

Abstract
Focal adhesion kinase (FAK), a cytoplasmic tyrosine kinase and scaffold protein localized to focal adhesions, is uniquely positioned at the convergence point of integrin and receptor tyrosine kinase signal transduction pathways. FAK is overexpressed in many tumor cells, hence various inhibitors targeting its activity have been tested for anti‐tumor activity. However, the direct effects of these pharmacologic agents on the endothelial cells of the vasculature have not been examined. Using primary human umbilical vein endothelial cells (HUVEC), we characterized the effects of two FAK inhibitors, PF‐573,228 and FAK Inhibitor 14 on essential processes for angiogenesis, such as migration, proliferation, viability and endothelial cell tube formation. We observed that treatment with either FAK Inhibitor 14 or PF‐573,228 resulted in reduced HUVEC viability, migration and tube formation in response to vascular endothelial growth factor (VEGF). Furthermore, we found that PF‐573,228 had the added ability to induce apoptosis of endothelial cells within 36 h post‐drug administration even in the continued presence of VEGF stimulation. FAK inhibitors also resulted in modification of the actin cytoskeleton within HUVEC, with observed increased stress fiber formation in the presence of drug. Given that endothelial cells were sensitive to FAK inhibitors at concentrations well below those reported to inhibit tumor cell migration, we confirmed their ability to inhibit endothelial‐derived FAK autophosphorylation and FAK‐mediated phosphorylation of recombinant paxillin at these doses. Taken together, our data indicate that small molecule inhibitors of FAK are potent anti‐angiogenic agents and suggest their utility in combinatorial therapeutic approaches targeting tumor angiogenesis.
Keywords: Endothelial cell, Focal adhesion kinase, Anti-angiogenic, FAK, Tyrosine kinase inhibitor
Highlights
FAK is overexpressed in tumors thus FAK inhibitors are being developed as novel anti‐tumor drugs.
FAK is critical for angiogenesis, thus we hypothesized that FAK inhibitors are anti‐angiogenic.
FAK inhibitors reduced viability, motility and tube formation in primary human endothelial cells.
Endothelial cells were more sensitive to FAK inhibitors than tumor cells.
FAK inhibitors are potent anti‐angiogenics warranting their use in anti‐angiogenic therapies.
Abbreviations
- ANOVA
analysis of variance
- DMSO
dimethyl sulfoxide
- ECM
extracellular matrix
- FAK
focal adhesion kinase
- FI14
FAK Inhibitor 14
- GST
glutathione-S-transferase
- HBSS
Hank's buffered saline solution
- HUVEC
human umbilical vein endothelial cells
- IB
immunoblot
- IGF-1R
insulin-like growth factor receptor 1
- IP
immunoprecipitation
- LB
Luria Bertani
- PBS
phosphate-buffered saline
- PF-228
PF-573,228
- TBST
tris-buffered saline/Tween-20
- TRITC
tetramethylrhodamine B isothiocyanate
- VEGF
vascular endothelial growth factor
- VEGFR2
vascular endothelial growth factor receptor 2
1. Introduction
Angiogenesis, or the growth of new blood vessels arising from pre‐existing ones, is a complex process directed by growth factors, receptors, extracellular matrix (ECM)‐to‐cell and cell‐to‐cell interactions. Tumor‐associated angiogenesis is necessary for sustaining tumor growth beyond 1 mm3 (Gimbrone et al., 1972; Hanahan and Weinberg, 2000). Due to its central role in tumor growth, therapeutic targeting of angiogenesis has become a major focus in recent years. Although angiogenesis can be modulated by various growth factors, vascular endothelial growth factor (VEGF) has been shown to play a predominant role in tumor‐associated angiogenesis (Smith et al., 2010; Veikkola et al., 2000). Thus, numerous agents targeting VEGF ligand or its receptors (VEGFR) have been developed and tested as anti‐cancer therapies alone or in combination in various cancer types (Ellis and Hicklin, 2008). Currently, there are four anti‐angiogenic agents approved for clinical use and many more being investigated in clinical trials, however, it is clear that many patients do not initially respond to and others acquire resistance to these modalities (Ebos and Kerbel, 2011). Resistance to VEGF pathway inhibitors, can arise from either evasive resistance (e.g. where alternate angiogenic signaling pathways are invoked) or intrinsic resistance (e.g. where redundant angiogenic signaling exists) (Bergers and Hanahan, 2008). Given these observations and clinical challenges, other targets involved in angiogenesis need to be examined to realize the full benefits of anti‐angiogenic therapy.
Focal adhesion kinase (FAK) is a 125‐kDa non‐receptor tyrosine kinase, which acts as a scaffold at sites of cell attachment to the extracellular matrix (ECM) and is activated following binding of integrins to ECM or upon growth factor stimulation including that mediated by VEGF (Abedi and Zachary, 1997; Mitra and Schlaepfer, 2006). FAK has been implicated as an important modulator of angiogenesis, as transgenic mouse models have indicated that endothelial FAK expression and activity are essential for the formation of new blood vessel networks during embryonic development (Braren et al., 2006; Ilic et al., 2003; Shen et al., 2005). More recently, using a tissue‐restricted knockout mouse model, it was demonstrated that endothelial FAK was essential for tumor growth and tumor‐associated angiogenesis, as mice lacking endothelial‐specific FAK expression exhibited reduced tumor angiogenesis and hence reduced tumor growth in vivo (Tavora et al., 2010). FAK activity is also modulated following the activation of growth factor receptors including VEGFR2, which upon activation by VEGF ligand can recruit and activate Src kinase which subsequently phosphorylates focal adhesion kinase (FAK) at tyrosine 861 and modulates endothelial cell migration and survival (Abu‐Ghazaleh et al., 2001). In addition to its putative role in angiogenesis, altered FAK activity and expression have been directly linked to tumorigenesis and metastasis since interference with FAK signaling led to decreased metastasis in a variety of tumor models, including breast and lung cancer (Golubovskaya et al., 2009; Zhao and Guan, 2009).
