumorigenesis in flies. Cell. 133, 1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias, M. , Glover, D.M. , 2007. Centrosome biogenesis and function: centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol.. 8, 451–463. [DOI] [PubMed] [Google Scholar]
- Birkbak, N.J. , 2011. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, J.R. , 1998. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J.. 17, 3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornens, M. , 2002. Centrosome composition and microtubule anchoring mechanisms. Curr. Opin. Cell Biol.. 14, 25–34. [DOI] [PubMed] [Google Scholar]
- Boveri, T. , 1929. The Origin of Malignant Tumors Williams and Wilkins; Baltimore, MD: [Google Scholar]
- Boyarchuk, Y. , Salic, A. , Dasso, M. , Arnaoutov, A. , 2007. Bub1 is essential for assembly of the functional inner centromere. J. Cell Biol.. 176, 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer, M. , 2010. HURP permits MTOC sorting for robust meiotic spindle bipolarity, similar to extra centrosome clustering in cancer cells. J. Cell Biol.. 191, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkley, B.R. , 2001. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol.. 11, 18–21. [DOI] [PubMed] [Google Scholar]
- Brito, D.A. , Rieder, C.L. , 2009. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motil. Cytoskeleton. 66, 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena, M. , Earnshaw, W.C. , 2003. The cellular geography of aurora kinases. Nat. Rev. Mol. Cell. Biol.. 4, 842–854. [DOI] [PubMed] [Google Scholar]
- Carter, S.L. , 2006. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet.. 38, 1043–1048. [DOI] [PubMed] [Google Scholar]
- Castellanos, E. , Dominguez, P. , Gonzalez, C. , 2008. Centrosome dysfunction in Drosophila neural stem cell causes tumors that are not due to genome instability. Curr. Biol.. 18, 1209–1214. [DOI] [PubMed] [Google Scholar]
- Caussinus, E. , Gonzalez, C. , 2005. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster . Nat. Genet.. 37, 1125–1129. [DOI] [PubMed] [Google Scholar]
- Chang, J.L. , 2006. Borealin/Dasra B is a cell cycle-regulated chromosomal passenger protein and its nuclear accumulation is linked to poor prognosis for human gastric cancer. Exp. Cell Res.. 312, 962–973. [DOI] [PubMed] [Google Scholar]
- Charrasse, S. , 1995. Characterization of the cDNA and pattern of expression of a new gene over-expressed in human hepatomas and colonic tumors. Eur. J. Biochem.. 234, 406–413. [DOI] [PubMed] [Google Scholar]
- Chen, J.G. , Horwitz, S.B. , 2002. Differential mitotic responses to microtubule-stabilizing and - destabilizing drugs. Cancer Res.. 62, 1935–1938. [PubMed] [Google Scholar]
- Dai, J. , Kataneva, A.V. , Higgins, J.M.G. , 2009. Studies of haspin-depleted cells reveal that spindle-pole integrity in mitosis requires chromosome cohesion. J. Cell Sci.. 122, 4168–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De, S. , Cipriano, R. , Jackson, M.W. , Stark, G.R. , 2009. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res.. 69, 8035–8042. [DOI] [PubMed] [Google Scholar]
- Diaz-Rodriguez, E. , Sotillo, R. , Schvartzman, J.-M. , Benezra, R. , 2008. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo . Proc. Natl. Acad. Sci. USA. 105, 16719–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson, H. , 2004. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J.. 23, 3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson, H. , Wheatley, S.P. , Morrison, C.G. , 2007. Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell Cycle. 6, 364–370. [DOI] [PubMed] [Google Scholar]
- Doxsey, S. , 2001. Re-evaluating centrosome function. Nat. Rev. Mol. Cell Biol.. 2, 688–698. [DOI] [PubMed] [Google Scholar]
