

Abstract
Human cancers are characterized by the presence of genomic instability. Recently, two studies have catalogued the presence of a specific class of genomic aberrations, large deletions and insertions, in a few thousand human cancers and reported that most of the prevalent recurrent focal deletions targeted common fragile sites and large genes. In various experimental systems, deletions in common fragile sites and large genes have been linked to the presence of DNA replication stress. Thus, taken together, these results suggest the presence of DNA replication stress in human cancers, consistent with the recently proposed oncogene‐induced DNA damage model for cancer development.
Keywords: Genomic deletions, Copy number changes, DNA replication stress, Common fragile sites, Large genes
Highlights
High throughput studies characterizing genomic instability in human cancers were reviewed.
The most frequent deletions in human cancers target the common fragile sites.
DNA replication stress induces deletions in common fragile sites.
Oncogenes induce DNA replication stress.
Thus, oncogene‐induced DNA replication stress induces genomic instability in human cancers.
1. Introduction
Genomic instability is a feature of most human cancers and is thought to play an important role both in cancer development and in the response to therapy (Murga and Fernandez‐Capetillo, 2007; Negrini et al., 2010). One can distinguish several types of genomic instabilities.
One type predisposes to accumulation of point mutations. For example, inheritance of a defective form of the base excision repair gene MYH leads to somatic G:C‐T:A mutations in colorectal cancers (Al‐Tassan et al., 2002). Additionally, germline mutations in nucleotide excision repair genes facilitate accumulation of UV light‐induced mutations, leading to skin cancer (Cleaver, 2005). And recent evidence suggests that even repair of DNA double‐strand breaks (DSBs) by homologous recombination (HR) may be associated with the accumulation of point mutations (Hicks et al., 2010; Deem et al., 2011).
A second type of genomic instability leads to microdeletions and microduplications. Microsatellite instability (MIN) is such a type (Lengauer et al., 1997). MIN leads to changes in the number of dinucleotide or trinucleotide repeats present at specific polymorphic sites in the genome, referred to as microsatellite repeats. Most patients, whose cancers have MIN, have inherited a defective copy of one of the genes that function in mismatch DNA repair (Fishel et al., 1993; Leach et al., 1993).
A third type of genomic instability, referred to as chromosomal instability (CIN), predisposes to large genomic deletions and amplifications (in the order of hundreds of kilobases in length) and/or chromosomal translocations. DNA DSBs are likely to be involved at some level in the formation of these lesions, either as initiators of CIN or as reaction intermediates. CIN is the most prevalent type of genomic instability in non‐inherited human cancers (Nowell, 1976; Lengauer et al., 1997).
In this review we focus on large deletions associated with CIN. Two recent studies catalogued such deletions in large cohorts of human cancers (Beroukhim et al., 2010; Bignell et al., 2010). The results of these studies suggest that many recurrent deletions associated with human cancers are due to DNA replication stress.
2. Somatic copy number changes (CNCs) in human cancers
Two recent studies have significantly broadened our knowledge regarding the types of large deletions and amplifications present in human cancers (Beroukhim et al., 2010; Bignell et al., 2010). The strength of the first study is the inclusion of thousands of samples of primary human cancer tissues, whereas the strength of the second study is the discrimination of homozygous from hemizygous deletions. Although these two studies reach very similar conclusions, we will first summarize their findings separately and then suggest mechanisms that could help explain the presence of CNCs in human cancers.
2.1. Distinct types of genes targeted by genomic deletions
Beroukhim et al. (2010) examined 3131 cancer samples, of which 2520 were cancer tissues isolated from human patients, 541 established cancer cell lines and 70 short‐term melanoma cultures. CNCs were determined using Affymetrix 250 K single nucleotide polymorphism (SNP) arrays. These arrays contain probes for approximately 250,000 SNPs, which corresponds to an average density of 1 SNP per 12,000 base pairs in the human genome.
The CNCs identified in this study included both amplifications and deletions. About 75% of the CNCs were focal in nature, affecting between 50,000–300,000 base pairs, while the remaining 25% involved an entire arm of a chromosome or the entire chromosome. It is reasonable to argue that different mechanisms are likely to be responsible for the two types of CNCs. We will review here the focal CNCs, because they represent the majority of the observed CNCs and because their focal nature provides mechanistic insights regarding their development.