Given that FAK has been shown to have aberrant activity and/or expression in many cancers [reviewed in (McLean et al., 2005)], it has been described as a “druggable” target. Hence, there has been a surge in the discovery and preclinical development of pharmacological inhibitors of FAK activity, such as NVP‐TAE‐226, PF‐562,271, PF‐573,228 and FAK Inhibitor 14 (also known as Y15) [reviewed in (Schultze and Fiedler, 2010; Schwock et al., 2010)]. To date the effectiveness of these inhibitors has predominantly been examined in cancer cell lines and murine tumor models, where FAK inhibitor treatment resulted in reductions in tumor growth and metastatic burden (Bagi et al., 2008; Beierle et al., 2008). However, little consideration has been given to the effect that these inhibitors may have on normal cells in the tumor microenvironment, such as endothelial cells. We thus investigated the direct effects of FAK inhibitors on various processes important to angiogenesis, namely endothelial cell viability, survival, migration and vessel formation. To this end, we examined the direct effects of two FAK inhibitors, PF‐573,228 (PF‐228) and FAK Inhibitor 14 (FI14) on primary human endothelial cells. We present results suggesting that both of these FAK inhibitors have direct potent anti‐angiogenic activities, and inhibit endothelial cell viability, migration and sprout formation along with the added ability to induce endothelial cell apoptosis in the case of PF‐228. Thus, their observed efficacy in tumor models may in part be a result of their ability to potently inhibit tumor‐associated angiogenesis.
2. Materials and methods
2.1. Reagents and cells
All chemical reagents were obtained from Sigma (Oakville, ON) or Fisher Scientific (Ottawa, ON) unless otherwise stated. The FAK inhibitors, PF‐573,228 (PF‐228) and FAK Inhibitor 14 (FI14), both from Tocris Bioscience (Ellisville, MO), were dissolved in dimethyl sulfoxide (DMSO) and then subsequently diluted to the indicated concentrations. Recombinant human vascular endothelial growth factor (VEGF) (rhVEGF165; R&D Systems, Minneapolis, MN) was reconstituted according to the manufacturer's instructions. Human umbilical vein endothelial cells (HUVEC; Cambrex/Lonza, Allendale, NJ) were cultured in endothelial cell growth media (Singlequot‐supplemented EGM2 media; Cambrex/Lonza) and used from passages 6–10. All cells were grown at 37 °C and 5% CO2.
2.2. Proliferation/viability assay
HUVEC were seeded at 5 × 103 cells/well in a 96‐well plate. The following day, cells were washed once with MCDB‐131 (Invitrogen, Burlington, ON) and then incubated in MCDB‐131 + 1% FBS containing either PF‐228 or FI14 at various concentrations in the presence of 50 ng/ml VEGF. Cells treated with equivalent volumes of DMSO were used as a vehicle control in these experiments. After 72 h, media was removed and replaced with MCDB‐131 + 1% FBS + 10% alamarBlue (AbD Serotec, Raleigh, NC). Plates were read (excitation 530 nm/emission 590 nm) using a Fluoroscan fluorescence plate reader (Thermo Scientific, Rockford, IL) 6 h post addition of alamarBlue.
2.3. Recombinant protein purification
Overnight cultures of glutathione‐S‐transferase (GST)‐tagged fusion protein were grown from DH5α bacteria in 3 mL of Luria Bertani (LB) media with 50 μg/mL ampicillin at 37 °C and diluted 1 in 10 next day. Diluted cultures were then grown for 1 h (at 37 °C) prior to being induced for 2 h (at 37 °C) by the addition of 1 mM isopropyl‐beta‐d‐thiogalactopyranoside and collected via centrifugation at 8000× g for 15 min. Bacterial pellets were lysed in RIPA lysis buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P‐40) with phosphatase inhibitors, sonicated and left on ice for 15 min. Lysates were cleared by centrifugation (15,000× g, 10 min) and inverted with glutathione sepharose beads (GE Healthcare Life Sciences, Baie d'Urfe, QC) for 30 min at room temperature. Beads were recovered by pulse centrifugation at maximum speed and washed 4× in NETN (20 mM Tris–HCl pH 8.0, 1 mM EDTA, 200 mM NaCl, 0.5% Nonidet P‐40) buffer before being used in other assays.
2.4. In vitro FAK kinase assay and immunoblots
FAK was immunoprecipitated (IP) by inverting 200 μg of total HUVEC lysate in RIPA lysis buffer with 2.5 μg/IP of anti‐FAK antibodies (BD Bioscience, Mississauga, ON), and 25 μl Protein A sepharose beads (GE Healthcare Life Sciences) for 2 h at 4 °C. Prior to washing 4× in NETN, approximately 1 μg of GST‐fusion paxillin protein was added to the respective reactions. In vitro kinase assays were then performed in the presence of γ‐P32‐ATP as previously described (Sabourin and Rudnicki, 1999), with the following modifications: the addition of 5 μM PF‐228, 5 μM FI14 or DMSO (vehicle control) 20 min prior to the initiation of the assay, and kinase reactions were incubated at 37 °C for 1 h. Kinase reactions were halted by the addition of 4× SDS sample buffer and resolved using 10% acrylamide gels and SDS‐polyacrylamide gel electrophoresis followed by transfer to PVDF membranes (Millipore, Billerica, MA). Subsequent exposure of the membranes to X‐ray film (Kodak, Toronto, ON) at −80 °C was used to visualize the radioactive signal from FAK kinase‐mediated phosphorylation events. The membrane was later probed with anti‐FAK or anti‐paxillin (both from BD Bioscience) primary antibodies overnight at 4 °C. After 3 washes in Tris‐buffered saline with 1% Tween‐20 (TBST), blots were incubated with horse radish peroxidise‐conjugated secondary antibody (Bio‐Rad, Mississauga, ON) for 1 h at room temperature, followed by 3 additional washes in TBST. Membranes were incubated with Western Lightning Chemiluminescent solution (Perkin–Elmer, Woodbridge, ON) and exposed to film (Kodak). Blots were stripped with 2× Re‐blot solution (Millipore) for 10 min at room temperature prior to re‐probing with additional antibodies.
2.5. Flow cytometric analyses of apoptosis and cell cycle
HUVEC (4 × 105) were seeded onto 60 mm dishes. The following day, cells were washed with HEPES‐buffered saline solution (HBSS) to remove non‐adherent cells and then cells were incubated with MCDB‐131 media containing 5% fetal bovine serum (unstimulated), or MCDB‐131 media with 5% fetal bovine serum supplemented with 50 ng/mL VEGF alone or in the presence of PF‐228 or FI14. Cells were incubated for an additional 48 h. Non‐adherent cells were harvested and pooled with trypsinized adherent cells which were then centrifuged (300× g), washed twice with phosphate‐buffered saline (PBS) and then resuspended in ice‐cold 70% ethanol. Cell suspensions were incubated at −20 °C for a minimum of 24 h. For analysis of the cell cycle status, cells were washed twice with PBS and resuspended in 500 μl of propidium iodide solution (48 μg/ml propidium iodide, 40 μg/ml RNase A in PBS) followed by a 30 min incubation at room temperature. Samples were then analyzed using a Coulter EPICS XL flow cytometer (Beckman‐Coulter, Mississauga, ON) on the FL2 channel. The percentage of apoptotic cells was calculated by examining cells with less then 2N DNA content using FCS Express flow cytometry analysis software (De Novo Software, Los Angeles, CA). The proportion of cells in G1 and G2/M was determined using ModFit LT (Verity Software House, Topsham, ME).