- Duncan, A.W. , 2010. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 467, 707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarson, M.B. , Cukierman, E. , Compton, D.A. , Golemis, E.A. , 2004. Human enhancer of invasion-cluster, a coiled-coil protein required for passage through mitosis. Mol. Cell. Biol.. 24, 3957–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, S.S. , Andrea, J. , Joshi, S. , Mantel, N. , 1967. Hepatocarcinogenicity of griseofulvin following parenteral administration to infant mice. Cancer Res.. 27, 1900–1906. [PubMed] [Google Scholar]
- Ferretti, C. , 2010. Expression of the kinetochore protein Hec1 during the cell cycle in normal and cancer cells and its regulation by the pRb pathway. Cell Cycle. 9, 4147–4182. [DOI] [PubMed] [Google Scholar]
- Fielding, A.B. , Lim, S. , Montgomery, K. , Dobreva, I. , Dedhar, S. , 2011. A critical role of integrin-linked kinase, ch-TOG and TACC3 in centrosome clustering in cancer cells. Oncogene. 30, 521–534. [DOI] [PubMed] [Google Scholar]
- Fukasawa, K. , 2007. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer. 7, 911–924. [DOI] [PubMed] [Google Scholar]
- Ganem, N.J. , Godinho, S.A. , Pellman, D. , 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature. 460, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg-Rashi, H. , 2009. The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain. Clin. Cancer Res.. 15, 1755–1761. [DOI] [PubMed] [Google Scholar]
- Grisham, L.M. , Wilson, L. , Bensch, K.G. , 1973. Antimitotic action of griseofulvin does not involve disruption of microtubules. Nature. 244, 294–296. [DOI] [PubMed] [Google Scholar]
- Guidotti, J.-E. , 2003. Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J. Biol. Chem.. 278, 19095–19101. [DOI] [PubMed] [Google Scholar]
- Gull, K. , Trinci, A.P.J. , 1973. Griseofulvin inhibits fungal mitosis. Nature. 244, 292–294. [DOI] [PubMed] [Google Scholar]
- Halazonetis, T.D. , 2008. An oncogene-induced DNA damage model for cancer development. Science. 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hannigan, G. , Troussard, A.A. , Dedhar, S. , 2005. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat. Rev. Cancer. 5, 51–63. [DOI] [PubMed] [Google Scholar]
- Hayama, S. , 2006. Activation of CDCA1-KNTC2, members of centromere protein complex, involved in pulmonary carcinogenesis. Cancer Res.. 66, 10339–10348. [DOI] [PubMed] [Google Scholar]
- Hori, T. , 2008. CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell. 135, 1039–1052. [DOI] [PubMed] [Google Scholar]
- Huang, H. , 2007. Tripin/hSgo2 recruits MCAK to the inner centromere to correct defective kinetochore attachments. J. Cell Biol.. 177, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indjeian, V.B. , Stern, B.M. , Murray, A.W. , 2005. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science. 307, 130–133. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K. , 2008. Mitotic centromere-associated kinesin is a novel marker for prognosis and lymph node metastasis in colorectal cancer. Br. J. Cancer. 98, 1824–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, S.P. , Bartek, J. , 2009. The DNA damage response in human biology and disease. Nature. 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra, J. , 2009. QLT0267, a small molecule inhibitor targeting integrin-linked kinase (ILK), and docetaxel can combine to produce synergistic interactions linked to enhanced cytotoxicity, reductions in P-AKT levels, altered F-actin architecture and improved treatment outcomes in an orthotopic breast cancer model. Breast Cancer Res.. 11, R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karna, P. , 2011. A novel microtubule-modulating noscapinoid triggers apoptosis by inducing spindle multipolarity via centrosome amplification and declustering. Cell Death Differ.. 18, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima, S.A. , Tsukahara, T. , Langegger, M. , Hauf, S. , Kitajima, T.S. , Watanabe, Y. , 2007. Shugoshin enables tension-generating attachment of kinetochores by loading Aurora to centromeres. Genes Dev.. 21, 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima, S.A. , Yamagishi, Y. , Honda, T. , Ishiguro, K. , Watanabe, Y. , 2010. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science. 327, 172–177. [DOI] [PubMed] [Google Scholar]