Analysis of the entire sample of 3131 cancers led to the identification of 76 focal amplifications and 82 focal deletions. Of the 76 focal amplifications, 25 targeted validated oncogenes, while for the remaining 51 amplifications it was not clear what type of genes were targeted (Figure. 1). Among the 10 most frequent focal amplifications (of the total of 76), all 10 targeted oncogenes. This suggests that the focal amplifications target primarily oncogenes and raises the possibility that novel oncogenes may be discovered in the less frequently amplified loci.
Figure 1.
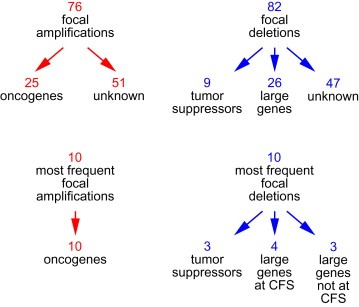
Spectrum of copy number changes in human cancers, according to Beroukhim et al. (2010). CFS, common fragile sites.
Of the 82 focal deletions, 9 targeted validated tumor suppressor genes and 26 targeted large genes (Beroukhim et al., 2010). The latter genes, defined in this study as genes having a length greater than 750 kilobases, appear to lack tumor suppressor function, raising the question why deletions targeting these genes are so common.
To gain insights into the mechanisms leading to deletions in human cancers, we examined the list of the 10 most frequent recurrent deletions, as reported by Beroukhim et al. (2010). Of these 10 deletions, 3 targeted tumor suppressor genes and 7 targeted large genes (Figure. 1).
The most frequent recurrent deletion targeted two tumor suppressor genes that are physically close to each other: cyclin‐dependent kinase inhibitor 2A (CDKN2A) and cyclin‐dependent kinase inhibitor 2B (CDKN2B) (Figure. 2). These two genes encode the cell cycle inhibitors p16INK4A/ARF and p15INK4B, respectively (Quelle et al., 1995; Krimpenfort et al., 2007). The seventh and ninth most common deletions also targeted tumor suppressor genes: the retinoblastoma 1 (Planas‐Silva and Weinberg, 1997) and FAT tumor suppressor homolog 1 (Mahoney et al., 1991; Hou et al., 2006) genes, respectively. However, all the remaining focal deletions in the top ten list did not target tumor suppressor genes (Figure. 2). Instead, the targeted genes that had diverse functions (Table 1); their only common feature being that they were large genes. Several of these genes also mapped to common fragile sites (Figure. 2; Table 1).
Figure 2.

List of the ten most frequent focal deletions in human cancers, according to Beroukhim et al. (2010). For each deletion, the name of the targeted gene and specific properties of the deletion and/or gene are indicated. TSG, known tumor suppressor gene; CFS, common fragile site; RS, replication stress.
Table 1.
List of the genes that are frequently targeted by focal deletions in human cancers.
| Gene symbol | Gene name | TSG | CFS | Comments and references |
|---|---|---|---|---|
| CDKN2A | Cyclin‐dependent kinase inhibitor 2A | YES | NO | CDKN2A encodes the cell cycle inhibitors p16INK4A and ARF (Quelle et al., 1995). |
| CDKN2B | Cyclin‐dependent kinase inhibitor 2B | YES | NO | CDKN2B encodes the cell cycle inhibitor p15INK4B (Krimpenfort et al., 2007). |
| FHIT | Fragile histidine triad | DEBATED | FRA3B | FHIT encodes a diadenosine triphosphate hydrolase, whose putative tumor suppressor activity is still being debated (Le Beau et al., 1998; Saldivar et al., 2010). It is the largest gene on chromosome 3. |
| WWOX | WW domain containing oxidoreductase | DEBATED | FRA16D | WWOX is expressed at high levels in the distal convoluted tubules of nephrons and its targeted deletion in mice leads to kidney failure and death within the first few weeks of life (Aqeilan et al., 2007; Ludes‐Meyers et al., 2009; Watanabe et al., 2003). |
| PTPRD | Protein tyrosine phosphatase receptor type delta | NO | NO | PTPRD is normally expressed in the brain and in B and T lymphocytes and appears to play a role in learning and memory (Uetani et al., 2000). |