2.6. Scratch wound assay
HUVEC were seeded at 4 × 105 cells/well into a 6‐well plate. The following day, confluent monolayers were scratched to create a “wound” using a sterile plastic tool. Cells were washed with HBSS and incubated with Singlequot‐supplemented EGM2 growth media containing PF‐228, FI14 or DMSO as a control. Twelve images/well were acquired with a digital camera (Nikon, Mississauga, ON) at 0 h and 24 h time points using a 4× objective attached to an Eclipse TE2000‐U microscope (Nikon). Wound diameters in images were measured and percentage wound closure was calculated as follows: [(wound diameter at 24 h‐wound diameter at 0 h)/wound diameter at 0 h] × 100.
2.7. Immunofluorescence
HUVEC were seeded at 1 × 105 cells/well in a 6‐well dish containing sterile coverslips (Fisher Scientific). Cells were treated with varying concentrations of PF‐228 or FI14 or DMSO as the vehicle control. After 24 h, cells were fixed with 4% paraformaldehyde in PBS (20 min at 37 °C). Next cells were washed with PBS and permeabilized with 0.2% Triton X‐100 and 1% BSA in PBS (20 min at room temperature). Cells were washed with PBS and then incubated with tetramethylrhodamine B isothiocyanate (TRITC)‐labeled phalloidin (0.5 μg/ml; 30 min; Sigma). Cells were washed three times with PBS followed by incubation with 1 μg/ml bisBenzimide Hoechst 33258 (Sigma) in 1% BSA in PBS. Coverslips were mounted onto slides using fluorescent mounting medium (KPL Inc., Gaithersburg, MD). Images were acquired using a 63× objective on a Zeiss Observer Z1 microscope and AxioVision software (Carl Zeiss, Toronto, ON).
2.8. Endothelial cell sprouting assays
Tissue culture dishes (60 mm) were coated with renatured collagen I (PureCol; Advanced BioMatrix, San Diego, CA) to form fibrillar collagen gels as previously described (Addison et al., 2005). Briefly, cold acidified collagen was diluted to 1.5 mg/ml, neutralized using 10× PBS and 0.1 N NaOH to approximately pH 7.4, and evenly distributed on the plate surface. Plates were then incubated at 37 °C overnight to permit gel formation. Afterward, plates were washed with HBSS, and incubated in EGM2 for 2 h to equilibrate gels before cells were added. A total of 2 × 105 HUVEC were seeded onto the surface of each collagen I gel. The following day, cells were washed twice with HBSS and stimulated with EGM2 supplemented with 50 ng/ml VEGF, in the presence or absence (DMSO as vehicle control) of the two FAK inhibitors, PF‐228 and FI14 at various concentrations. The number of vessel sprouts per high power field was counted daily for 8 days. Fresh supplemented media containing VEGF and FAK inhibitors, was replaced every 48 h. On day 8, images were acquired with a Nikon digital camera attached to an Eclipse TE2000‐U microscope (Nikon) using a 4× objective.
2.9. Statistical analyses
All statistical analyses were performed using Prism 3.0 (GraphPad, La Jolla, CA).
3. Results
3.1. FAK inhibitors impair endothelial cell viability
The FAK inhibitors PF‐228 and FI14 had recently been shown to inhibit tumor growth in xenograft models in vivo (Golubovskaya et al., 2008; Hochwald et al., 2009; Slack‐Davis et al., 2007), however their direct effect on the tumor endothelium was not specifically addressed. We were thus interested in examining the direct anti‐angiogenic effects of these previously described FAK small molecule inhibitors on various endothelial cell processes important for angiogenesis. We tested the ability of each drug to inhibit viability of primary HUVEC, by exposing cells to various concentrations of FAK inhibitors or equivalent amounts of DMSO as a vehicle control for 72 h, at which time cell viability was assessed using alamarBlue assays. A dose‐dependent decrease in HUVEC viability was observed for both PF‐228 (Figure 1A) and FI14 (Figure 1B). In contrast to what had been observed in tumor cells, HUVEC were sensitive to these drugs at relatively low concentrations, with significant inhibition of cell viability at doses as low as 0.5 μM for PF‐228 and at 4 μM FI14. At the higher doses of 10 μM PF‐228 or 8–10 μM FI14 which were reported to have some proliferative inhibitory activity in the tumor cell studies (Golubovskaya et al., 2008; Slack‐Davis et al., 2007), endothelial cells were completely killed. These results suggest that, endothelial cells are more sensitive than tumor cells to FAK drugs at relatively low doses.
Figure 1.
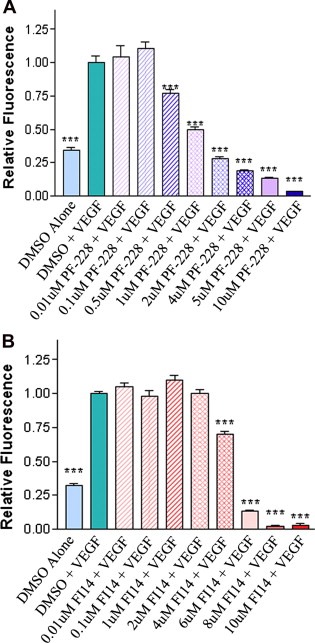
FAK inhibitors decrease number of viable endothelial cells in a dose‐dependent manner. HUVEC were incubated with various concentrations of either PF‐228 (A) or FI14 (B) in the presence of VEGF. Cells treated with vehicle (DMSO) were used as controls. Media containing alamarBlue was added 72 h post drug addition, to assess number of viable cells. Fluorescence was measured 6 h post‐alamarBlue addition. HUVEC treated with increasing concentrations of PF‐228 (A) or FI14 (B) showed reduced cell viability compared to DMSO controls. Experiments shown are representative of three or more experiments done in replicates of 8 wells per condition. Asterisks denote statistically significant differences (***p < 0.001) as determined by unpaired Student's t test.