- Kelly, A.E. , Ghenoiu, C. , Xue, J.Z. , Zierhut, C. , Kimura, H. , Funabiki, H. , 2010. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase aurora B. Science. 330, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffa, M.D. , Casanova, C.M. , Santarella, R. , Kocher, T. , Wilm, M. , Mattaj, I.W. , 2006. HURP is part of a Ran-dependent complex involved in spindle formation. Curr. Biol.. 16, 743–754. [DOI] [PubMed] [Google Scholar]
- Kops, G.J. , Foltz, D.R. , Cleveland, D.W. , 2004. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA. 101, 8699–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsami, M.K. , 2006. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J. Pathol.. 209, 512–521. [DOI] [PubMed] [Google Scholar]
- Krämer, A. , 2003. Centrosome aberrations as a possible mechanism for chromosomal instability in non-Hodgkin's lymphoma. Leukemia. 17, 2207–2213. [DOI] [PubMed] [Google Scholar]
- Krämer, A. , 2004. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol.. 6, 884–891. [DOI] [PubMed] [Google Scholar]
- Krämer, A. , Neben, K. , Ho, A.D. , 2002. Centrosome replication, genomic instability and cancer. Leukemia. 16, 767–775. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski, N. , 2010. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat. Chem. Biol.. 6, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, M. , 2008. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev.. 22, 2189–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawo, S. , 2009. HAUS, the 8-subunit complex, regulates centrosome and spindle integrity. Curr. Biol.. 19, 1–11. [DOI] [PubMed] [Google Scholar]
- Leber, B. , 2010. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med.. 2, 32–38. [DOI] [PubMed] [Google Scholar]
- Levine, D.S. , Sanchez, C.A. , Rabinovitch, P.S. , Reid, B.J. , 1991. Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc. Natl. Acad. Sci. USA. 88, 6427–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle, W.L. , Salisbury, J.L. , 1999. Altered centrosome structure is associated with abnormal mitoses in human breast tumors. Am. J. Pathol.. 155, 1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle, W.L. , Lutz, W.H. , Ingle, J.N. , Maihle, N.J. , Salisbury, J.L. , 1998. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc. Natl. Acad. Sci. USA. 95, 2950–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D. , Vader, G. , Vromans, M.J.M. , Lampson, M.A. , Lens, S.M.A. , 2009. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science. 323, 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler, H. , 2007. DNA damage-induced accumulation of centrosomal Chk1 contributes to its checkpoint function. Cell Cycle. 6, 2541–2548. [DOI] [PubMed] [Google Scholar]
- Loncarek, J. , Hergert, P. , Khodjakov, A. , 2010. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr. Biol.. 20, 1277–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo, D.S. , 2006. Systemic antifungal agents: an update of established and new therapies. Adv. Dermatol.. 22, 101–124. [DOI] [PubMed] [Google Scholar]
- Nakamura, Y. , 2007. Clinicopathological and biological significance of mitotic centromere-associated kinesin overexpression in human gastric cancer. Br. J. Cancer. 97, 543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neben, K. , Giesecke, C. , Schweizer, S. , Ho, A.D. , Krämer, A. , 2003. Centrosome aberrations in acute myeloid leukemia are correlated with cytogenetic risk profile. Blood. 101, 289–291. [DOI] [PubMed] [Google Scholar]
- Nguyen, H.G. , 2009. Deregulated Aurora-B induced tetraploidy promotes tumorigenesis. FASEB J.. 23, 2741–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, M.-H. , 2010. Phosphorylation and activation of cell division cycle associated 5 by mitogen-activated protein kinase play a crucial role in human lung carcinogenesis. Cancer Res.. 70, 5337–5347. [DOI] [PubMed] [Google Scholar]