| MACROD2 | MACRO domain containing 2 | NO | NO | The function of MACROD2 is unclear, but the presence of the MACRO domain suggests that its protein product binds poly‐ ADP‐ribose (Bradley et al., 2010). |
| PARK2 | Parkinson protein 2 | NO | FRA6E | Mutations in PARK2 are associated with autosomal recessive juvenile Parkinson's disease (Kitada et al., 1998). |
| LRP1B | Low density lipoprotein‐related protein 1B | NO | FRA2F | LRP1B, a member of the low density lipoprotein receptor family, is expressed preferentially in the brain; its inactivation in mice does not lead to any obvious phenotype (Marschang et al., 2004). |
| RB1 | Retinoblastoma 1 | YES | NO | RB1 is a tumor suppressor that regulates the G1‐S transition (Planas‐Silva and Weinberg, 1997). |
| FAT1 | FAT tumor suppressor homolog 1 | YES | NO | FAT1 encodes a cadherin that coordinates cell–cell adhesion to cellular growth; it was initially discovered as a tumor suppressor gene in Drosophila (Hou et al., 2006; Mahoney et al., 1991). |
| PDE4D | Phosphodiesterase 4D | NO | NO | PDE4D regulates cAMP levels and contraction in the airway smooth muscle cells and in the heart muscle (Hansen et al., 2000; Lehnart et al., 2005). |
| IMMP2L | Inner mitochondrial membrane peptidase 2 like | NO | FRA7K | The major phenotype of IMMP2L‐deficient mice is infertility (Lu et al., 2008). |
| FAM190A | Family with sequence similarity 190, member A | NO | FRA4F | FAM190A is expressed in the cerebellum and may be linked to attention deficit disorder (Lantieri et al., 2010). |
| GRID2 | Glutamate receptor, ionotropic, delta 2 | NO | FRA4F | GRID2 is associated with cerebellar ataxia (Lalouette et al., 1998). |
| A2BP1 | Ataxin 2 binding protein 1 | NO | NO | A2BP1 regulates alternative splicing of neuronal transcripts (Lee et al., 2009). |
| UNKNOWN chr9(12 Mb) | – | NO | NO | No TSG or large genes at this locus. |
| CNTNAP2 | Contactin‐associated protein‐like 2 | NO | FRA7I | CNTNAP2 may play a role in speech development (Smith et al., 2006; Newbury and Monaco, 2010). |
| LSAMP | Limbic system‐associated membrane protein | NO | NO | LSAMP regulates the behavioral response to stress (Catania et al., 2008). |
TSG, tumor suppressor gene; CFS, common fragile site.
2.2. Homozygous and hemizygous deletions
Bignell et al. (2010) analyzed 746 human cancer cell lines using Affymetrix 6.0 SNP arrays. These arrays contain approximately 1.8 million SNP and copy number probes, providing an average density of about 1 probe per 1700 bp in the human genome. Because cancer cell lines are devoid of normal tissue DNA, it was possible in this study to distinguish homozygous deletions (affecting all alleles) from hemizygous deletions (affecting only one allele).
A total of 206 recurrent deletions were identified. Of these, 14 mapped within known tumor suppressor genes, 19 within common fragile sites, 5 within regions that are normally rearranged in B cells and T cells, such as, for example, the T cell receptor genes, and the remaining 168 recurrent deletions were “unexplained” (Figure 3). We prepared a list of the ten most common focal deletions taking into account both homozygous and hemizygous deletions (using data taken from Supplementary Table 7; columns ws and HD; Bignell et al., 2010). According to the authors, 1 of these deletions mapped to a tumor suppressor gene, 5 mapped to common fragile sites and 4 were “unexplained” (Figure 3).
Figure 3.
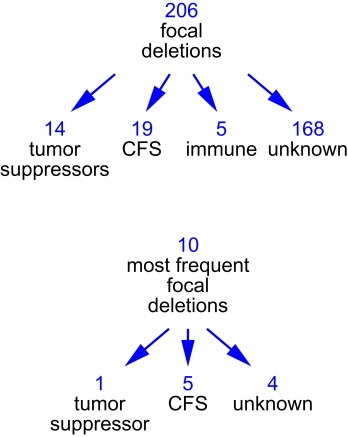
Spectrum of focal deletions in human cancers, according to Bignell et al. (2010). CFS, common fragile sites; immune, regions normally rearranged in B cells and T cells.