3.2. Endothelial FAK kinase autophosphorylation and phosphorylation of target substrates is blocked by FAK inhibitors
Given the observed differences in the effective inhibitory concentration of FAK drugs on HUVEC viability compared to that previously reported in tumor cells, we wanted to ensure that FAK activity was blocked in endothelial cells by these lower doses of inhibitors, particularly since previous studies in tumor cells indicated that inhibition of FAK autophosphorylation did not occur until doses in excess of 8–10 μM (Golubovskaya et al., 2008; Slack‐Davis et al., 2007). We thus assessed the ability of FAK inhibitors to block endothelial‐derived FAK activity using in vitro kinase activity assays. Endothelial FAK was immunoprecipitated from HUVEC and was subsequently pre‐incubated with FAK inhibitors or vehicle control (DMSO) prior to incubation with radiolabeled ATP in the presence or absence of exogenous recombinant GST‐paxillin as a target substrate. Kinase reactions were incubated and proteins subsequently resolved by SDS‐PAGE and transferred to membranes. Membranes were exposed to film to develop the autoradiography signal from incorporated P32 in the phosphorylation reactions (Figure 2, autorad panels), and were then subsequently subjected to western blot analysis for total FAK and total recombinant paxillin to ensure equal loading (Figure 2, IB panels). FAK autophosphorylation was significantly inhibited by the presence of either FI14 or PF‐228 as compared to DMSO (Figure 2, top panel, lanes 2 and 3 compared to 1, and lanes 5 and 6 compared to 4) irrespective of the addition of exogenous paxillin [a known target of FAK (Bellis et al., 1995)] to the kinase reaction. Furthermore, FAK kinase activity against target substrates, in this case exogenously added recombinant paxillin, was also significantly reduced by the presence of either FI14 or PF‐228 (Figure 2, paxillin autorad panel, lanes 2 and 3 compared to 1). Equivalent levels of FAK and exogenously added paxillin in the kinase reactions were additionally confirmed by immunoblot analysis for each specific protein. Thus it would appear that the small molecule FAK inhibitors are able to effectively inhibit endothelial cell‐derived FAK autophosphorylation and phosphorylation of kinase targets at lower concentrations than previously reported for other cell types.
Figure 2.
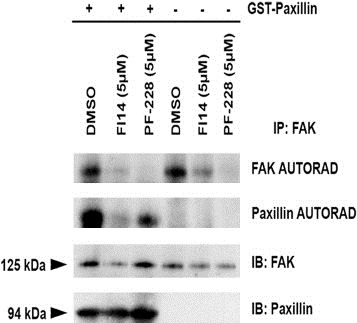
FAK inhibitors FI14 and PF‐228 inhibit FAK kinase activity in vitro. FAK was immunoprecipitated from HUVEC total protein lysates and in vitro kinase assays were performed as described in Section 2.4, in the presence or absence of exogenously added recombinant paxillin as a kinase target substrate. Membranes were exposed to film to reveal the autoradiographic signals for phospho‐FAK and phospho‐paxillin (indicated by autorad panels). Membranes were subsequently analyzed by western blot for total FAK and paxillin as a loading control (indicated by IB panels). The amount of FAK autophosphorylation and paxillin target substrate phosphorylation by FAK was reduced by the presence of FAK inhibitors compared to vehicle control.
3.3. The FAK inhibitor PF‐573,228 induces endothelial cell apoptosis
As our initial study assessed viable cell numbers, the reduction in cell viability we observed could be attributable to a decrease in proliferation or an increase in apoptosis. We thus measured apoptotic cells and the proportion of cells in various stages of the cell cycle by flow cytometric (FACS) analysis of propidium iodide stained cells. HUVEC were incubated with each FAK inhibitor at various concentrations in the presence of 50 ng/ml VEGF for 48 h, at which time cells were fixed, permeabilized and stained with propidium iodide for FACS analysis. We observed that exposure to PF‐228 led to an increase in the number of apoptotic HUVEC in a dose‐dependent manner as measured by the proportion of cells in the subG1 stage of the cell cycle, as compared to vehicle controls (Figure 3A). Interestingly, no increase in apoptosis was observed following treatment with FI14 at similar concentrations (Figure 3A). With respect to the proportion of cells in the G1 phase of the cell cycle, there was a trend for decreases in the G1 content in cells treated with 5 μM PF‐228 which was concomitant with the observed increases in apoptotic cells (Figure 3B). In contrast, no significant changes in the proportion of cells in G1 were observed following FI14 treatment (Figure 3B). We also examined the proportion of cells in the G2/M phase of the cell cycle, and observed dose‐dependent increases following treatment with PF‐228 and a slight trend for an increased proportion of cells in G2/M following FI14 treatment (Figure 3C). As the results suggested a possible inhibitor‐induced G2 arrest for both drugs, followed by induction of apoptosis in the case of PF‐228, we performed a time course analysis for HUVEC treated with VEGF in combination with either 5 μM PF‐228, 4 μM FI14 or vehicle control. When the percentage of apoptotic cells or those in each phase of the cell cycle were plotted as a function of time, we observed early increases in G2 and decreases in G1 for all three conditions, likely as a result of stimulation of cell proliferation and survival in response to VEGF treatment (Figure 3D–F). By 72 h, increases in apoptotic cells as a result of serum starvation were observed for vehicle control or FI14‐treated cells (Figure 3D & F). However, in comparison, HUVEC incubated with 5 μM PF‐228 showed a dramatic increase in the percentage of apoptotic cells and a concomitant decrease in the number of cells in the G2 phase of the cell cycle as early as 36 h post‐stimulation with drug (Figure 3E). Taken together, these results suggest that FI14 and PF‐228 induce marked G2 arrest, with subsequent induction of apoptosis occurring in PF‐228‐treated HUVEC, which in part, may account for the previously observed reduction in endothelial cell viability.
Figure 3.