- Nguyen, C.L. , McLaughlin-Drubin, M.E. , Münger, K. , 2008. Delocalization of the microtubule motor dynein from mitotic spindles by the human papillomavirus E7 oncoprotein is not sufficient for induction of multipolar mitoses. Cancer Res.. 68, 8715–8722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg, E.A. , 2002. Centrosome aberrations: cause or consequence of cancer progression?. Nat. Rev. Cancer. 2, 815–825. [DOI] [PubMed] [Google Scholar]
- Panda, D. , Rathinasamy, K. , Santra, M.K. , Wilson, L. , 2005. Kinetic suppression of microtubule dynamic instability by griseofulvin: implications for its possible use in the treatment of cancer. Proc. Natl. Acad. Sci. USA. 102, 9878–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvin, J.D. , 2009. The BRCA1-dependent ubiquitin ligase, γ-tubulin, and centrosomes. Environ. Mol. Mutagen.. 50, 649–653. [DOI] [PubMed] [Google Scholar]
- Peset, I. , Vernos, I. , 2008. The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol.. 18, 379–388. [DOI] [PubMed] [Google Scholar]
- Pihan, G.A. , Purohit, A. , Wallace, J. , 1998. Centrosome defects and genetic instability in malignant tumors. Cancer Res.. 58, 3974–3985. [PubMed] [Google Scholar]
- Pihan, G.A. , Purohit, A. , Wallace, J. , Malhotra, R. , Liotta, L. , Doxsey, S.J. , 2001. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res.. 61, 2212–2219. [PubMed] [Google Scholar]
- Putkey, F.R. , 2002. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev. Cell. 3, 351–365. [DOI] [PubMed] [Google Scholar]
- Quintyne, N.J. , Reing, J.E. , Hoffelder, D.R. , Gollin, S.M. , Saunders, W.S. , 2005. Spindle multipolarity is prevented by centrosomal clustering. Science. 307, 127–129. [DOI] [PubMed] [Google Scholar]
- Rebacz, B. , 2007. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res.. 67, 6342–6350. [DOI] [PubMed] [Google Scholar]
- Ring, D. , Hubble, R. , Kirschner, M. , 1982. Mitosis in a cell with multiple centrioles. J. Cell Biol.. 94, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, H.M.R. , Black, E.J. , Brown, R. , Gillespie, D.A.F. , 2007. DNA mismatch repair and Chk1-dependent centrosome amplification in response to DNA alkylation damage. Cell Cycle. 6, 982–992. [DOI] [PubMed] [Google Scholar]
- Roobol, A. , Gull, K. , Pogson, C.I. , 1977. Evidence that griseofulvin binds to a microtubule associated protein. FEBS Lett.. 75, 149–153. [DOI] [PubMed] [Google Scholar]
- Ruchaud, S. , Carmena, M. , Earnshaw, W.C. , 2007. Chromosomal passengers: conducting cell division. Nat. Rev. Mol. Cell Biol.. 8, 798–812. [DOI] [PubMed] [Google Scholar]
- Schmitz, J. , Watrin, E. , Lénárt, P. , Mechtler, K. , Peters, J.-M. , 2007. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr. Biol.. 17, 630–636. [DOI] [PubMed] [Google Scholar]
- Schvartzman, J.-M. , Sotillo, R. , Benezra, R. , 2010. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev. Cancer. 10, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, S.P. , 2009. Mutational evolution in a lobular breast tumor profiled at single nucleotide resolution. Nature. 461, 809–813. [DOI] [PubMed] [Google Scholar]
- Shao, S. , 2010. Centrosomal Nlp is an oncogenic protein that is gene-amplified in human tumors and causes spontaneous tumorigenesis in transgenic mice. J. Clin. Invest.. 120, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimo, A. , 2007. Elevated expression of protein regulator of cytokinesis 1, involved in the growth of breast cancer cells. Cancer Sci.. 98, 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth, W.T. , Nardi, I.K. , Scholl, L.M. , Cimini, D. , 2009. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation. in cancer cells. PLoS One. 4, e6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillje, H.H.W. , Nagel, S. , Körner, R. , Nigg, E.A. , 2006. HURP is a Ran-importin β-related protein that stabilizes kinetochore microtubules in the vincinity of chromosomes. Curr. Biol.. 16, 731–742. [DOI] [PubMed] [Google Scholar]