The most common deletion targeted the CDKN2A tumor suppressor gene. The preponderance of deletions targeting CDKN2A were homozygous, consistent with the need to completely inactivate the function of this gene (Figure 4).
Figure 4.

List of the ten most frequent focal deletions in human cancers, according to Bignell et al. (2010). For each deletion, the name of the targeted gene and specific properties of the deletion and/or gene are indicated. TSG, known tumor suppressor gene; CFS, common fragile site; Homo, homozygous deletion; Hemi, hemizygous deletion; RS, replication stress; FAM, FAM190A; chr9:12 Mb, chromosome 9:position 12 Mb on Build 36.3 of the human genome.
Five of the ten most common deletions targeted genes that map to common fragile sites. These genes were: FHIT, WWOX, IMMP2L, FAM190A‐GRID2 and CNTNAP2, which map to the common fragile sites FRA3B, FRA16D, FRA7K, FRA4F and FRA7I, respectively (Figure 4; Table 1). The majority of the deletions targeting these genes were hemizygous, indicating that gene function was maintained. The only common feature of these genes was their large size (Table 1).
The remaining four of the ten most common deletions, which were considered “unexplained” by Bignell et al. (2010), had features similar to the deletions that targeted the common fragile sites. They were hemizygous and did not target tumor suppressor genes. Three of the four “unexplained” deletions targeted the large genes MACROD2, A2BP1 and LSAMP (Figure 4; Table 1), while the fourth deletion targeted a region of chromosome 9 (around position 12 Mb) that contains no obvious tumor suppressor genes nor any large genes.
3. Driver and passenger mutations
Both studies described above identified two classes of focal deletions in human cancers (Beroukhim et al., 2010; Bignell et al., 2010). The deletions in the first class tended to be homozygous and targeted tumor suppressor genes, while the deletions in the second class were predominantly hemizygous and targeted large genes and common fragile sites.
The two classes of deletions described above are reminiscent of the two classes of point mutations described in human cancers: driver and passenger mutations (Sjoblom et al., 2006; Greenman et al., 2007). It is well‐accepted that only a few of the many point mutations present in human cancers contribute to the transformed phenotype. These mutations, referred to as driver mutations, would be those that activate oncogenes or inactivate tumor suppressor genes. The second class of point mutations constitutes the majority of the mutations found in human cancers. These mutations do not contribute to the transformed phenotype, but instead are thought to have been acquired during cancer development and to have persisted due to the clonal nature of human cancers. These point mutations are referred to as passenger mutations.
The concept of driver and passenger mutations can, of course, be extended to the classes of deletions, described above (Figure 5). The homozygous deletions targeting known tumor suppressor genes would be the driver mutations. They probably arise at a low frequency, but are strongly selected for, because they confer a growth advantage. In contrast, the hemizygous nature of the deletions targeting large genes and common fragile sites suggests that these deletions are probably close to neutral in terms of selective advantage. Therefore, these deletions would be passenger mutations, indicative of the mechanisms leading to genomic instability in human cancers.
Figure 5.
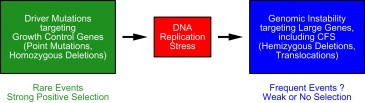
Driver mutations in growth control genes contribute to oncogenic transformation, but also induce DNA replication stress. Passenger mutations occur in response to DNA replication stress; they confer little or no selective advantage, but are indicative of the mechanisms underlying genomic instability in cancer.
4. Common fragile sites and large genes
As mentioned above, several of the deletions targeting large genes in human cancers map to common fragile sites. Common fragile sites have been defined cytogenetically as gaps or breaks on metaphase chromosomes in cells that have been treated with inhibitors of DNA replication (Glover et al., 1984; Durkin and Glover, 2007).