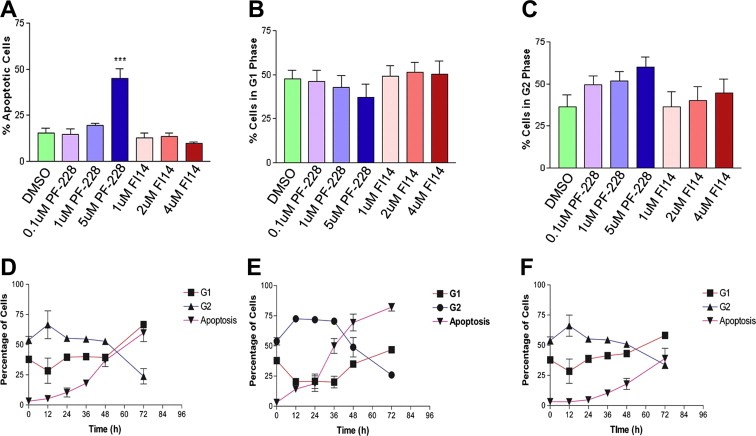
FAK inhibitors induce cell cycle arrest and apoptosis in treated endothelial cells. Seeded HUVEC were treated with DMSO as vehicle control or varying concentrations of PF‐228 or FI14 in the presence of 50 ng/mL VEGF for 48 h. Cells were then harvested and analyzed by flow cytometry as described in Section 2.5. The percentage of cells undergoing apoptosis (A) was quantified by examining cells with less than 2N DNA content following flow cytometric analysis on the FL2 channel. DNA content was used to calculate percentage of cells in G1 (B) and G2 (C) as described in Section 2.5. For the time course experiment, seeded HUVEC were incubated with VEGF (50 ng/ml) and DMSO as a vehicle control (D), or VEGF (50 ng/ml) in the presence of 5 μM PF‐228 (E) or 4 μM FI 14 (F). Cells were harvested at 12, 24, 36, 48 and 72 h post treatment, stained with propidium iodide and analyzed by flow cytometry as described in Section 2.5. Experiments shown are pooled results from three independent experiments performed in duplicate. Asterisks denote statistically significant differences (*p < 0.05, ***p < 0.001) as determined by unpaired Student's t test.
3.4. FAK inhibitors impair endothelial cell migration and sprout formation
As endothelial cell migration and sprout formation are requirements for angiogenesis, we also assessed the ability of the FAK inhibitors to impair these processes. For migration, HUVEC monolayers were scratched as described in Section 2.6, and following wounding, were treated with PF‐228, FI14 or DMSO as control. When comparing the images taken at the time of initial wounding (0 h) with those taken 24 h later, HUVEC treated with FAK inhibitors had migrated significantly less than DMSO vehicle control treated cells, as noted by the larger remaining wound width (Figure 4A). As expected, when percent wound closure was measured, a significant dose‐dependent inhibition of cell migration into the wound area was observed in FAK inhibitor‐treated cells (Figure 4B), with PF‐228 being a slightly more potent inhibitor of cell migration. We also examined the effects of the FAK inhibitors on the actin cytoskeleton, whose remodeling is known to be modulated by FAK during cell migration. HUVEC were thus treated with either PF‐228 or FI14 for 24 h and were fixed, permeabilized and stained with TRITC‐labeled phalloidin to bind polymerized actin (Figure 4C). Cells treated with FAK inhibitors exhibited increased actin stress fiber formation suggesting that inhibition of FAK activity prevented the dynamic remodeling of the actin cytoskeleton thus inhibiting migration.
Figure 4.
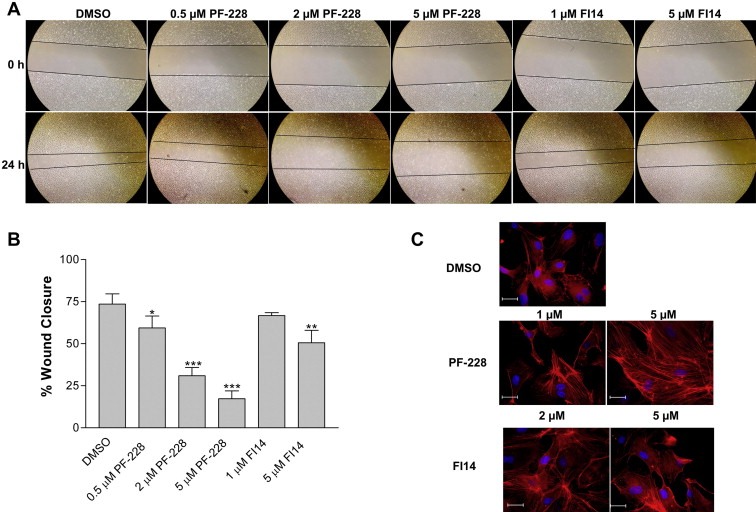
FAK inhibitors impede endothelial cell migration and alter the cellular actin cytoskeleton. HUVEC monolayers were wounded as described in Section 2.6, and cells were subsequently incubated with various concentrations of either PF‐228, FI14 or vehicle control (DMSO). Images were acquired at time of wounding (0 h) and 24 h post‐wounding (A, with lines indicating wound fronts) and percent wound closure was measured (B). Asterisks denote statistically significant differences (*p < 0.05, **p < 0.01, ***p < 0.001) as determined by unpaired Student's t test. HUVEC were grown on coverslips and treated with FAK inhibitors at the indicated concentrations for 24 h, prior to being stained as described in Section 2.7 with TRITC‐labeled phalloidin (red) to visualize actin filaments, and bisBenzimide Hoechst 33258 (blue) to visualize nuclei (C). More condensed phalloidin positive actin filaments were visualized in cells incubated with FAK inhibitors suggesting increased stress fiber formation. Experiments shown are representative of two independently performed experiments. Scale bar, 200 μM.
In addition to cell migration, cell organization into vessel structures is also an important feature of angiogenesis, hence we tested the ability of FAK inhibitors to impede this process. VEGF‐induced sprout formation in a collagen I sprouting assay was examined in the presence or absence of FAK inhibitors at various concentrations. In this assay, HUVEC sprout only under continued stimulation by VEGF, and over time, significant increases in the number of sprouts can be observed under these conditions (Figure 5A & B). Compared to VEGF plus vehicle control, treatment with either FAK inhibitor resulted in significant dose‐dependent decreases in the number of VEGF‐induced sprouts over time (Figure 5A & B). However, it should be noted that PF‐228 was far more effective in inhibiting endothelial cell sprout formation than FI14, and inhibited sprout formation at the lowest concentration used in the assay (0.1 μM) to a similar extent to that observed with the highest concentration used for FI14 (Figure 5A). Although we observed some endothelial cell sprouting of HUVEC treated with 1 μM PF‐228 at early time points, this quickly dwindled as cell viability decreased over time with continued drug administration (Figure 5B). The significant impact on cell viability was also observed at the highest concentration of PF‐228 used, as these cells never sprouted and subsequently died despite the presence and the continued administration of sprout‐inducing doses of VEGF (Figure 5B). These results clearly demonstrate the need for FAK activity in sprout formation by endothelial cells, and the potent efficacy of FAK inhibitors to block this process thereby essentially blocking angiogenesis.
Figure 5.