- Sluder, G. , Thompson, E.A. , Miller, F.J. , Hayes, J. , Rieder, C.L. , 1997. The checkpoint control for anaphase onset does not monitor excess numbers of spindle poles or bipolar spindle symmetry. J. Cell Sci.. 110, 421–429. [DOI] [PubMed] [Google Scholar]
- Sotillo, R. , 2007. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 11, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S. , Peters, J.M. , 2008. Polo and Aurora kinases: lessons derived from chemical biology. Curr. Opin. Cell Biol.. 20, 77–84. [DOI] [PubMed] [Google Scholar]
- Thery, M. , 2005. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol.. 7, 947–953. [DOI] [PubMed] [Google Scholar]
- Tsou, A.P. , 2003. Identification of a novel cell cycle regulated gene, HURP, overexpressed in human hepatocellular carcinoma. Oncogene. 22, 298–307. [DOI] [PubMed] [Google Scholar]
- Uehara, R. , 2009. The augmin complex plays a critical role in spindle microtubule generation for mitotic progression and cytokinesis in human cells. Proc. Natl. Acad. Sci. USA. 106, 6998–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse, V. , Prykhozhij, S. , Hardwick, K.G. , 2007. Shugoshin 2 regulates localization of the chromosomal passenger proteins in fission yeast mitosis. Mol. Biol. Cell. 18, 1657–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hansemann, D. , 1890. Über asymmetrische Zellteilung in Epithelkrebsen und deren biologische Bedeutung. Virchows Arch. Patholog. Anat.. 119, 299–326. (in German) [Google Scholar]
- Wang, X. , 2009. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 461, 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F. , 2010. Histone H3 Thr-3 phosphorylation by haspin positions aurora B at centromeres in mitosis. Science. 330, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, B.A. , Silk, A.D. , Montagna, C. , Verdier-Pinard, P. , Cleveland, D.W. , 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 11, 25–36. [DOI] [PubMed] [Google Scholar]
- Weber, K. , Wehland, J. , Herzog, W. , 1976. Griseofulvin interacts with microtubules both in vitro and in vivo . J. Mol. Biol.. 102, 817–829. [DOI] [PubMed] [Google Scholar]
- Wehland, J. , Herzog, W. , Weber, K. , 1977. Interaction of griseofulvin with microtubules, microtubule protein and tubulin. J. Mol. Biol.. 111, 329–342. [DOI] [PubMed] [Google Scholar]
- Wei, R.R. , Al-Bassam, J. , Harrison, S.C. , 2007. The Ndc80/HEC1 complex is a contact point for kinetochore-microtubule attachment. Nat. Struct. Mol. Biol.. 14, 54–59. [DOI] [PubMed] [Google Scholar]
- Wilkinson, R.W. , 2007. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res.. 13, 3682–3688. [DOI] [PubMed] [Google Scholar]
- Wong, J. , Fang, G. , 2006. HURP controls spindle dynamics to promote proper interkinetochore tension and efficient kinetochore capture. J. Cell Biol.. 173, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , 2008. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res.. 68, 8393–8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Lin, Y.T. , Wei, R. , Chen, Y. , Shan, Z. , Lee, W.H. , 2008. Hice1, a novel microtubule-associated protein required for maintenance of spindle integrity and chromosomal stability in human cells. Mol. Cell. Biol.. 28, 3652–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi, Y. , Honda, T. , Tanno, Y. , Watanabe, Y. , 2010. Two histone marks establish the inner centromere and chromosome bi-orientation. Science. 330, 239–243. [DOI] [PubMed] [Google Scholar]
- Yamashita, Y.M. , Mahowald, A.P. , Perlin, J.R. , Fuller, M.T. , 2007. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science. 315, 518–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Loncarek, J. , Khodjakov, A. , Rieder, C.L. , 2008. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat. Cell Biol.. 10, 748–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , 2007. Centrosomal localization of DNA damage checkpoint proteins. J. Biol. Chem.. 101, 451–465. [DOI] [PubMed] [Google Scholar]