Practically all the characterized common fragile sites map within large genes (Helmrich et al., 2006, 2007, 2007, 2006). Thus, the question arises whether deletions targeting large genes that have not yet been associated with common fragile sites are also causally related to DNA replication stress. To address this question, we recently characterized by array comparative genomic hybridization the deletions induced by aphidicolin in human chromosome 3 (Pasi et al., 2011). We observed deletions targeting the established common fragile sites, primarily FRA3B, but also other sites that have not yet been described as common fragile sites. All these latter sites corresponded to large genes. Of particular interest, one of these sites mapped to position 118 Mb of chromosome 3, corresponding to the LSAMP gene. A similar analysis by another group also identified deletions within the LSAMP gene and other large genes in cells treated with aphidicolin (Arlt et al., 2009). Interestingly, the LSAMP gene was identified by Bignell et al. (2010) as a site frequently deleted in human cancers (Figure 4). Because the deletions at this site tended to be hemizygous, Bignell et al. (2010) treated cells with aphidicolin, but did not observe breaks or gaps at this position on metaphase chromosomes, suggesting that this site is not a common fragile site. However, analysis of aphidicolin‐treated cells indicates that this site is predisposed to deletions after treatment with aphidicolin (Arlt et al., 2009; Pasi et al., 2011). Thus, not all deletions induced by partial inhibition of DNA replication manifest themselves as common fragile sites on metaphase chromosomes and, therefore, deletions in large genes, irrespective of whether they map to common fragile sites or not, indicate the presence of DNA replication stress.
Based on the above argument, revisiting the lists of most frequent focal deletions, as identified by Beroukhim et al. (2010) and Bignell et al. (2010), reveals that the vast majority of these deletions in human cancers may be passenger mutations induced by DNA replication stress (2, 4).
5. Conclusion
Many of the common focal deletions in human cancers have features suggesting that they are passenger mutations: they are hemizygous and target genes that, in their majority, appear to lack tumor suppressor function (Beroukhim et al., 2010; Bignell et al., 2010). It is assumed that there is little positive or negative selection pressure for passenger mutations in human cancers (Sjoblom et al., 2006; Greenman et al., 2007). Accordingly, the nature of these mutations can help provide insights into the mechanisms leading to genomic instability in human cancers.
A few years ago, we and others proposed a model to explain the high frequency of p53 mutations in human cancers (Bartkova et al., 2005; Gorgoulis et al., 2005; Halazonetis et al., 2008). This model, referred to as the oncogene‐induced DNA damage model for cancer development, proposes that deregulation of growth control genes induces DNA replication stress and DNA damage, which in turn activate the ATM‐p53 pathway, leading to cell cycle arrest, apoptosis and/or senescence. Progression of precancerous lesions to cancer is associated with inactivation of the ATM‐p53 pathway, most often by mutations targeting the p53 gene. Initial evidence that activated oncogenes induced DNA replication stress was provided by analysis of allelic imbalances (loss of heterozygosity) in a small number of precancerous lesions. This analysis showed preferential targeting of common fragile sites, consistent with the DNA damage being due to DNA replication stress (Bartkova et al., 2005; Gorgoulis et al., 2005). Further studies have supported these initial observations (Tsantoulis et al., 2008). In addition, evidence has been obtained in various experimental systems that activated oncogenes induce collapse of DNA replication forks and replication‐dependent induction of DNA DSBs (Bartkova et al., 2006; Di Micco et al., 2006).
The recent high‐throughput genomic analyses of cancer specimens provides a very large sample size to interrogate the presence of DNA replication stress in human cancers (Beroukhim et al., 2010; Bignell et al., 2010). In one study, seven of the ten most common recurrent deletions in human cancers targeted common fragile sites and large genes (Figure. 2). In the other study, eight of the ten most common recurrent deletions can be attributed to DNA replication stress (Figure 4). Taken together, these findings point to the presence of DNA replication stress in human cancers, a key prediction of the oncogene‐induced DNA damage model (Halazonetis et al., 2008). Given that the number of human cancer specimens analyzed in these studies is in the thousands, we believe that these results provide strong justification for studies aiming to elucidate the mechanisms by which oncogenes perturb DNA replication. In addition, novel therapeutic approaches could be developed that capitalize on the presence of DNA replication stress in cancer, but not normal, cells (Toledo et al., 2011).
Acknowledgments
The authors thank the European FP7 Project GENICA and the Swiss National Foundation for financial support.
Dereli‐Öza Aygül, Versini Gwennaelle, and Halazonetis Thanos D., (2011), Studies of genomic copy number changes in human cancers reveal signatures of DNA replication stress, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.05.002.