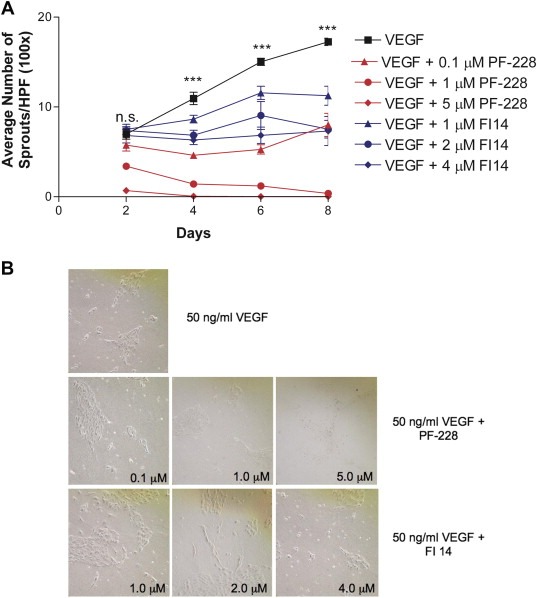
FAK inhibitors block HUVEC sprouting on collagen I gels. For assessing formation of sprouts, HUVEC were seeded onto collagen I gel‐coated plates and treated with 50 ng/ml VEGF in the absence (with added DMSO as a vehicle control) or presence of increasing concentrations of FAK inhibitors. HUVEC sprouting assay as a function of time (A). Sprouts were counted every 2 days in 10 random fields of view at 100× magnification. Mean number of sprouts per high powered field (HPF) and its associated standard error from duplicate dishes in each of two independent experiments are shown over time. Asterisks denote statistically significant differences (***p < 0.001) as determined by one‐way ANOVA analysis between cells treated with each of the drugs and vehicle. Sample images from day 8 of a representative experiment are shown (B). The effect of FAK inhibitors on sprout formation on collagen I was dose‐dependent.
4. Discussion
The two FAK inhibitors we used in this study, have been previously extensively characterized for their kinase specificity and their anti‐tumor activity (Golubovskaya et al., 2008; Hochwald et al., 2009; Slack‐Davis et al., 2007), however these studies did not evaluate their direct effects on endothelial cells or angiogenesis. In our present study, we have demonstrated the FAK inhibitors PF‐228 and FI14 potently inhibit a variety of processes in endothelial cells that are essential for angiogenesis, hence pharmacological inhibition of FAK activity is an extremely potent anti‐angiogenic therapeutic strategy. In endothelial cells, we observed that both FAK inhibitors impaired VEGF‐induced proliferation in a dose‐dependent manner. In its initial characterization in tumor cells, PF‐228 did not inhibit tumor cell growth until the highest concentrations used in that study (10 μM) which the authors attributed to potential off‐target effects, as at that concentration there was also some inhibition of the cyclin‐dependent kinases (CDK) 1 and 7 (Slack‐Davis et al., 2007). Interestingly in our study, the viability of VEGF‐stimulated HUVEC became compromised at doses of PF‐228 as low as 0.5 μM, which although it is still ∼2‐fold higher than the reported IC50 for inhibition of FAK autophosphorylation in tumor cells by this drug, is 20 times lower than that at which tumor cell viability was impaired, suggesting that endothelial cells are significantly more sensitive to FAK inhibition. Similarly, FI14 was previously shown to inhibit tumor cell growth at approximately 10 μM (Golubovskaya et al., 2008; Hochwald et al., 2009), however HUVEC viability was decreased by treatment at half this concentration (4–6 μM) FI14. The reductions in FAK autophosphorylation/activity in the presence of both compounds observed in the kinase assay also support the notion that endothelial FAK activity is significantly impaired even at these lower concentrations of drug.
Unlike what has been reported in tumor cells, we also observed that HUVEC incubated with increasing concentrations of PF‐228 accumulated in G2/M phase and subsequently underwent apoptosis. Similarly for HUVEC treated with FI14, there was a tendency for cells to accumulate in G2/M. These observations suggest that preventing FAK activity seriously perturbs the cell cycle, at least in primary endothelial cells. Although there have been no prior reports of the ability of these drugs to induce G2/M arrests or apoptosis in treated tumor cells, tumor cells are less dependent on attachment to substrate, while endothelial cells are critically dependent on cell attachment to a substratum (Meredith et al., 1993). Hence, it is highly likely that inhibition of FAK activity by these drugs in endothelial cells results in failure to convey appropriate cell attachment signals, and hence they undergo cell death by anoikis. Interestingly, PF‐228 induced apoptosis of endothelial cells, while FI14 only resulted in an apparent cell cycle arrest. As the kinase specificities of these two drugs differ in the respect that PF‐228 also effectively inhibits the kinase activity of the closely related FAK family member Pyk2, while FI14 does not target Pyk2 (Golubovskaya et al., 2008; Slack‐Davis et al., 2007), it is tempting to hypothesize that it is the blockade of Pyk2 by PF‐228 that promotes endothelial cell apoptosis. This is supported by recent studies in transgenic animals which have suggested that endothelial expressed Pyk2 can compensate for FAK in animals with vascular targeted FAK deletions, and thus Pyk2 activity may also compensate for FAK blockade in the presence of FI14 in endothelial cells resulting in the slightly reduced efficacy of this drug as compared to PF‐228 observed in our studies.
Treatment of HUVEC with either PF‐228 or FI14 also significantly reduced endothelial cell migration and sprout formation, key processes in angiogenesis. Our results corroborate previous work demonstrating a reduction in haptotactic (and chemotactic) migration in tumor cell lines treated with PF‐228 (Slack‐Davis et al., 2007). However, again HUVEC were significantly more sensitive to FAK inhibition than were tumor cells, as endothelial cell migration was impaired by concentrations of PF‐228 as low as 0.5 μM. With respect to FI14, the experiments described herein are the first to show an effect of this drug on cell migration, as previous studies had only observed defects in tumor cell adhesion and attachment (Golubovskaya et al., 2008; Hochwald et al., 2009). We also noted increases in the number of actin stress fibers in endothelial cells treated with FAK inhibitors. Although this phenotype was not examined in previous studies that treated tumor cells with these drugs, the aberrant actin formations we observed in FAK inhibitor‐treated HUVEC are similar to those previously observed in FAK knockout cells or in endothelial cells lacking FAK expression (Ilic et al., 1995; Tavora et al., 2010). Taken together, these data suggest that pharmacological inhibition of FAK impairs its ability to dynamically modulate the actin cytoskeleton and facilitate migration and sprout formation in endothelial cells, processes absolutely required for angiogenesis to occur.