References
- Al-Tassan, N. , Chmiel, N.H. , Maynard, J. , Fleming, N. , Livingston, A.L. , Williams, G.T. , Hodges, A.K. , Davies, D.R. , David, S.S. , Sampson, J.R. , Cheadle, J.P. , 2002. Inherited variants of MYH associated with somatic G: C-T: a mutations in colorectal tumors. Nat. Genet.. 30, 227–232. [DOI] [PubMed] [Google Scholar]
- Aqeilan, R.I. , Trapasso, F. , Hussain, S. , Costinean, S. , Marshall, D. , Pekarsky, Y. , Hagan, J.P. , Zanesi, N. , Kaou, M. , Stein, G.S. , 2007. Targeted deletion of Wwox reveals a tumor suppressor function. Proc. Natl. Acad. Sci. U.S.A.. 104, 3949–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt, M.F. , Mulle, J.G. , Schaibley, V.M. , Ragland, R.L. , Durkin, S.G. , Warren, S.T. , Glover, T.W. , 2009. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet.. 84, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova, J. , Horejsi, Z. , Koed, K. , Kramer, A. , Tort, F. , Zieger, K. , Guldberg, P. , Sehested, M. , Nesland, J.M. , Lukas, C. , 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Rezaei, N. , Liontos, M. , Karakaidos, P. , Kletsas, D. , Issaeva, N. , Vassiliou, L.V. , Kolettas, E. , Niforou, K. , Zoumpourlis, V.C. , 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 444, 633–637. [DOI] [PubMed] [Google Scholar]
- Beroukhim, R. , Mermel, C.H. , Porter, D. , Wei, G. , Raychaudhuri, S. , Donovan, J. , Barretina, J. , Boehm, J.S. , Dobson, J. , Urashima, M. , 2010. The landscape of somatic copy-number alteration across human cancers. Nature. 463, 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell, G.R. , Greenman, C.D. , Davies, H. , Butler, A.P. , Edkins, S. , Andrews, J.M. , Buck, G. , Chen, L. , Beare, D. , Latimer, C. , 2010. Signatures of mutation and selection in the cancer genome. Nature. 463, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, W.E. , Raelson, J.V. , Dubois, D.Y. , Godin, E. , Fournier, H. , Prive, C. , Allard, R. , Pinchuk, V. , Lapalme, M. , Paulussen, R.J. , Belouchi, A. , 2010. Hotspots of large rare deletions in the human genome. PLoS One. 5, e9401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania, E.H. , Pimenta, A. , Levitt, P. , 2008. Genetic deletion of Lsamp causes exaggerated behavioral activation in novel environments. Behav. Brain Res.. 188, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver, J.E. , 2005. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer. 5, 564–573. [DOI] [PubMed] [Google Scholar]
- Deem, A. , Keszthelyi, A. , Blackgrove, T. , Vayl, A. , Coffey, B. , Mathur, R. , Chabes, A. , Malkova, A. , 2011. Break-induced replication is highly inaccurate. PLoS Biol.. 9, e1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco, R. , Fumagalli, M. , Cicalese, A. , Piccinin, S. , Gasparini, P. , Luise, C. , Schurra, C. , Garre, M. , Nuciforo, P.G. , Bensimon, A. , 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 444, 638–642. [DOI] [PubMed] [Google Scholar]
- Durkin, S.G. , Glover, T.W. , 2007. Chromosome fragile sites. Annu. Rev. Genet.. 41, 169–192. [DOI] [PubMed] [Google Scholar]
- Fishel, R. , Lescoe, M.K. , Rao, M.R. , Copeland, N.G. , Jenkins, N.A. , Garber, J. , Kane, M. , Kolodner, R. , 1993. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 75, 1027–1038. [DOI] [PubMed] [Google Scholar]
- Glover, T.W. , Berger, C. , Coyle, J. , Echo, B. , 1984. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet.. 67, 136–142. [DOI] [PubMed] [Google Scholar]
- Gorgoulis, V.G. , Vassiliou, L.V. , Karakaidos, P. , Zacharatos, P. , Kotsinas, A. , Liloglou, T. , Venere, M. , Ditullio, R.A. , Kastrinakis, N.G. , Levy, B. , 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 434, 907–913. [DOI] [PubMed] [Google Scholar]
- Greenman, C. , Stephens, P. , Smith, R. , Dalgliesh, G.L. , Hunter, C. , Bignell, G. , Davies, H. , Teague, J. , Butler, A. , Stevens, C. , 2007. Patterns of somatic mutation in human cancer genomes. Nature. 446, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis, T.D. , Gorgoulis, V.G. , Bartek, J. , 2008. An oncogene-induced DNA damage model for cancer development. Science. 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hansen, G. , Jin, S. , Umetsu, D.T. , Conti, M. , 2000. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc. Natl. Acad. Sci. USA. 97, 6751–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich, A. , Stout-Weider, K. , Hermann, K. , Schrock, E. , Heiden, T. , 2006. Common fragile sites are conserved features of human and mouse chromosomes and relate to large active genes. Genome Res.. 16, 1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks, W.M. , Kim, M. , Haber, J.E. , 2010. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 329, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, R. , Liu, L. , Anees, S. , Hiroyasu, S. , Sibinga, N.E. , 2006. The Fat1 cadherin integrates vascular smooth muscle cell growth and migration signals. J. Cell Biol.. 173, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada, T. , Asakawa, S. , Hattori, N. , Matsumine, H. , Yamamura, Y. , Minoshima, S. , Yokochi, M. , Mizuno, Y. , Shimizu, N. , 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Krimpenfort, P. , Ijpenberg, A. , Song, J.Y. , van der Valk, M. , Nawijn, M. , Zevenhoven, J. , Berns, A. , 2007. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 448, 943–946. [DOI] [PubMed] [Google Scholar]
- Lalouette, A. , Guenet, J.L. , Vriz, S. , 1998. Hotfoot mouse mutations affect the delta 2 glutamate receptor gene and are allelic to lurcher. Genomics. 50, 9–13. [DOI] [PubMed] [Google Scholar]
- Lantieri, F. , Glessner, J.T. , Hakonarson, H. , Elia, J. , Devoto, M. , 2010. Analysis of GWAS top hits in ADHD suggests association to two polymorphisms located in genes expressed in the cerebellum. Am. J. Med. Genet. B. Neuropsychiatr. Genet.. 153B, 1127–1133. [DOI] [PubMed] [Google Scholar]
- Le Beau, M.M. , Drabkin, H. , Glover, T.W. , Gemmill, R. , Rassool, F.V. , McKeithan, T.W. , Smith, D.I. , 1998. An FHIT tumor suppressor gene?. Genes Chrom. Cancer. 21, 281–289. [DOI] [PubMed] [Google Scholar]
- Leach, F.S. , Nicolaides, N.C. , Papadopoulos, N. , Liu, B. , Jen, J. , Parsons, R. , Peltomaki, P. , Sistonen, P. , Aaltonen, L.A. , Nystrom-Lahti, M. , 1993. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 75, 1215–1225. [DOI] [PubMed] [Google Scholar]
- Lee, J.A. , Tang, Z.Z. , Black, D.L. , 2009. An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev.. 23, 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart, S.E. , Wehrens, X.H. , Reiken, S. , Warrier, S. , Belevych, A.E. , Harvey, R.D. , Richter, W. , Jin, S.L. , Conti, M. , Marks, A.R. , 2005. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 123, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer, C. , Kinzler, K.W. , Vogelstein, B. , 1997. Genetic instability in colorectal cancers. Nature. 386, 623–627. [DOI] [PubMed] [Google Scholar]
- Ludes-Meyers, J.H. , Kil, H. , Parker-Thornburg, J. , Kusewitt, D.F. , Bedford, M.T. , Aldaz, C.M. , 2009. Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS One. 4, e7775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, B. , Poirier, C. , Gaspar, T. , Gratzke, C. , Harrison, W. , Busija, D. , Matzuk, M.M. , Andersson, K.E. , Overbeek, P.A. , Bishop, C.E. , 2008. A mutation in the inner mitochondrial membrane peptidase 2-like gene (Immp2l) affects mitochondrial function and impairs fertility in mice. Biol. Reprod.. 78, 601–610. [DOI] [PubMed] [Google Scholar]