In support of our findings, preclinical studies with a different FAK inhibitor, PF‐562,271, in murine tumor xenograft models demonstrated that tumor burden was decreased with an accompanying reduction in microvascular density following treatment with this drug (Roberts et al., 2008). Although the authors speculated on the possible anti‐angiogenic activity of this drug, they did not provide any direct evidence of this. As the FAK inhibitor‐treated tumors were smaller in size compared to control treated tumors to begin with, the reduced vasculature could have simply been a general result of reduced tumor burden. It was also demonstrated that Matrigel‐induced tube formation and neovascularization in a xenograft transplantation model were inhibited by the drug NVP‐TAE‐226, a dual‐specificity inhibitor that targets both FAK and insulin‐like growth factor 1 receptor (IGF‐1R) (Schultze et al., 2010). The fact that this inhibitor also targets IGF‐1R however, complicates the interpretation of the direct role of FAK inhibition in the measured angiogenic phenotypes. Like FAK, IGF‐1R is abundant in endothelial cells and is a potent mediator of the IGF‐1‐induced angiogenic effects (Nakao‐Hayashi et al., 1992; Nitert et al., 2005). Thus, the effects described by Schultze et al. (2010), could have resulted from inhibition of FAK or IGF‐1R or both, as the drug‐specific inhibition of the target kinases were not analyzed in their study (Schultze et al., 2010). Our work is thus the first to clearly demonstrate that human endothelial cells themselves are extremely sensitive to FAK inhibitors used as single modalities and supports the notion that the ability of FAK inhibitors to effectively impair tumor growth in vivo may in part be due to their ability to function as potent anti‐angiogenic agents. Our results also suggest that the effects of potential anti‐tumor agents, like FAK inhibitors, on normal cells, such as endothelial cells, should be considered in the development and characterization of these novel agents for treatment of pathological diseases.
Single targeted agent therapies appear somewhat ineffective in clinical settings, thus a move toward multi‐targeted approaches for anti‐tumor therapies is required. Given its ability to impair tumor invasion, and our demonstrated ability to significantly impair angiogenic processes in human endothelial cells, combination of FAK inhibitors with other pharmacologic agents will likely lead to enhanced therapeutic efficacy. An example of such a strategy suggested that the FAK inhibitor PF‐562,271 when combined with sunitinib (SU11248), an inhibitor of multiple angiogenic receptor tyrosine kinases, may be more beneficial than sunitinib alone (Bagi et al., 2009). Oddly, this particular study did not examine the effects of PF‐562,271 alone, and hence even though they did examine vessel flow in their study, direct effects of PF‐562,271 on this parameter could not be ascertained. Further studies with specific receptor tyrosine kinase inhibitors or other anti‐cancer drugs are warranted to pursue this hypothesis. Furthermore, given that our previous work demonstrated diminished efficacy of anti‐angiogenic compounds in the presence of different tumor‐associated ECM proteins such as collagen or fibronectin (Addison et al., 2005; Delaney et al., 2006), the use of FAK inhibitors to block ECM‐integrin signals in combination with other anti‐angiogenic compounds may be useful to overcome this potential mechanism of resistance and increase the efficacy of current anti‐angiogenic drugs in a patient setting.
5. Conclusion
In summary, we have demonstrated that the angiogenic activity of primary endothelial cells can be significantly inhibited following administration of the FAK tyrosine kinase inhibitors PF‐228 and FI14. Endothelial cells appear to be more sensitive than tumor cells to these inhibitors as significantly lower concentrations of inhibitors showed substantial deleterious effects on endothelial cell viability, migration and tube formation. Thus, future studies of FAK tyrosine kinase inhibitors, alone or in combination with other anti‐tumor or anti‐angiogenic drugs, in preclinical models are warranted. Moreover, the effects of these drugs on multiple cellular compartments should be investigated further given the demonstrated central role of FAK in normal and tumor cells.
Acknowledgments
The authors wish to thank Grant Howe for critically reviewing the paper and Huijun Zhao for expert technical assistance. Funding for this research was provided through a Canadian Institutes of Health Research grant (MOP#84369) awarded to C.L. Addison. J.L. Quizi was the recipient of a Doctoral Fellowship from the Canadian Breast Cancer Foundation.
Cabrita Miguel A., Jones Laura M., Quizi Jennifer L., Sabourin Luc A., McKay Bruce C. and Addison Christina L., (2011), Focal adhesion kinase inhibitors are potent anti‐angiogenic agents, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.10.004.
References
- Abedi, H. , Zachary, I. , 1997. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. The Journal of Biological Chemistry. 272, 15442–15451. [DOI] [PubMed] [Google Scholar]
- Abu-Ghazaleh, R. , Kabir, J. , Jia, H. , Lobo, M. , Zachary, I. , 2001. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. The Biochemical Journal. 360, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addison, C.L. , Nor, J.E. , Zhao, H. , Linn, S.A. , Polverini, P.J. , Delaney, C.E. , 2005. The response of VEGF-stimulated endothelial cells to angiostatic molecules is substrate-dependent. BMC Cell Biology. 6, 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagi, C.M. , Christensen, J. , Cohen, D.P. , Roberts, W.G. , Wilkie, D. , Swanson, T. , Tuthill, T. , Andresen, C.J. , 2009. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biology & Therapy. 8, 856–865. [DOI] [PubMed] [Google Scholar]
- Bagi, C.M. , Roberts, G.W. , Andresen, C.J. , 2008. Dual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors: implications for bone metastases. Cancer. 112, 2313–2321. [DOI] [PubMed] [Google Scholar]
- Beierle, E.A. , Trujillo, A. , Nagaram, A. , Golubovskaya, V.M. , Cance, W.G. , Kurenova, E.V. , 2008. TAE226 inhibits human neuroblastoma cell survival. Cancer Investigation. 26, 145–151. [DOI] [PubMed] [Google Scholar]