- Mahoney, P.A. , Weber, U. , Onofrechuk, P. , Biessmann, H. , Bryant, P.J. , Goodman, C.S. , 1991. The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell. 67, 853–868. [DOI] [PubMed] [Google Scholar]
- Marschang, P. , Brich, J. , Weeber, E.J. , Sweatt, J.D. , Shelton, J.M. , Richardson, J.A. , Hammer, R.E. , Herz, J. , 2004. Normal development and fertility of knockout mice lacking the tumor suppressor gene LRP1b suggest functional compensation by LRP1. Mol. Cell. Biol.. 24, 3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAvoy, S. , Ganapathiraju, S.C. , Ducharme-Smith, A.L. , Pritchett, J.R. , Kosari, F. , Perez, D.S. , Zhu, Y. , James, C.D. , Smith, D.I. , 2007. Non-random inactivation of large common fragile site genes in different cancers. Cytogenet. Genome Res.. 118, 260–269. [DOI] [PubMed] [Google Scholar]
- Murga, M. , Fernandez-Capetillo, O. , 2007. Genomic instability: on the birth and death of cancer. Clin. Transl. Oncol.. 9, 216–220. [DOI] [PubMed] [Google Scholar]
- Negrini, S. , Gorgoulis, V.G. , Halazonetis, T.D. , 2010. Genomic instability – an evolving hallmark of cancer. Nat. Rev. Mol. Cell. Biol.. 11, 220–228. [DOI] [PubMed] [Google Scholar]
- Newbury, D.F. , Monaco, A.P. , 2010. Genetic advances in the study of speech and language disorders. Neuron. 68, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell, P.C. , 1976. The clonal evolution of tumor cell populations. Science. 194, 23–28. [DOI] [PubMed] [Google Scholar]
- Pasi, C.E. , Dereli-Oz, A. , Negrini, S. , Friedli, M. , Fragola, G. , Lombardo, A. , Van Houwe, G. , Naldini, L. , Casola, S. , Testa, G. , 2011. Genomic instability in induced stem cells. Cell Death Differ.. 18, 745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas-Silva, M.D. , Weinberg, R.A. , 1997. The restriction point and control of cell proliferation. Curr. Opin. Cell Biol.. 9, 768–772. [DOI] [PubMed] [Google Scholar]
- Quelle, D.E. , Zindy, F. , Ashmun, R.A. , Sherr, C.J. , 1995. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 83, 993–1000. [DOI] [PubMed] [Google Scholar]
- Saldivar, J.C. , Shibata, H. , Huebner, K. , 2010. Pathology and biology associated with the fragile FHIT gene and gene product. J. Cell. Biochem.. 109, 858–865. [DOI] [PubMed] [Google Scholar]
- Sjoblom, T. , Jones, S. , Wood, L.D. , Parsons, D.W. , Lin, J. , Barber, T.D. , Mandelker, D. , Leary, R.J. , Ptak, J. , Silliman, N. , 2006. The consensus coding sequences of human breast and colorectal cancers. Science. 314, 268–274. [DOI] [PubMed] [Google Scholar]
- Smith, D.I. , McAvoy, S. , Zhu, Y. , Perez, D.S. , 2007. Large common fragile site genes and cancer. Semin. Cancer Biol.. 17, 31–41. [DOI] [PubMed] [Google Scholar]
- Smith, D.I. , Zhu, Y. , McAvoy, S. , Kuhn, R. , 2006. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett.. 232, 48–57. [DOI] [PubMed] [Google Scholar]
- Toledo, L.I. , Murga, M. , Fernandez-Capetillo, O. , 2011. Targeting ATR and Chk1 kinases for cancer treatment: a new model for old and new drugs. Mol. Onc.. (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantoulis, P.K. , Kotsinas, A. , Sfikakis, P.P. , Evangelou, K. , Sideridou, M. , Levy, B. , Mo, L. , Kittas, C. , Wu, X.R. , Papavassiliou, A.G. , Gorgoulis, V.G. , 2008. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 27, 3256–3264. [DOI] [PubMed] [Google Scholar]
- Uetani, N. , Kato, K. , Ogura, H. , Mizuno, K. , Kawano, K. , Mikoshiba, K. , Yakura, H. , Asano, M. , Iwakura, Y. , 2000. Impaired learning with enhanced hippocampal long-term potentiation in PTPdelta-deficient mice. EMBO J.. 19, 2775–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, A. , Hippo, Y. , Taniguchi, H. , Iwanari, H. , Yashiro, M. , Hirakawa, K. , Kodama, T. , Aburatani, H. , 2003. An opposing view on WWOX protein function as a tumor suppressor. Cancer Res.. 63, 8629–8633. [PubMed] [Google Scholar]