- Bellis, S.L. , Miller, J.T. , Turner, C.E. , 1995. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. Journal of Biological Chemistry. 270, 17437–17441. [DOI] [PubMed] [Google Scholar]
- Bergers, G. , Hanahan, D. , 2008. Modes of resistance to anti-angiogenic therapy. Nature Reviews. 8, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braren, R. , Hu, H. , Kim, Y.H. , Beggs, H.E. , Reichardt, L.F. , Wang, R. , 2006. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. The Journal of Cell Biology. 172, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney, C.E. , Weagant, B.T. , Addison, C.L. , 2006. The inhibitory effects of endostatin on endothelial cells are modulated by extracellular matrix. Experimental Cell Research. 312, 2476–2489. [DOI] [PubMed] [Google Scholar]
- Ebos, J.M. , Kerbel, R.S. , 2011. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nature Reviews Clinical Oncology. 8, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, L.M. , Hicklin, D.J. , 2008. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nature Reviews. 8, 579–591. [DOI] [PubMed] [Google Scholar]
- Gimbrone, M.A. , Leapman, S.B. , Cotran, R.S. , Folkman, J. , 1972. Tumor dormancy in vivo by prevention of neovascularization. The Journal of Experimental Medicine. 136, 261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya, V.M. , Nyberg, C. , Zheng, M. , Kweh, F. , Magis, A. , Ostrov, D. , Cance, W.G. , 2008. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. Journal of Medicinal Chemistry. 51, 7405–7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya, V.M. , Zheng, M. , Zhang, L. , Li, J.-L. , Cance, W.G. , 2009. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer. 9, 280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hochwald, S.N. , Nyberg, C. , Zheng, M. , Zheng, D. , Wood, C. , Massoll, N.A. , Magis, A. , Ostrov, D. , Cance, W.G. , Golubovskaya, V.M. , 2009. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 8, 2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic, D. , Furuta, Y. , Kanazawa, S. , Takeda, N. , Sobue, K. , Nakatsuji, N. , Nomura, S. , Fujimoto, J. , Okada, M. , Yamamoto, T. , 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377, 539–544. [DOI] [PubMed] [Google Scholar]
- Ilic, D. , Kovacic, B. , McDonagh, S. , Jin, F. , Baumbusch, C. , Gardner, D.G. , Damsky, C.H. , 2003. Focal adhesion kinase is required for blood vessel morphogenesis. Circulation Research. 92, 300–307. [DOI] [PubMed] [Google Scholar]
- McLean, G.W. , Carragher, N.O. , Avizienyte, E. , Evans, J. , Brunton, V.G. , Frame, M.C. , 2005. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nature Reviews Cancer. 5, 505–515. [DOI] [PubMed] [Google Scholar]
- Meredith, J.E. , Fazeli, B. , Schwartz, M.A. , 1993. The extracellular matrix as a cell survival factor. Molecular Biology of the Cell. 4, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra, S.K. , Schlaepfer, D.D. , 2006. Integrin-regulated FAK-Src signaling in normal and cancer cells. Current Opinion in Cell Biology. 18, 516–523. [DOI] [PubMed] [Google Scholar]
- Nakao-Hayashi, J. , Ito, H. , Kanayasu, T. , Morita, I. , Murota, S. , 1992. Stimulatory effects of insulin and insulin-like growth factor I on migration and tube formation by vascular endothelial cells. Atherosclerosis. 92, 141–149. [DOI] [PubMed] [Google Scholar]
- Nitert, M.D. , Chisalita, S.I. , Olsson, K. , Bornfeldt, K.E. , Arnqvist, H.J. , 2005. IGF-I/insulin hybrid receptors in human endothelial cells. Molecular and Cellular Endocrinology. 229, 31–37. [DOI] [PubMed] [Google Scholar]
- Roberts, W.G. , Ung, E. , Whalen, P. , Cooper, B. , Hulford, C. , Autry, C. , Richter, D. , Emerson, E. , Lin, J. , Kath, J. , Coleman, K. , Yao, L. , Martinez-Alsina, L. , Lorenzen, M. , Berliner, M. , Luzzio, M. , Patel, N. , Schmitt, E. , LaGreca, S. , Jani, J. , Wessel, M. , Marr, E. , Griffor, M. , Vajdos, F. , 2008. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Research. 68, 1935–1944. [DOI] [PubMed] [Google Scholar]
- Sabourin, L.A. , Rudnicki, M.A. , 1999. Induction of apoptosis by SLK, a Ste20-related kinase. Oncogene. 18, 7566–7575. [DOI] [PubMed] [Google Scholar]
- Schultze, A. , Decker, S. , Otten, J. , Horst, A.K. , Vohwinkel, G. , Schuch, G. , Bokemeyer, C. , Loges, S. , Fiedler, W. , 2010. TAE226-mediated inhibition of focal adhesion kinase interferes with tumor angiogenesis and vasculogenesis. Investigational New Drugs. 28, 825–833. [DOI] [PubMed] [Google Scholar]
- Schultze, A. , Fiedler, W. , 2010. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opinion on Investigational Drugs. 19, 777–788. [DOI] [PubMed] [Google Scholar]
- Schwock, J. , Dhani, N. , Hedley, D.W. , 2010. Targeting focal adhesion kinase signaling in tumor growth and metastasis. Expert Opinion on Therapeutic Targets. 14, 77–94. [DOI] [PubMed] [Google Scholar]
- Shen, T.-L. , Park, A.Y.J. , Alcaraz, A. , Peng, X. , Jang, I. , Koni, P. , Flavell, R.A. , Gu, H. , Guan, J.-L. , 2005. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. The Journal of Cell Biology. 169, 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis, J.K. , Martin, K.H. , Tilghman, R.W. , Iwanicki, M. , Ung, E.J. , Autry, C. , Luzzio, M.J. , Cooper, B. , Kath, J.C. , Roberts, W.G. , Parsons, J.T. , 2007. Cellular characterization of a novel focal adhesion kinase inhibitor. The Journal of Biological Chemistry. 282, 14845–14852. [DOI] [PubMed] [Google Scholar]
- Smith, N.R. , Baker, D. , James, N.H. , Ratcliffe, K. , Jenkins, M. , Ashton, S.E. , Sproat, G. , Swann, R. , Gray, N. , Ryan, A. , Jurgensmeier, J.M. , Womack, C. , 2010. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clinical Cancer Research. 16, 3548–3561. [DOI] [PubMed] [Google Scholar]
- Tavora, B. , Batista, S. , Reynolds, L.E. , Jadeja, S. , Robinson, S. , Kostourou, V. , Hart, I. , Fruttiger, M. , Parsons, M. , Hodivala-Dilke, K.M. , 2010. Endothelial FAK is required for tumour angiogenesis. EMBO Molecular Medicine. 2, 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veikkola, T. , Karkkainen, M. , Claesson-Welsh, L. , Alitalo, K. , 2000. Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Research. 60, 203–212. [PubMed] [Google Scholar]
- Zhao, J. , Guan, J.-L. , 2009. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Reviews. 28, 35–49. [DOI] [PubMed] [Google Scholar]